Abetalipoproteinemia
Apolipoproteins B
Hypobetalipoproteinemias
Hypolipoproteinemias
Apolipoproteins
Lipoproteins, HDL
Lipoproteins
Lipoproteins, LDL
Carrier Proteins
Lipoproteins, VLDL
Motor Skills Disorders
Hyperlipoproteinemia Type III
Handwriting
Encyclopedias as Topic
Liver-specific inactivation of the abetalipoproteinemia gene completely abrogates very low density lipoprotein/low density lipoprotein production in a viable conditional knockout mouse. (1/80)
Conventional knockout of the microsomal triglyceride transfer protein large subunit (lMTP) gene is embryonic lethal in the homozygous state in mice. We have produced a conditional lMTP knockout mouse by inserting loxP sequences flanking exons 5 and 6 by gene targeting. Homozygous floxed mice were born live with normal plasma lipids. Intravenous injection of an adenovirus harboring Cre recombinase (AdCre1) produced deletion of exons 5 and 6 and disappearance of lMTP mRNA and immunoreactive protein in a liver-specific manner. There was also disappearance of plasma apolipoprotein (apo) B-100 and marked reduction in apoB-48 levels. Wild-type mice showed no response, and heterozygous mice, an intermediate response, to AdCre1. Wild-type mice doubled their plasma cholesterol level following a high cholesterol diet. This hypercholesterolemia was abolished in AdCre1-treated lMTP-/- mice, the result of a complete absence of very low/intermediate/low density lipoproteins and a slight reduction in high density lipoprotein. Heterozygous mice showed an intermediate lipoprotein phenotype. The rate of accumulation of plasma triglyceride following Triton WR1339 treatment in lMTP-/- mice was <10% that in wild-type animals, indicating a failure of triglyceride-rich lipoprotein production. Pulse-chase experiments using hepatocytes isolated from wild-type and lMTP-/- mice revealed a failure of apoB secretion in lMTP-/- animals. Therefore, the liver-specific inactivation of the lMTP gene completely abrogates apoB-100 and very low/intermediate/low density lipoprotein production. These conditional knockout mice are a useful in vivo model for studying the role of MTP in apoB biosynthesis and the biogenesis of apoB-containing lipoproteins. (+info)Abetalipoproteinaemia. A case report with pathological studies. (2/80)
The clinical and pathological features of a case of abetalipoproteinaemia in a 38-year-old patient are described in detail. A feature not previously recorded was a marked reduction in the velocity of ocular horizontal saccadic movements. Pathological studies revealed an active chronic demyelinating process. The patient showed no response to large doses of vitamin E. The rationale for this therapy, and the possible reasons for its failure are discussed. (+info)Abetalipoproteinemia caused by maternal isodisomy of chromosome 4q containing an intron 9 splice acceptor mutation in the microsomal triglyceride transfer protein gene. (3/80)
Uniparental disomy (UPD), a rare inheritance of 2 copies of a single chromosome homolog or a region of a chromosome from one parent, can result in various autosomal recessive diseases. Abetalipoproteinemia (ABL) is a rare autosomal recessive deficiency of apoB-containing lipoproteins caused by a microsomal triglyceride transfer protein (MTP) deficiency. In this study, we describe a patient with ABL inherited as a homozygous intron 9 splice acceptor G(-1)-to-A mutation of the transfer protein gene. This mutation alters the splicing of the mRNA, resulting in a 36 amino acids, in-frame deletion of sequence encoded by exon 10. We analyzed chromosome 4, including MTP gene (4q22-24), using short tandem repeat markers. The proband has only his mother's genes in chromosome 4q spanning a 150-centimorgan region; ie, segmental maternal isodisomy 4q21-35, probably due to mitotic recombination. Nonpaternity between the proband and his father was excluded using 6 polymorphic markers from different chromosomes (paternity probability, 0.999). Maternal isodisomy (maternal UPD 4q) was the basis for homozygosity of the MTP gene mutation in this patient. (+info)Progress towards understanding the role of microsomal triglyceride transfer protein in apolipoprotein-B lipoprotein assembly. (4/80)
The microsomal triglyceride transfer protein (MTP) is necessary for the proper assembly of the apolipoprotein B containing lipoproteins, very low density lipoprotein and chylomicrons. Recent research has significantly advanced our understanding of the role of MTP in these pathways at the molecular and cellular level. Biochemical studies suggest that initiation of lipidation of the nascent apolipoprotein B polypeptide may occur through a direct association with MTP. This early lipidation may be required to allow the nascent polypeptide to fold properly and therefore avoid ubiquitination and degradation. Concerning the addition of core neutral lipids in the later stages of lipoprotein assembly, cell culture studies show that MTP lipid transfer activity is not required for this to occur for apolipoprotein B-100 containing lipoproteins. Likewise, MTP does not appear to directly mediate addition of core neutral lipid to nascent apoB-48 particles. However, new data indicate that MTP is required to produce triglyceride rich droplets in the smooth endoplasmic reticulum which may supply the core lipids for conversion of nascent, dense apoB-48 particles to mature VLDL. In addition, assembly of dense apolipoprotein B-48 containing lipoproteins has been observed in mouse liver in the absence of MTP. As a result of these new data, an updated model for the role of MTP in lipoprotein assembly is proposed. (+info)Novel mutations in the microsomal triglyceride transfer protein gene causing abetalipoproteinemia. (5/80)
Abetalipoproteinemia (ABL) is an inherited disease characterized by the virtual absence of apolipoprotein B (apoB)-containing lipoproteins from plasma. Only limited numbers of families have been screened for mutations in the microsomal triglyceride transfer protein (MTP) gene. To clarify the genetic basis of clinical diversity of ABL, mutations of the MTP gene have been screened in 4 unrelated patients with ABL. Three novel mutations have been identified: a frameshift mutation caused by a single adenine deletion at position 1389 of the cDNA, and a missense mutation, Asn780Tyr, each in homozygous forms; and a splice site mutation, 2218-2A-->G, in a compound heterozygous form. The frameshift and splice site mutations are predicted to encode truncated forms of MTP. When transiently expressed in Cos-1 cells, the Asn780Tyr mutant MTP bound protein disulfide isomerase (PDI) but displayed negligible MTP activity. It is of interest that the patient having the Asn780Tyr mutation, a 27-year-old male, has none of the manifestations characteristic of classic ABL even though his plasma apoB and vitamin E were virtually undetectable. These results indicated that defects of the MTP gene are the proximal cause of ABL. (+info)Familial defective apolipoprotein B-100: a lesson from homozygous and heterozygous patients. (6/80)
Familial defective apolipoprotein B-100 (FDB) is a genetic disorder caused by a substitution of glutamine for arginine at residue 3500 of the apolipoprotein B-100 molecule. We have identified 23 heterozygotes and one homozygote for FDB (frequency 1:20) in a group of 510 patients with hypercholesterolemia. Mean age of the patients (18 females and 6 males) was 46 years. The diagnosis of FDB was based on point mutation PCR analysis of exon 26 of the apo B gene. Plasma lipids in heterozygous patients were: total cholesterol 8.76+/-1.2 mmol/l, triglycerides 1.42+/-0.5 mmol/l, HDL-cholesterol 1.43+/-0.3 mmol/l, LDL-cholesterol 6.69+/-1.2 mmol/l, apoB 1.69+/-0.4 g/l, Lp(a) 0.26+/-0.2 g/l. The most frequent apoE genotype was 3/3 (19 patients), apoE 3/4 genotype was found in 3 patients and one person had apoE 2/3. Xanthelasma palpebrarum was present in 4 patients and tendon xanthomas in 3 patients including the homozygote. Premature manifestation of coronary heart disease was revealed in 3 patients. Sixteen patients were treated with statins, a combination of statin and resin was used in 2 patients (including the homozygote), whereas six patients were treated with the diet only. We conclude that although the plasma lipid levels of total and LDL cholesterol in FDB patients are lower than in patients with familial hypercholesterolemia, the patients with FDB suffer from premature atherosclerosis. The therapeutic approach to FDB individuals and patients with familial hypercholesterolemia is very similar. (+info)A study of the abnormal lipoproteins in abetalipoproteinemia. (7/80)
The serum lipoproteins of five patients with abetalipoproteinemia (ABL) were separated by ultracentrifugation and then analyzed either intact or after delipidation. In accord with previous findings, all of the patients lacked serum particles with the characteristics of normal low-density lipoproteins (LDL) and of the LDL apoprotein as assessed by immunochemical methods. Each patient exhibited on every examination an abnormal particle, "LDL", which had the flotational properties of LDL, the polypeptide makeup of high-density lipoproteins HDL, the spectral and morphological characteristics of neither LDL nor HDL, and a relatively low content of cholesteryl esters. The HDL were abnormal in having a marked decrease in their total plasma content, an altered proportion of the subclasses HDL2 and HDL3, and a peculiar polypeptide distribution, comprising both normal and additional components, usually not seen in normal controls. The patients also exhibited a decrease of plasma lecithin-cholesterol acyl transferase (LCAT) activity which probably accounted for the low content of cholesteryl esters in both "LDL" and HDL, and in turn for the unusual appearance of "LDL" on electron microscopy. It is concluded that ABL is a disorder affecting all serum lipoprotein classes. Whether the abetalipoproteinemia previously described and noted in the current studies is related to or independent of the abnormalities observed in the other lipoproteins was not established. How the deficiency of LCAT activity, observed in all patients studied, contributed to some of the observed structural lipoprotein abnormalities also remained undetermined. (+info)Measurement of human high density lipoprotein apolipoprotein A-1 in serum by radioimmunoassay. (8/80)
A sensitive and specific double antibody radioimmunoassay for the major apolipoprotein (apo A-I) of human serum high density lipoprotein (HDL) was developed. Initial studies indicated that direct measurements of apo A-I concentration in whole untreated sera or isolated high density lipoprotein fractions yielded variable results, which were lower than those obtained in the corresponding samples which had been subjected to delipidation. Subsequently, it was observed that heating diluted sera or HDL for 3 hr at 52 degrees C prior to assay resulted in maximal increases in apo A-I immunoreactivity to levels comparable to those found in the delipidated specimens. This simple procedure permitted multiple sera to be assayed efficiently with full recovery of apo A-I. (+info)Abetalipoproteinemia is a rare inherited genetic disorder that affects the way the body absorbs and metabolizes fats and fat-soluble vitamins. It is caused by mutations in the genes responsible for producing proteins involved in the formation and transport of beta-lipoproteins, which are necessary for the absorption of dietary fats and cholesterol from the intestines.
Individuals with abetalipoproteinemia are unable to produce adequate levels of these lipoproteins, leading to a deficiency in fat-soluble vitamins (A, D, E, and K) and an accumulation of fats in the intestines. This results in various symptoms such as steatorrhea (fatty, foul-smelling stools), malabsorption, diarrhea, failure to thrive, and neurological issues due to vitamin E deficiency.
The disorder is typically diagnosed in infancy or early childhood and requires lifelong dietary management, including a low-fat diet and supplementation with fat-soluble vitamins. Early intervention can help prevent the progression of neurological symptoms and improve overall prognosis.
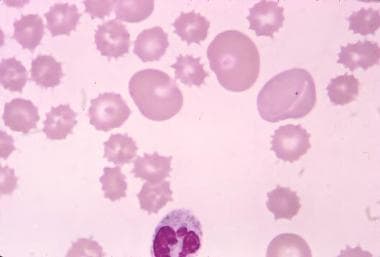
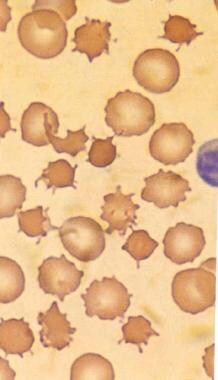
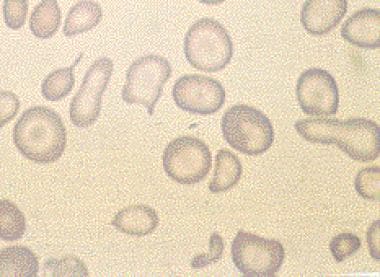
Acanthocytes are irregularly shaped red blood cells that have thorny or spiculated projections on their surface. These abnormal red blood cells are often seen in various medical conditions, including abetalipoproteinemia, malabsorption syndromes, liver diseases, and neuroacanthocytosis. The presence of acanthocytes can indicate abnormalities in lipid metabolism or membrane structure, which can lead to hemolysis and anemia. A blood film or smear is typically used to identify acanthocytes under a microscope.
Apolipoprotein B (ApoB) is a type of protein that plays a crucial role in the metabolism of lipids, particularly low-density lipoprotein (LDL) or "bad" cholesterol. ApoB is a component of LDL particles and serves as a ligand for the LDL receptor, which is responsible for the clearance of LDL from the bloodstream.
There are two main forms of ApoB: ApoB-100 and ApoB-48. ApoB-100 is found in LDL particles, very low-density lipoprotein (VLDL) particles, and chylomicrons, while ApoB-48 is only found in chylomicrons, which are produced in the intestines and responsible for transporting dietary lipids.
Elevated levels of ApoB are associated with an increased risk of cardiovascular disease (CVD), as they indicate a higher concentration of LDL particles in the bloodstream. Therefore, measuring ApoB levels can provide additional information about CVD risk beyond traditional lipid profile tests that only measure total cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides.
Hypobetalipoproteinemias are a group of genetic disorders characterized by low levels of betalipoproteins, including low-density lipoprotein (LDL) and/or apolipoprotein B (apoB), in the blood. These conditions can lead to decreased absorption and transportation of dietary fats and fat-soluble vitamins, such as vitamin E and A.
There are two main types of hypobetalipoproteinemias:
1. Type I (also known as Abetalipoproteinemia): This is a rare autosomal recessive disorder caused by mutations in the microsomal triglyceride transfer protein (MTTP) gene. It results in almost undetectable levels of LDL, apoB, and chylomicrons in the blood. Symptoms typically appear in infancy or early childhood and include fat malabsorption, steatorrhea (fatty stools), and failure to thrive. Additionally, individuals with type I hypobetalipoproteinemia may develop neurological symptoms such as ataxia, neuropathy, and retinitis pigmentosa due to vitamin E deficiency.
2. Type II (also known as Homozygous or Compound Heterozygous Hypobetalipoproteinemia): This is a less severe form of the disorder caused by mutations in the APOB gene, which encodes apolipoprotein B. It leads to reduced levels of LDL and apoB but not as dramatically low as in type I. Symptoms may include mild fat malabsorption, decreased blood cholesterol levels, and an increased risk of developing fatty liver disease (hepatic steatosis). Neurological symptoms are less common than in type I hypobetalipoproteinemia.
Early diagnosis and treatment of hypobetalipoproteinemias, particularly type I, are crucial to prevent severe complications associated with fat-soluble vitamin deficiencies and neurological damage. Treatment typically involves dietary modifications, including supplementation with high doses of fat-soluble vitamins (A, D, E, and K).
Hypolipoproteinemias are a group of genetic disorders characterized by low levels of lipoproteins in the blood. Lipoproteins are complex particles composed of proteins and lipids that play a crucial role in the transport and metabolism of fat molecules, such as cholesterol and triglycerides, in the body.
There are several types of hypolipoproteinemias, each associated with deficiencies in specific lipoproteins:
1. Hypobetalipoproteinemia: This disorder is characterized by low levels of beta-lipoproteins, also known as low-density lipoproteins (LDL), or "bad" cholesterol. It can lead to decreased absorption of fat-soluble vitamins and an increased risk of fatty liver disease.
2. Abetalipoproteinemia: This is a rare autosomal recessive disorder characterized by the absence of beta-lipoproteins and apolipoprotein B, which results in very low levels of LDL cholesterol and high-density lipoproteins (HDL), or "good" cholesterol. It can lead to fat malabsorption, neurological symptoms, and retinal degeneration.
3. Tangier disease: This disorder is caused by a deficiency in apolipoprotein A-I and results in low levels of HDL cholesterol. It can cause enlarged orange-colored tonsils, neuropathy, and an increased risk of coronary artery disease.
4. Familial hypoalphalipoproteinemia: This disorder is characterized by low levels of HDL cholesterol due to a deficiency in apolipoprotein A-I or A-II. It can increase the risk of premature coronary artery disease.
It's important to note that while some hypolipoproteinemias are associated with an increased risk of cardiovascular disease, others may actually protect against it due to reduced levels of atherogenic lipoproteins. Treatment for these disorders typically involves dietary modifications and supplementation of fat-soluble vitamins and essential fatty acids. In some cases, medication may be necessary to manage symptoms or prevent complications.
Apolipoproteins are a group of proteins that are associated with lipids (fats) in the body and play a crucial role in the metabolism, transportation, and regulation of lipids. They are structural components of lipoprotein particles, which are complexes of lipids and proteins that transport lipids in the bloodstream.
There are several types of apolipoproteins, including ApoA, ApoB, ApoC, ApoD, ApoE, and others. Each type has a specific function in lipid metabolism. For example, ApoA is a major component of high-density lipoprotein (HDL), often referred to as "good cholesterol," and helps remove excess cholesterol from cells and tissues and transport it to the liver for excretion. ApoB, on the other hand, is a major component of low-density lipoprotein (LDL), or "bad cholesterol," and plays a role in the delivery of cholesterol to cells and tissues.
Abnormal levels of apolipoproteins or dysfunctional forms of these proteins have been linked to various diseases, including cardiovascular disease, Alzheimer's disease, and metabolic disorders such as diabetes. Therefore, measuring apolipoprotein levels in the blood can provide valuable information for diagnosing and monitoring these conditions.
High-Density Lipoproteins (HDL) are a type of lipoprotein that play a crucial role in the transportation and metabolism of cholesterol in the body. They are often referred to as "good" cholesterol because they help remove excess cholesterol from cells and carry it back to the liver, where it can be broken down and removed from the body. This process is known as reverse cholesterol transport.
HDLs are composed of a lipid core containing cholesteryl esters and triglycerides, surrounded by a shell of phospholipids, free cholesterol, and apolipoproteins, primarily apoA-I. The size and composition of HDL particles can vary, leading to the classification of different subclasses of HDL with varying functions and metabolic fates.
Elevated levels of HDL have been associated with a lower risk of developing cardiovascular diseases, while low HDL levels increase the risk. However, it is essential to consider that HDL function and quality may be more important than just the quantity in determining cardiovascular risk.
Lipoproteins are complex particles composed of multiple proteins and lipids (fats) that play a crucial role in the transport and metabolism of fat molecules in the body. They consist of an outer shell of phospholipids, free cholesterols, and apolipoproteins, enclosing a core of triglycerides and cholesteryl esters.
There are several types of lipoproteins, including:
1. Chylomicrons: These are the largest lipoproteins and are responsible for transporting dietary lipids from the intestines to other parts of the body.
2. Very-low-density lipoproteins (VLDL): Produced by the liver, VLDL particles carry triglycerides to peripheral tissues for energy storage or use.
3. Low-density lipoproteins (LDL): Often referred to as "bad cholesterol," LDL particles transport cholesterol from the liver to cells throughout the body. High levels of LDL in the blood can lead to plaque buildup in artery walls and increase the risk of heart disease.
4. High-density lipoproteins (HDL): Known as "good cholesterol," HDL particles help remove excess cholesterol from cells and transport it back to the liver for excretion or recycling. Higher levels of HDL are associated with a lower risk of heart disease.
Understanding lipoproteins and their roles in the body is essential for assessing cardiovascular health and managing risks related to heart disease and stroke.
Low-density lipoproteins (LDL), also known as "bad cholesterol," are a type of lipoprotein that carry cholesterol and other fats from the liver to cells throughout the body. High levels of LDL in the blood can lead to the buildup of cholesterol in the walls of the arteries, which can increase the risk of heart disease and stroke.
Lipoproteins are complex particles composed of proteins (apolipoproteins) and lipids (cholesterol, triglycerides, and phospholipids) that are responsible for transporting fat molecules around the body in the bloodstream. LDL is one type of lipoprotein, along with high-density lipoproteins (HDL), very low-density lipoproteins (VLDL), and chylomicrons.
LDL particles are smaller than HDL particles and can easily penetrate the artery walls, leading to the formation of plaques that can narrow or block the arteries. Therefore, maintaining healthy levels of LDL in the blood is essential for preventing cardiovascular disease.
Carrier proteins, also known as transport proteins, are a type of protein that facilitates the movement of molecules across cell membranes. They are responsible for the selective and active transport of ions, sugars, amino acids, and other molecules from one side of the membrane to the other, against their concentration gradient. This process requires energy, usually in the form of ATP (adenosine triphosphate).
Carrier proteins have a specific binding site for the molecule they transport, and undergo conformational changes upon binding, which allows them to move the molecule across the membrane. Once the molecule has been transported, the carrier protein returns to its original conformation, ready to bind and transport another molecule.
Carrier proteins play a crucial role in maintaining the balance of ions and other molecules inside and outside of cells, and are essential for many physiological processes, including nerve impulse transmission, muscle contraction, and nutrient uptake.
VLDL (Very Low-Density Lipoproteins) are a type of lipoprotein that play a crucial role in the transport and metabolism of fat molecules, known as triglycerides, in the body. They are produced by the liver and consist of a core of triglycerides surrounded by a shell of proteins called apolipoproteins, phospholipids, and cholesterol.
VLDL particles are responsible for delivering fat molecules from the liver to peripheral tissues throughout the body, where they can be used as an energy source or stored for later use. During this process, VLDL particles lose triglycerides and acquire more cholesterol, transforming into intermediate-density lipoproteins (IDL) and eventually low-density lipoproteins (LDL), which are also known as "bad" cholesterol.
Elevated levels of VLDL in the blood can contribute to the development of cardiovascular disease due to their association with increased levels of triglycerides and LDL cholesterol, as well as decreased levels of high-density lipoproteins (HDL), which are considered "good" cholesterol.
Motor skills disorders are conditions that affect a person's ability to perform coordinated movements. These movements can be simple, such as buttoning a shirt, or complex, such as playing a musical instrument. Motor skills disorders can make it difficult for a person to perform everyday activities and can impact their quality of life.
There are two main types of motor skills: fine motor skills and gross motor skills. Fine motor skills involve the small movements of the hands, fingers, and wrists, such as writing or using utensils. Gross motor skills involve larger movements of the arms, legs, and torso, such as crawling, walking, or running.
Motor skills disorders can affect either fine or gross motor skills, or both. Some common types of motor skills disorders include:
* Developmental coordination disorder (DCD): a condition that affects a child's ability to perform coordinated movements and is often diagnosed in early childhood. Children with DCD may have difficulty with tasks such as tying their shoes, buttoning their clothes, or using scissors.
* Cerebral palsy: a group of disorders that affect movement and muscle tone, caused by damage to the brain before, during, or after birth. Cerebral palsy can cause stiff or floppy muscles, uncontrolled movements, and difficulty with balance and coordination.
* Dyspraxia: a condition that affects a person's ability to plan and perform coordinated movements. People with dyspraxia may have difficulty with tasks such as writing, buttoning their clothes, or playing sports.
* Ataxia: a group of disorders that affect coordination and balance, caused by damage to the cerebellum (the part of the brain that controls movement). Ataxia can cause unsteady gait, poor coordination, and difficulty with fine motor tasks.
Motor skills disorders can be caused by a variety of factors, including genetics, injury, illness, or developmental delays. Treatment for motor skills disorders may include physical therapy, occupational therapy, speech therapy, and medication. In some cases, surgery may also be necessary to treat the underlying cause of the disorder.
Hyperlipoproteinemia Type III, also known as Broad Beta Disease or Remnant Hyperlipidemia, is a genetic disorder characterized by an increased level of chylomicron remnants and intermediate-density lipoproteins (IDL) in the blood. This results in elevated levels of both low-density lipoprotein (LDL), or "bad" cholesterol, and triglycerides, and decreased levels of high-density lipoprotein (HDL), or "good" cholesterol. The condition can lead to premature atherosclerosis and an increased risk for cardiovascular disease. It is caused by mutations in the APOE gene, which encodes the apolipoprotein E protein, leading to abnormal clearance of lipoproteins from the blood.
I'm sorry for any confusion, but "handwriting" is not a term that has a specific medical definition. Handwriting refers to the personal style or manner in which a person writes by hand. It can vary from person to person and can be influenced by factors such as an individual's fine motor skills, cultural background, and personal preferences.
However, in a broader context, there are certain medical conditions that can affect handwriting, such as neurological disorders (like stroke, Parkinson's disease, or cerebral palsy) or orthopedic issues (like arthritis or fractures). In these cases, healthcare professionals might assess a person's handwriting as part of the diagnostic process or to monitor the progression or treatment response of a condition. But again, handwriting itself is not a medical term with a specific definition.
An encyclopedia is a comprehensive reference work containing articles on various topics, usually arranged in alphabetical order. In the context of medicine, a medical encyclopedia is a collection of articles that provide information about a wide range of medical topics, including diseases and conditions, treatments, tests, procedures, and anatomy and physiology. Medical encyclopedias may be published in print or electronic formats and are often used as a starting point for researching medical topics. They can provide reliable and accurate information on medical subjects, making them useful resources for healthcare professionals, students, and patients alike. Some well-known examples of medical encyclopedias include the Merck Manual and the Stedman's Medical Dictionary.
Steatorrhea is a medical condition characterized by the excessive amount of fat in stools, which can make them appear greasy, frothy, and foul-smelling. This occurs due to poor absorption of dietary fats in the intestines, a process called malabsorption. The most common causes of steatorrhea include conditions that affect the pancreas, such as cystic fibrosis or chronic pancreatitis, celiac disease, and other gastrointestinal disorders. Symptoms associated with steatorrhea may include abdominal pain, bloating, diarrhea, weight loss, and vitamin deficiencies due to malabsorption of fat-soluble vitamins (A, D, E, K). The diagnosis typically involves testing stool samples for fat content and further investigations to determine the underlying cause. Treatment is focused on addressing the underlying condition and providing dietary modifications to manage symptoms.
 Abetalipoproteinemia
Abetalipoproteinemia Abetalipoproteinemia: MedlinePlus Genetics
Abetalipoproteinemia: MedlinePlus Genetics Abetalipoproteinemia | Rare Diseases | RareGuru
Abetalipoproteinemia | Rare Diseases | RareGuru Spur Cell Anemia: Practice Essentials, Pathophysiology, Etiology
Spur Cell Anemia: Practice Essentials, Pathophysiology, Etiology Abetalipoproteinemia: Video, Anatomy & Definition | Osmosis
Abetalipoproteinemia: Video, Anatomy & Definition | Osmosis Abetalipoproteinaemia | Amedes Genetics
Abetalipoproteinaemia | Amedes Genetics EU
EU List of Rare Diseases | A-Z Database | NORD
List of Rare Diseases | A-Z Database | NORD Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US urofacial syndrome - Ontology Browser - Rat Genome Database
urofacial syndrome - Ontology Browser - Rat Genome Database What Happens if You Don't Get Enough Vitamin E?
What Happens if You Don't Get Enough Vitamin E? Neuroacanthocytosis - wikidoc
Neuroacanthocytosis - wikidoc Hypolipidemia - Hormonal and Metabolic Disorders - MSD Manual Consumer Version
Hypolipidemia - Hormonal and Metabolic Disorders - MSD Manual Consumer Version Diagnostic Pathology Nonneoplastic Pediatrics, 2nd edition - Angelica R. Putnam - 1020 - ELSEVIER HEALTH SCIENCES -...
Diagnostic Pathology Nonneoplastic Pediatrics, 2nd edition - Angelica R. Putnam - 1020 - ELSEVIER HEALTH SCIENCES -... Vitamin E succinate and cancer treatment: a vitamin E prototype for selective antitumour activity | British Journal of Cancer
Vitamin E succinate and cancer treatment: a vitamin E prototype for selective antitumour activity | British Journal of Cancer International Classification of Diseases - Endocrine, Nutritional and Metabolic Diseases, and Immunity Disorders
International Classification of Diseases - Endocrine, Nutritional and Metabolic Diseases, and Immunity Disorders Abhd10 Mouse Gene Details | abhydrolase domain containing 10 | International Mouse Phenotyping Consortium
Abhd10 Mouse Gene Details | abhydrolase domain containing 10 | International Mouse Phenotyping Consortium Pediatric gastrointestinal pathology - Libre Pathology
Pediatric gastrointestinal pathology - Libre Pathology Lipid disorders
Lipid disorders Diseases and Conditions
Diseases and Conditions Namespace
Namespace