Anemia, Sickle Cell
Hemoglobin, Sickle
Erythrocytes, Abnormal
Antisickling Agents
Hemoglobin SC Disease
Fetal Hemoglobin
Anemia, Hemolytic
Anemia, Aplastic
Hemoglobins
Fanconi Anemia
Hydroxyurea
Thalassemia
Anemia, Hemolytic, Autoimmune
Anemia, Hypochromic
beta-Thalassemia
Erythrocytes
Anemia, Macrocytic
Anemia, Pernicious
Hemoglobinopathies
Jamaica
Acute Chest Syndrome
alpha-Thalassemia
Blood Transfusion
Hemoglobin C Disease
Anemia, Sideroblastic
Hemolysis
Hemoglobin A
Anemia, Megaloblastic
Infectious Anemia Virus, Equine
Anemia, Refractory
Erythrocyte Indices
Hemoglobins, Abnormal
Erythrocyte Deformability
Hematocrit
Exchange Transfusion, Whole Blood
Hemoglobin C
Leg Ulcer
Globins
Pregnancy Complications, Hematologic
Erythrocyte Aging
Erythropoietin
Iron
Erythrocyte Transfusion
Equine Infectious Anemia
Reticulocytes
Pain
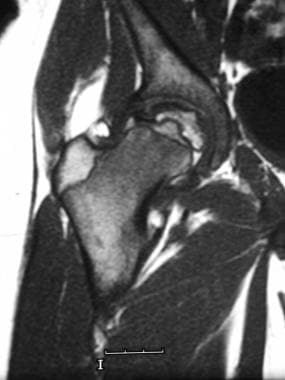
Splenic Infarction
Erythrocyte Membrane
Chicken anemia virus
Anemia, Dyserythropoietic, Congenital
Erythropoiesis
Anemia, Diamond-Blackfan
Vascular Diseases
Blood Viscosity
Fanconi Anemia Complementation Group Proteins
Iron Overload
Ferritins
Oxygen
Erythrocyte Aggregation
gamma-Globins
Nigeria
Reticulocyte Count
Oxyhemoglobins
Hemorheology
beta-Globins
Dimethyl Adipimidate
Anemia, Neonatal
Prevalence
Splenic Diseases
Lutheran Blood-Group System
Hematinics
Anemia, Refractory, with Excess of Blasts
Kidney Papillary Necrosis
Glucosephosphate Dehydrogenase Deficiency
Priapism
Blood Cell Count
Osmotic Fragility
2,3-Diphosphoglycerate
Neonatal Screening
Fanconi Anemia Complementation Group C Protein
Fanconi Anemia Complementation Group D2 Protein
Retinal Diseases
Fanconi Anemia Complementation Group A Protein
Hemoglobin A2
Hypertension, Pulmonary
Premarital Examinations
Heinz Bodies
Risk Factors
Methemoglobin
Diphosphoglyceric Acids
Enuresis
Mediterranean Islands
Spherocytosis, Hereditary
Prospective Studies
Anemia, Hemolytic, Congenital Nonspherocytic
Retrospective Studies
Hemoglobinuria
Treatment Outcome
Polymer structure and solubility of deoxyhemoglobin S in the presence of high concentrations of volume-excluding 70-kDa dextran. Effects of non-s hemoglobins and inhibitors. (1/2332)
Earlier observations indicated that volume exclusion by admixed non-hemoglobin macromolecules lowered the polymer solubility ("Csat") of deoxyhemoglobin (Hb) S, presumably by increasing its activity. In view of the potential usefulness of these observations for in vitro studies of sickling-related polymerization, we examined the ultrastructure, solubility behavior, and phase distributions of deoxygenated mixtures of Hb S with 70-kDa dextran, a relatively inert, low ionic strength space-filling macromolecule. Increasing admixture of dextran progressively lowered the Csat of deoxyHb S. With 12 g/dl dextran, a 5-fold decrease in apparent Csat ("dextran-Csat") was obtained together with acceptable sensitivity and proportionality with the standard Csat when assessing the effects of non-S Hb admixtures (A, C, and F) or polymerization inhibitors (alkylureas or phenylalanine). The volume fraction of dextran excluding Hb was 70-75% of total deoxyHb-dextran (12 g/dl) volumes. Electron microscopy showed polymer fibers and fiber-to-crystal transitions indistinguishable from those formed without dextran. Thus when Hb quantities are limited, as with genetically engineered recombinant Hbs or transgenic sickle mice, the dextran-Csat provides convenient and reliable screening of effects of Hb S modifications on polymerization under near-physiological conditions, avoiding problems of high ionic strength. (+info)Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. (2/2332)
High levels of fetal hemoglobin (Hb F) protect from many of the complications of sickle cell disease and lead to improved survival. Butyrate and other short chain fatty acids were previously shown to increase Hb F production in erythroid cells in vitro and in animal models in vivo. However, butyrates are also known to inhibit the proliferation of many cell types, including erythroid cells. Experience with the use of butyrate in animal models and in early clinical trials demonstrated that the Hb F response may be lost after prolonged administration of high doses of butyrate. We hypothesized that this loss of response may be a result of the antiproliferative effects of butyrate. We designed a regimen consisting of intermittent or pulse therapy in which butyrate was administered for 4 days followed by 10 to 24 days with no drug exposure. This pulse regimen induced fetal globin gene expression in 9 of 11 patients. The mean Hb F in this group increased from 7.2% to 21.0% (P <.002) after intermittent butyrate therapy for a mean duration of 29.9 weeks. This was associated with a parallel increase in the number of F cells and F reticulocytes. The total hemoglobin levels also increased from a mean of 7.8 g/dL to a mean of 8.8 g/dL (P <.006). The increased levels of Hb F were sustained in all responders, including 1 patient who has been on pulse butyrate therapy for more than 28 months. This regimen, which resulted in a marked and sustained increase in Hb F levels in more than two thirds of the adult sickle cell patients enrolled in this study, was well tolerated without adverse side effects. These encouraging results require confirmation along with an appropriate evaluation of clinical outcomes in a larger number of patients with sickle cell disease. (+info)In vivo blood flow abnormalities in the transgenic knockout sickle cell mouse. (3/2332)
The accepted importance of circulatory impairment to sickle cell anemia remains to be verified by in vivo experimentation. Intravital microscopy studies of blood flow in patients are limited to circulations that can be viewed noninvasively and are restricted from deliberate perturbations of the circulation. Further knowledge of sickle blood flow abnormalities has awaited an animal model of human sickle cell disease. We compared blood flow in the mucosal-intestinal microvessels of normal mice with that in transgenic knockout sickle cell mice that have erythrocytes containing only human hemoglobin S and that exhibit a degree of hemolytic anemia and pathological complications similar to the human disease. In sickle cell mice, in addition to seeing blood flow abnormalities such as sludging in all microvessels, we detected decreased blood flow velocity in venules of all diameters. Flow responses to hyperoxia in both normal and sickle cell mice were dramatic, but opposite: Hyperoxia promptly slowed or halted flow in normal mice but markedly enhanced flow in sickle cell mice. Intravital microscopic studies of this murine model provide important insights into sickle cell blood flow abnormalities and suggest that this model can be used to evaluate the causes of abnormal flow and new approaches to therapy of sickle cell disease. (+info)Candida dubliniensis candidemia in patients with chemotherapy-induced neutropenia and bone marrow transplantation. (4/2332)
The recently described species Candida dubliniensis has been recovered primarily from superficial oral candidiasis in HIV-infected patients. No clinically documented invasive infections were reported until now in this patient group or in other immunocompromised patients. We report three cases of candidemia due to this newly emerging Candida species in HIV-negative patients with chemotherapy-induced immunosuppression and bone marrow transplantation. (+info)Development of viral vectors for gene therapy of beta-chain hemoglobinopathies: optimization of a gamma-globin gene expression cassette. (5/2332)
Progress toward gene therapy of beta-chain hemoglobinopathies has been limited in part by poor expression of globin genes in virus vectors. To derive an optimal expression cassette, we systematically analyzed the sequence requirements and relative strengths of the Agamma- and beta-globin promoters, the activities of various erythroid-specific enhancers, and the importance of flanking and intronic sequences. Expression was analyzed by RNase protection after stable plasmid transfection of the murine erythroleukemia cell line, MEL585. Promoter truncation studies showed that the Agamma-globin promoter could be deleted to -159 without affecting expression, while deleting the beta-globin promoter to -127 actually increased expression compared with longer fragments. Expression from the optimal beta-globin gene promoter was consistently higher than that from the optimal Agamma-globin promoter, regardless of the enhancer used. Enhancers tested included a 2.5-kb composite of the beta-globin locus control region (termed a muLCR), a combination of the HS2 and HS3 core elements of the LCR, and the HS-40 core element of the alpha-globin locus. All three enhancers increased expression from the beta-globin gene to roughly the same extent, while the HS-40 element was notably less effective with the Agamma-globin gene. However, the HS-40 element was able to efficiently enhance expression of a Agamma-globin gene linked to the beta-globin promoter. Inclusion of extended 3' sequences from either the beta-globin or the Agamma-globin genes had no significant effect on expression. A 714-bp internal deletion of Agamma-globin intron 2 unexpectedly increased expression more than twofold. With the combination of a -127 beta-globin promoter, an Agamma-globin gene with the internal deletion of intron 2, and a single copy of the HS-40 enhancer, gamma-globin expression averaged 166% of murine alpha-globin mRNA per copy in six pools and 105% in nine clones. When placed in a retrovirus vector, this cassette was also expressed at high levels in MEL585 cells (averaging 75% of murine alpha-globin mRNA per copy) without reducing virus titers. However, recombined provirus or aberrant splicing was observed in 5 of 12 clones, indicating a significant degree of genetic instability. Taken together, these data demonstrate the development of an optimal expression cassette for gamma-globin capable of efficient expression in a retrovirus vector and form the basis for further refinement of vectors containing this cassette. (+info)Osteonecrosis of the hip in sickle-cell disease associated with tuberculous arthritis. A review of 15 cases. (6/2332)
We report a study of 15 cases of tuberculous hips with sickle-cell disease who presented during 1991-1993. Although the osteonecrosis was long-standing, biopsy was nearly always required to reveal the more recent tuberculous infection. Management consisted of 6 months of anti-tuberculous chemotherapy with appropriate palliative surgery 5-8 weeks after the start of drug treatment. The operative techniques which we used are described. The results were good both post-operatively, and in 12 patients followed-up at an average of 3 years. We recommend this combined management for the treatment of secondary tuberculous infections of hips previously damaged by sickle-cell disease. (+info)Large cerebral vessel disease in sickle cell anaemia. (7/2332)
An 18 year old male with documented sickle cell disease was admitted to the hospital for the final time in coma. Cerebral angiography revealed multiple stenotic lesions of the large cerebral vessels. The pathology of this large vessel involvement is demonstrated and the potential contribution of large as opposed to small cerebral vessel disease in the neurological manifestations of sickle cell anaemia is discussed. (+info)Perceived stress factors and coping mechanisms among mothers of children with sickle cell disease in western Nigeria. (8/2332)
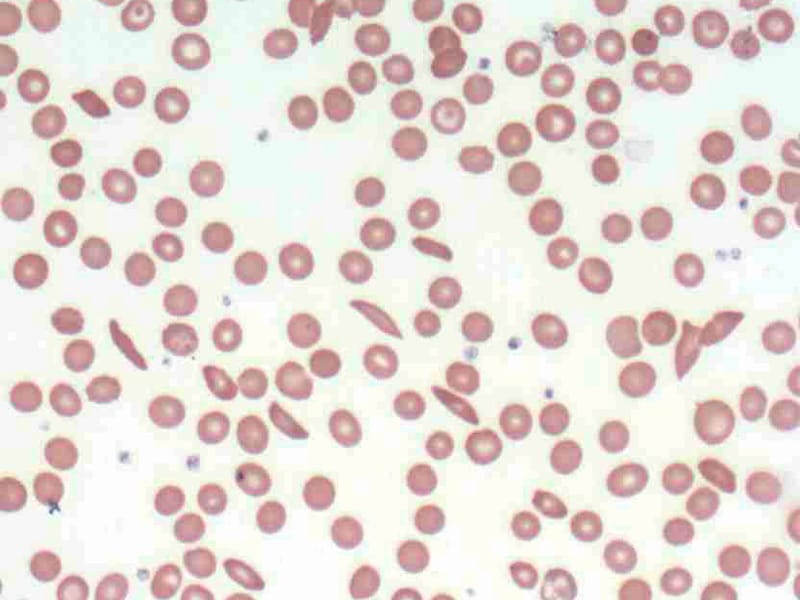
While many studies have looked at the stressful effects of chronic illness of those who suffer such conditions, less is known about the effects on caregivers, especially in developing countries. Mothers in particular must bear the brunt of care and stress for children who have sickle cell disease (SCD). A sample of 200 mothers attending six SCD clinics in both public and private hospitals in the Ibadan-Ibarapa Health Zone of Oyo State, Nigeria, were interviewed. Stress levels were measured using an instrument comprised of stressors listed by mothers themselves in focus group discussions that preceded the survey. Higher levels of stress were associated with less educated and older women, as well as non-married women and those in polygamous households. Stress levels were also greater when there was more than one child with SCD in the family and when the index child was of school age. Coping mechanisms varied according to the category of stressor. Financial stress and disease factors were met with confrontation while family sources of stress were either complained about, accepted or avoided. Knowledge of the different types of mothers who experience more stress and of their preferred coping mechanisms can be useful in designing clinic-based counseling. (+info)Sickle cell anemia is a genetic disorder that affects the hemoglobin in red blood cells. Hemoglobin is responsible for carrying oxygen throughout the body. In sickle cell anemia, the hemoglobin is abnormal and causes the red blood cells to take on a sickle shape, rather than the normal disc shape. These sickled cells are stiff and sticky, and they can block blood vessels, causing tissue damage and pain. They also die more quickly than normal red blood cells, leading to anemia.
People with sickle cell anemia often experience fatigue, chronic pain, and jaundice. They may also have a higher risk of infections and complications such as stroke, acute chest syndrome, and priapism. The disease is inherited from both parents, who must both be carriers of the sickle cell gene. It primarily affects people of African descent, but it can also affect people from other ethnic backgrounds.
There is no cure for sickle cell anemia, but treatments such as blood transfusions, medications to manage pain and prevent complications, and bone marrow transplantation can help improve quality of life for affected individuals. Regular medical care and monitoring are essential for managing the disease effectively.
Sickle cell trait is a genetic condition where an individual inherits one abnormal gene for hemoglobin S (HbS) from one parent and one normal gene for hemoglobin A (HbA) from the other parent. Hemoglobin is a protein in red blood cells that carries oxygen throughout the body.
People with sickle cell trait do not have sickle cell disease, but they can pass the abnormal HbS gene on to their children. In certain situations, such as high altitude, low oxygen levels, or intense physical exertion, individuals with sickle cell trait may experience symptoms similar to those of sickle cell disease, such as fatigue, pain, and shortness of breath. However, these symptoms are typically milder and less frequent than in people with sickle cell disease.
It is important for individuals who know they have sickle cell trait to inform their healthcare providers, especially if they become pregnant or plan to engage in activities that may cause low oxygen levels, such as scuba diving or high-altitude climbing.
Anemia is a medical condition characterized by a lower than normal number of red blood cells or lower than normal levels of hemoglobin in the blood. Hemoglobin is an important protein in red blood cells that carries oxygen from the lungs to the rest of the body. Anemia can cause fatigue, weakness, shortness of breath, and a pale complexion because the body's tissues are not getting enough oxygen.
Anemia can be caused by various factors, including nutritional deficiencies (such as iron, vitamin B12, or folate deficiency), blood loss, chronic diseases (such as kidney disease or rheumatoid arthritis), inherited genetic disorders (such as sickle cell anemia or thalassemia), and certain medications.
There are different types of anemia, classified based on the underlying cause, size and shape of red blood cells, and the level of hemoglobin in the blood. Treatment for anemia depends on the underlying cause and may include dietary changes, supplements, medication, or blood transfusions.
Hemoglobin S (HbS) is a genetic variant of hemoglobin, which is the oxygen-carrying protein in red blood cells. This abnormal form of hemogllobin results from a mutation in the beta-globin gene, leading to the substitution of valine for glutamic acid at position six of the beta-globin chain.
In individuals with sickle cell disease (a group of inherited red blood cell disorders), both copies of their beta-globin genes carry this mutation, causing the majority of their hemoglobin to be HbS. When deoxygenated, HbS molecules have a tendency to polymerize and form long, rigid rods within the red blood cells, distorting their shape into a characteristic sickle or crescent form.
These sickled red blood cells are less flexible and more prone to rupture (hemolysis), leading to chronic anemia, vaso-occlusive crises, and other disease complications. Sickle cell disease primarily affects people of African, Mediterranean, Middle Eastern, and Indian ancestry, but it can also be found in other populations worldwide.
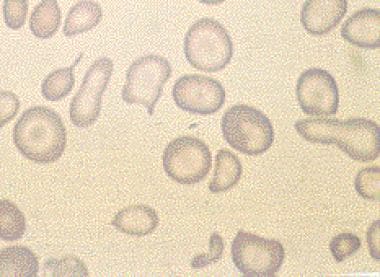
Abnormal erythrocytes refer to red blood cells that have an abnormal shape, size, or other characteristics. This can include various types of abnormalities such as:
1. Anisocytosis: Variation in the size of erythrocytes.
2. Poikilocytosis: Variation in the shape of erythrocytes, including but not limited to teardrop-shaped cells (dacrocytes), crescent-shaped cells (sickle cells), and spherical cells (spherocytes).
3. Anemia: A decrease in the total number of erythrocytes or a reduction in hemoglobin concentration, which can result from various underlying conditions such as iron deficiency, chronic disease, or blood loss.
4. Hemoglobinopathies: Abnormalities in the structure or function of hemoglobin, the protein responsible for carrying oxygen in erythrocytes, such as sickle cell anemia and thalassemia.
5. Inclusion bodies: Abnormal structures within erythrocytes, such as Heinz bodies (denatured hemoglobin) or Howell-Jolly bodies (nuclear remnants).
These abnormalities can be detected through a complete blood count (CBC) and peripheral blood smear examination. The presence of abnormal erythrocytes may indicate an underlying medical condition, and further evaluation is often necessary to determine the cause and appropriate treatment.
Antisickling agents are medications or substances that help prevent or reduce the sickling of red blood cells in individuals with sickle cell disease. Sickling is a pathological process where the normally disc-shaped red blood cells become crescent-shaped due to abnormal hemoglobin (HbS). This change in shape can lead to blockages in small blood vessels, causing tissue damage and various complications such as pain crises, acute chest syndrome, and stroke.
Antisickling agents work by either inhibiting the polymerization of HbS or improving the oxygen-carrying capacity of red blood cells. The most commonly used antisickling agent is hydroxyurea, which increases the production of fetal hemoglobin (HbF) in red blood cells. HbF has a higher affinity for oxygen than HbS and can prevent the polymerization of HbS, thereby reducing sickling. Other antisickling agents include:
1. L-glutamine: An amino acid that helps maintain the structural integrity of red blood cells and reduces oxidative stress.
2. Arginate: A salt of arginine, an amino acid that helps improve nitric oxide production and vasodilation, reducing sickling.
3. Senicapoc: A drug that inhibits the formation of HbS polymers by blocking the interaction between HbS molecules.
4. Voxelotor (Oxbryta): A medication that binds to HbS and stabilizes it in its oxygenated state, reducing sickling.
These antisickling agents can help alleviate symptoms, decrease the frequency of pain crises, and improve the quality of life for individuals with sickle cell disease. However, they should be used under the supervision of a healthcare professional, as each has its benefits, risks, and potential side effects.
Hemoglobin SC disease, also known as sickle cell-C disease or SC disorder, is a genetic blood disorder that is a variant of sickle cell anemia. It is caused by the presence of both hemoglobin S (HbS) and hemoglobin C (HbC) in the red blood cells.
Hemoglobin is the protein in red blood cells that carries oxygen throughout the body. In Hemoglobin SC disease, the abnormal HbS and HbC proteins can cause the red blood cells to become rigid, sticky, and C-shaped (sickled), which can lead to blockages in small blood vessels.
Symptoms of Hemoglibin SC disease may include anemia, fatigue, jaundice, episodes of pain (known as sickle cell crises), and an increased risk of infection. The severity of the symptoms can vary widely from person to person. Treatment typically focuses on managing symptoms and preventing complications, and may include medications, blood transfusions, and sometimes a bone marrow transplant.
Fetal hemoglobin (HbF) is a type of hemoglobin that is produced in the fetus and newborn babies. It is composed of two alpha-like globin chains and two gamma-globin chains, designated as α2γ2. HbF is the primary form of hemoglobin during fetal development, replacing the embryonic hemoglobin (HbG) around the eighth week of gestation.
The unique property of HbF is its higher affinity for oxygen compared to adult hemoglobin (HbA), which helps ensure adequate oxygen supply from the mother to the developing fetus. After birth, as the newborn starts breathing on its own and begins to receive oxygen directly, the production of HbF gradually decreases and is usually replaced by HbA within the first year of life.
In some genetic disorders like sickle cell disease and beta-thalassemia, persistence of HbF into adulthood can be beneficial as it reduces the severity of symptoms due to its higher oxygen-carrying capacity and less polymerization tendency compared to HbS (in sickle cell disease) or unpaired alpha chains (in beta-thalassemia). Treatments like hydroxyurea are used to induce HbF production in these patients as a therapeutic approach.
Hemolytic anemia is a type of anemia that occurs when red blood cells are destroyed (hemolysis) faster than they can be produced. Red blood cells are essential for carrying oxygen throughout the body. When they are destroyed, hemoglobin and other cellular components are released into the bloodstream, which can lead to complications such as kidney damage and gallstones.
Hemolytic anemia can be inherited or acquired. Inherited forms of the condition may result from genetic defects that affect the structure or function of red blood cells. Acquired forms of hemolytic anemia can be caused by various factors, including infections, medications, autoimmune disorders, and certain medical conditions such as cancer or blood disorders.
Symptoms of hemolytic anemia may include fatigue, weakness, shortness of breath, pale skin, jaundice (yellowing of the skin and eyes), dark urine, and a rapid heartbeat. Treatment for hemolytic anemia depends on the underlying cause and may include medications, blood transfusions, or surgery.
Aplastic anemia is a medical condition characterized by pancytopenia (a decrease in all three types of blood cells: red blood cells, white blood cells, and platelets) due to the failure of bone marrow to produce new cells. It is called "aplastic" because the bone marrow becomes hypocellular or "aplastic," meaning it contains few or no blood-forming stem cells.
The condition can be acquired or inherited, with acquired aplastic anemia being more common. Acquired aplastic anemia can result from exposure to toxic chemicals, radiation, drugs, viral infections, or autoimmune disorders. Inherited forms of the disease include Fanconi anemia and dyskeratosis congenita.
Symptoms of aplastic anemia may include fatigue, weakness, shortness of breath, pale skin, easy bruising or bleeding, frequent infections, and fever. Treatment options for aplastic anemia depend on the severity of the condition and its underlying cause. They may include blood transfusions, immunosuppressive therapy, and stem cell transplantation.
Hemoglobin (Hb or Hgb) is the main oxygen-carrying protein in the red blood cells, which are responsible for delivering oxygen throughout the body. It is a complex molecule made up of four globin proteins and four heme groups. Each heme group contains an iron atom that binds to one molecule of oxygen. Hemoglobin plays a crucial role in the transport of oxygen from the lungs to the body's tissues, and also helps to carry carbon dioxide back to the lungs for exhalation.
There are several types of hemoglobin present in the human body, including:
* Hemoglobin A (HbA): This is the most common type of hemoglobin, making up about 95-98% of total hemoglobin in adults. It consists of two alpha and two beta globin chains.
* Hemoglobin A2 (HbA2): This makes up about 1.5-3.5% of total hemoglobin in adults. It consists of two alpha and two delta globin chains.
* Hemoglobin F (HbF): This is the main type of hemoglobin present in fetal life, but it persists at low levels in adults. It consists of two alpha and two gamma globin chains.
* Hemoglobin S (HbS): This is an abnormal form of hemoglobin that can cause sickle cell disease when it occurs in the homozygous state (i.e., both copies of the gene are affected). It results from a single amino acid substitution in the beta globin chain.
* Hemoglobin C (HbC): This is another abnormal form of hemoglobin that can cause mild to moderate hemolytic anemia when it occurs in the homozygous state. It results from a different single amino acid substitution in the beta globin chain than HbS.
Abnormal forms of hemoglobin, such as HbS and HbC, can lead to various clinical disorders, including sickle cell disease, thalassemia, and other hemoglobinopathies.
Fanconi anemia is a rare, inherited disorder that affects the body's ability to produce healthy blood cells. It is characterized by bone marrow failure, congenital abnormalities, and an increased risk of developing certain types of cancer. The condition is caused by mutations in genes responsible for repairing damaged DNA, leading to chromosomal instability and cell death.
The classic form of Fanconi anemia (type A) is typically diagnosed in childhood and is associated with various physical abnormalities such as short stature, skin pigmentation changes, thumb and radial ray anomalies, kidney and genitourinary malformations, and developmental delays. Other types of Fanconi anemia (B-G) may have different clinical presentations but share the common feature of bone marrow failure and cancer predisposition.
Bone marrow failure in Fanconi anemia results in decreased production of all three types of blood cells: red blood cells, white blood cells, and platelets. This can lead to anemia (low red blood cell count), neutropenia (low white blood cell count), and thrombocytopenia (low platelet count). These conditions increase the risk of infections, fatigue, and bleeding.
Individuals with Fanconi anemia have a significantly higher risk of developing various types of cancer, particularly acute myeloid leukemia (AML) and solid tumors such as squamous cell carcinomas of the head, neck, esophagus, and anogenital region.
Treatment for Fanconi anemia typically involves managing symptoms related to bone marrow failure, such as transfusions, growth factors, and antibiotics. Hematopoietic stem cell transplantation (HSCT) is the only curative treatment option for bone marrow failure but carries risks of its own, including graft-versus-host disease and transplant-related mortality. Regular cancer surveillance is essential due to the increased risk of malignancies in these patients.
Hydroxyurea is an antimetabolite drug that is primarily used in the treatment of myeloproliferative disorders such as chronic myelogenous leukemia (CML), essential thrombocythemia, and polycythemia vera. It works by interfering with the synthesis of DNA, which inhibits the growth of cancer cells.
In addition to its use in cancer therapy, hydroxyurea is also used off-label for the management of sickle cell disease. In this context, it helps to reduce the frequency and severity of painful vaso-occlusive crises by increasing the production of fetal hemoglobin (HbF), which decreases the formation of sickled red blood cells.
The medical definition of hydroxyurea is:
A hydantoin derivative and antimetabolite that inhibits ribonucleoside diphosphate reductase, thereby interfering with DNA synthesis. It has been used as an antineoplastic agent, particularly in the treatment of myeloproliferative disorders, and more recently for the management of sickle cell disease to reduce the frequency and severity of painful vaso-occlusive crises by increasing fetal hemoglobin production.
Thalassemia is a group of inherited genetic disorders that affect the production of hemoglobin, a protein in red blood cells responsible for carrying oxygen throughout the body. The disorder results in less efficient or abnormal hemoglobin, which can lead to anemia, an insufficient supply of oxygen-rich red blood cells.
There are two main types of Thalassemia: alpha and beta. Alpha thalassemia occurs when there is a problem with the alpha globin chain production, while beta thalassemia results from issues in beta globin chain synthesis. These disorders can range from mild to severe, depending on the number of genes affected and their specific mutations.
Severe forms of Thalassemia may require regular blood transfusions, iron chelation therapy, or even a bone marrow transplant to manage symptoms and prevent complications.
Hemolytic anemia, autoimmune is a type of anemia characterized by the premature destruction of red blood cells (RBCs) in which the immune system mistakenly attacks and destroys its own RBCs. This occurs when the body produces autoantibodies that bind to the surface of RBCs, leading to their rupture (hemolysis). The symptoms may include fatigue, weakness, shortness of breath, and dark colored urine. The diagnosis is made through blood tests that measure the number and size of RBCs, reticulocyte count, and the presence of autoantibodies. Treatment typically involves suppressing the immune system with medications such as corticosteroids or immunosuppressive drugs, and sometimes removal of the spleen (splenectomy) may be necessary.
Hypochromic anemia is a type of anemia characterized by the presence of red blood cells that have lower than normal levels of hemoglobin and appear paler in color than normal. Hemoglobin is a protein in red blood cells that carries oxygen from the lungs to the rest of the body. In hypochromic anemia, there may be a decrease in the production or increased destruction of red blood cells, leading to a reduced number of red blood cells and insufficient oxygen supply to the tissues.
Hypochromic anemia can result from various underlying medical conditions, including iron deficiency, thalassemia, chronic inflammation, lead poisoning, and certain infections or chronic diseases. Treatment for hypochromic anemia depends on the underlying cause and may include iron supplements, dietary changes, medications, or blood transfusions.
Beta-thalassemia is a genetic blood disorder that affects the production of hemoglobin, a protein in red blood cells that carries oxygen throughout the body. Specifically, beta-thalassemia is caused by mutations in the beta-globin gene, which leads to reduced or absent production of the beta-globin component of hemoglobin.
There are two main types of beta-thalassemia:
1. Beta-thalassemia major (also known as Cooley's anemia): This is a severe form of the disorder that typically becomes apparent in early childhood. It is characterized by a significant reduction or absence of beta-globin production, leading to anemia, enlarged spleen and liver, jaundice, and growth retardation.
2. Beta-thalassemia intermedia: This is a milder form of the disorder that may not become apparent until later in childhood or even adulthood. It is characterized by a variable reduction in beta-globin production, leading to mild to moderate anemia and other symptoms that can range from nonexistent to severe.
Treatment for beta-thalassemia depends on the severity of the disorder and may include blood transfusions, iron chelation therapy, and/or bone marrow transplantation. In some cases, genetic counseling and prenatal diagnosis may also be recommended for families with a history of the disorder.
Erythrocytes, also known as red blood cells (RBCs), are the most common type of blood cell in circulating blood in mammals. They are responsible for transporting oxygen from the lungs to the body's tissues and carbon dioxide from the tissues to the lungs.
Erythrocytes are formed in the bone marrow and have a biconcave shape, which allows them to fold and bend easily as they pass through narrow blood vessels. They do not have a nucleus or mitochondria, which makes them more flexible but also limits their ability to reproduce or repair themselves.
In humans, erythrocytes are typically disc-shaped and measure about 7 micrometers in diameter. They contain the protein hemoglobin, which binds to oxygen and gives blood its red color. The lifespan of an erythrocyte is approximately 120 days, after which it is broken down in the liver and spleen.
Abnormalities in erythrocyte count or function can lead to various medical conditions, such as anemia, polycythemia, and sickle cell disease.
Macrocytic anemia is a type of anemia in which the red blood cells are larger than normal in size (macrocytic). This condition can be caused by various factors such as deficiency of vitamin B12 or folate, alcohol abuse, certain medications, bone marrow disorders, and some inherited genetic conditions.
The large red blood cells may not function properly, leading to symptoms such as fatigue, weakness, shortness of breath, pale skin, and a rapid heartbeat. Macrocytic anemia can be diagnosed through a complete blood count (CBC) test, which measures the size and number of red blood cells in the blood.
Treatment for macrocytic anemia depends on the underlying cause. In cases of vitamin B12 or folate deficiency, supplements or dietary changes may be recommended. If the anemia is caused by medication, a different medication may be prescribed. In severe cases, blood transfusions or injections of vitamin B12 may be necessary.
Pernicious anemia is a specific type of vitamin B12 deficiency anemia that is caused by a lack of intrinsic factor, a protein made in the stomach that is needed to absorb vitamin B12. The absence of intrinsic factor leads to poor absorption of vitamin B12 from food and results in its deficiency.
Vitamin B12 is essential for the production of healthy red blood cells, which carry oxygen throughout the body. Without enough vitamin B12, the body cannot produce enough red blood cells, leading to anemia. Pernicious anemia typically develops slowly over several years and can cause symptoms such as fatigue, weakness, pale skin, shortness of breath, and a decreased appetite.
Pernicious anemia is an autoimmune disorder, which means that the body's immune system mistakenly attacks healthy cells in the stomach lining, leading to a loss of intrinsic factor production. It is more common in older adults, particularly those over 60 years old, and can also be associated with other autoimmune disorders such as type 1 diabetes, Hashimoto's thyroiditis, and Addison's disease.
Treatment for pernicious anemia typically involves vitamin B12 replacement therapy, either through oral supplements or injections of the vitamin. In some cases, dietary changes may also be recommended to ensure adequate intake of vitamin B12-rich foods such as meat, fish, poultry, and dairy products.
Hemoglobinopathies are a group of genetic disorders characterized by structural or functional abnormalities of the hemoglobin molecule in red blood cells. Hemoglobin is a complex protein that plays a crucial role in carrying oxygen throughout the body. The two most common types of hemoglobinopathies are sickle cell disease and thalassemia.
In sickle cell disease, a single mutation in the beta-globin gene results in the production of an abnormal form of hemoglobin called hemoglobin S (HbS). When deoxygenated, HbS molecules tend to aggregate and form long polymers, causing the red blood cells to become sickle-shaped, rigid, and fragile. These abnormally shaped cells can block small blood vessels, leading to tissue damage, chronic pain, organ dysfunction, and other serious complications.
Thalassemias are a heterogeneous group of disorders caused by mutations in the genes that regulate the production of alpha- or beta-globin chains. These mutations result in reduced or absent synthesis of one or more globin chains, leading to an imbalance in hemoglobin composition and structure. This imbalance can cause premature destruction of red blood cells (hemolysis), resulting in anemia, jaundice, splenomegaly, and other symptoms.
Hemoglobinopathies are typically inherited in an autosomal recessive manner, meaning that affected individuals have two copies of the abnormal gene – one from each parent. Carriers of a single abnormal gene usually do not show any signs or symptoms of the disorder but can pass the abnormal gene on to their offspring.
Early diagnosis and appropriate management of hemoglobinopathies are essential for improving quality of life, reducing complications, and increasing survival rates. Treatment options may include blood transfusions, iron chelation therapy, antibiotics, pain management, and, in some cases, bone marrow transplantation or gene therapy.
I'm sorry for any confusion, but "Jamaica" is not a medical term. It is a country located in the Caribbean Sea, known for its beautiful beaches, vibrant culture, and as the birthplace of reggae music. If you have any questions about medical terms or concepts, I would be happy to help answer those!
Acute chest syndrome (ACS) is a serious complication of sickle cell disease, characterized by the presence of new infiltrates on chest X-ray and at least one other clinical symptom such as fever, cough, chest pain, or difficulty breathing. It is often caused by infection, fat embolism, or lung tissue inflammation, leading to respiratory distress, hypoxemia, and potentially respiratory failure. Prompt diagnosis and treatment with antibiotics, analgesics, and sometimes blood transfusions or exchange transfusions are essential for managing ACS.
Alpha-thalassemia is a genetic disorder that affects the production of hemoglobin, a protein in red blood cells that carries oxygen throughout the body. It is caused by deletions or mutations in the genes that produce the alpha-globin chains of hemoglobin.
There are several types of alpha-thalassemia, ranging from mild to severe. The most severe form, called hydrops fetalis, occurs when all four alpha-globin genes are deleted or mutated. This can cause stillbirth or death shortly after birth due to heart failure and severe anemia.
Less severe forms of alpha-thalassemia can cause mild to moderate anemia, which may be asymptomatic or associated with symptoms such as fatigue, weakness, and jaundice. These forms of the disorder are more common in people from Mediterranean, Southeast Asian, and African backgrounds.
Treatment for alpha-thalassemia depends on the severity of the condition and may include blood transfusions, iron chelation therapy, or occasionally stem cell transplantation.
A blood transfusion is a medical procedure in which blood or its components are transferred from one individual (donor) to another (recipient) through a vein. The donated blood can be fresh whole blood, packed red blood cells, platelets, plasma, or cryoprecipitate, depending on the recipient's needs. Blood transfusions are performed to replace lost blood due to severe bleeding, treat anemia, support patients undergoing major surgeries, or manage various medical conditions such as hemophilia, thalassemia, and leukemia. The donated blood must be carefully cross-matched with the recipient's blood type to minimize the risk of transfusion reactions.
Hemoglobin C disease is a genetic disorder that affects the structure and function of hemoglobin, a protein in red blood cells responsible for carrying oxygen throughout the body. The disease is caused by a mutation in the gene that produces the beta-globin chain of hemoglobin, resulting in the production of an abnormal form of hemoglobin called Hemoglobin C (HbC).
People with Hemoglobin C disease inherit one copy of the HbC gene from each parent. This means they have two copies of the mutated gene and produce mostly Hemoglobin C, instead of the normal Hemoglobin A. The presence of Hemoglobin C can cause the red blood cells to become rigid and fragile, leading to a condition called hemolytic anemia.
Symptoms of Hemoglobin C disease may include fatigue, weakness, shortness of breath, pale skin, jaundice, and dark urine. The severity of the symptoms can vary widely from person to person, with some individuals experiencing mild symptoms and others having more severe complications.
Hemoglobin C disease is a chronic condition that requires ongoing medical management, including regular monitoring of hemoglobin levels, iron status, and other blood parameters. Treatment may include blood transfusions, folic acid supplementation, and medications to manage symptoms such as anemia and pain.
It's important to note that Hemoglobin C disease is not the same as sickle cell disease, which is another genetic disorder that affects hemoglobin structure and function. While both conditions can cause hemolytic anemia, they are caused by different mutations in the beta-globin gene and have distinct clinical features and management approaches.
Sideroblastic anemia is a type of anemia characterized by the presence of ringed sideroblasts in the bone marrow. Ringed sideroblasts are red blood cell precursors that have an abnormal amount of iron accumulated in their mitochondria, which forms a ring around the nucleus. This results in the production of abnormal hemoglobin and impaired oxygen transport.
Sideroblastic anemia can be classified as congenital or acquired. Congenital sideroblastic anemias are caused by genetic defects that affect heme synthesis or mitochondrial function, while acquired sideroblastic anemias are associated with various conditions such as myelodysplastic syndromes, chronic alcoholism, lead toxicity, and certain medications.
Symptoms of sideroblastic anemia may include fatigue, weakness, shortness of breath, and pallor. Diagnosis is typically made through a bone marrow aspiration and biopsy, which can identify the presence of ringed sideroblasts. Treatment depends on the underlying cause but may include iron chelation therapy, vitamin B6 supplementation, or blood transfusions.
Hemolysis is the destruction or breakdown of red blood cells, resulting in the release of hemoglobin into the surrounding fluid (plasma). This process can occur due to various reasons such as chemical agents, infections, autoimmune disorders, mechanical trauma, or genetic abnormalities. Hemolysis may lead to anemia and jaundice, among other complications. It is essential to monitor hemolysis levels in patients undergoing medical treatments that might cause this condition.
Hemoglobin A is the most common form of hemoglobin, which is the oxygen-carrying protein in red blood cells. Hemoglobin A is a tetramer composed of two alpha and two beta globin chains, each containing a heme group that binds to oxygen. It is typically measured in laboratory tests to assess for various medical conditions such as anemia or diabetes. In the context of diabetes, the measurement of hemoglobin A1c (a form of hemoglobin A that is glycated or bound to glucose) is used to monitor long-term blood sugar control.
Megaloblastic anemia is a type of macrocytic anemia, which is characterized by the presence of large, structurally abnormal, and immature red blood cells called megaloblasts in the bone marrow. This condition arises due to impaired DNA synthesis during erythropoiesis (the process of red blood cell production), often as a result of deficiencies in vitamin B12 or folate, or from the use of certain medications that interfere with DNA synthesis.
The hallmark feature of megaloblastic anemia is the presence of megaloblasts in the bone marrow, which exhibit an asynchrony between nuclear and cytoplasmic maturation. This means that although the cytoplasm of these cells may appear well-developed, their nuclei remain underdeveloped and fragmented. As a result, the peripheral blood shows an increase in mean corpuscular volume (MCV), reflecting the larger size of the red blood cells.
Additional hematological findings include decreased reticulocyte counts, neutrophil hypersegmentation, and occasionally thrombocytopenia or leukopenia. Neurological symptoms may also be present due to the involvement of the nervous system in vitamin B12 deficiency.
Megaloblastic anemia is typically treated with supplementation of the deficient vitamin (B12 or folate), which helps restore normal erythropoiesis and alleviate symptoms over time.
Erythrocyte count, also known as red blood cell (RBC) count, is a laboratory test that measures the number of red blood cells in a sample of blood. Red blood cells are important because they carry oxygen from the lungs to the rest of the body. A low erythrocyte count may indicate anemia, while a high count may be a sign of certain medical conditions such as polycythemia. The normal range for erythrocyte count varies depending on a person's age, sex, and other factors.
Equine Infectious Anemia (EIA) is a viral disease that affects horses and other equine animals. The causative agent of this disease is the Equine Infectious Anemia Virus (EIAV), which belongs to the family Retroviridae and genus Lentivirus. This virus is primarily transmitted through the transfer of infected blood, most commonly through biting insects such as horseflies and deerflies.
The EIAV attacks the immune system of the infected animal, causing a variety of symptoms including fever, weakness, weight loss, anemia, and edema. The virus has a unique ability to integrate its genetic material into the host's DNA, which can lead to a lifelong infection. Some animals may become chronic carriers of the virus, showing no signs of disease but remaining infectious to others.
There is currently no cure for EIA, and infected animals must be isolated to prevent the spread of the disease. Vaccines are available in some countries, but they do not provide complete protection against infection and may only help reduce the severity of the disease. Regular testing and monitoring of equine populations are essential to control the spread of this virus.
Refractory anemia is a type of anemia that does not respond to typical treatments, such as iron supplements or hormonal therapy. It is often associated with various bone marrow disorders, including myelodysplastic syndromes (MDS), a group of conditions characterized by abnormal blood cell production in the bone marrow.
In refractory anemia, the bone marrow fails to produce enough healthy red blood cells, leading to symptoms such as fatigue, weakness, shortness of breath, and pale skin. The condition can be difficult to treat, and treatment options may include more aggressive therapies such as immunosuppressive drugs, chemotherapy, or stem cell transplantation.
It is important to note that the term "refractory" in this context refers specifically to the lack of response to initial treatments, rather than a specific severity or type of anemia.
Hemolytic anemia, congenital is a type of anemia that is present at birth and characterized by the abnormal breakdown (hemolysis) of red blood cells. This can occur due to various genetic defects that affect the structure or function of the red blood cells, making them more susceptible to damage and destruction.
There are several types of congenital hemolytic anemias, including:
1. Congenital spherocytosis: A condition caused by mutations in genes that affect the shape and stability of red blood cells, leading to the formation of abnormally shaped and fragile cells that are prone to hemolysis.
2. G6PD deficiency: A genetic disorder that affects the enzyme glucose-6-phosphate dehydrogenase (G6PD), which is essential for protecting red blood cells from damage. People with this condition have low levels of G6PD, making their red blood cells more susceptible to hemolysis when exposed to certain triggers such as infections or certain medications.
3. Hereditary elliptocytosis: A condition caused by mutations in genes that affect the structure and flexibility of red blood cells, leading to the formation of abnormally shaped and fragile cells that are prone to hemolysis.
4. Pyruvate kinase deficiency: A rare genetic disorder that affects an enzyme called pyruvate kinase, which is essential for the production of energy in red blood cells. People with this condition have low levels of pyruvate kinase, leading to the formation of fragile and abnormally shaped red blood cells that are prone to hemolysis.
Symptoms of congenital hemolytic anemia can vary depending on the severity of the condition but may include fatigue, weakness, pale skin, jaundice, dark urine, and an enlarged spleen. Treatment may involve blood transfusions, medications to manage symptoms, and in some cases, surgery to remove the spleen.
Erythrocyte indices are a set of calculated values that provide information about the size and hemoglobin content of red blood cells (erythrocytes). These indices are commonly used in the complete blood count (CBC) test to help diagnose various types of anemia and other conditions affecting the red blood cells.
The three main erythrocyte indices are:
1. Mean Corpuscular Volume (MCV): This is the average volume of a single red blood cell, measured in femtoliters (fL). MCV helps to differentiate between microcytic, normocytic, and macrocytic anemia. Microcytic anemia is characterized by low MCV values (100 fL).
2. Mean Corpuscular Hemoglobin (MCH): This is the average amount of hemoglobin present in a single red blood cell, measured in picograms (pg). MCH helps to assess the oxygen-carrying capacity of red blood cells. Low MCH values may indicate hypochromic anemia, where the red blood cells have reduced hemoglobin content.
3. Mean Corpuscular Hemoglobin Concentration (MCHC): This is the average concentration of hemoglobin in a single red blood cell, measured as a percentage. MCHC reflects the hemoglobin concentration relative to the size of the red blood cells. Low MCHC values may indicate hypochromic anemia, while high MCHC values could suggest spherocytosis or other conditions affecting red blood cell shape and integrity.
These erythrocyte indices are calculated based on the red blood cell count, hemoglobin concentration, and hematocrit results obtained from a CBC test. They provide valuable information for healthcare professionals to diagnose and manage various hematological conditions.
Abnormal hemoglobins refer to variants of the oxygen-carrying protein found in red blood cells, which differ from the normal adult hemoglobin (HbA) in terms of their structure and function. These variations can result from genetic mutations that affect the composition of the globin chains in the hemoglobin molecule. Some abnormal hemoglobins are clinically insignificant, while others can lead to various medical conditions such as hemolytic anemia, thalassemia, or sickle cell disease. Examples of abnormal hemoglobins include HbS (associated with sickle cell anemia), HbC, HbE, and HbF (fetal hemoglobin). These variants can be detected through specialized laboratory tests, such as hemoglobin electrophoresis or high-performance liquid chromatography (HPLC).
Erythrocyte deformability refers to the ability of red blood cells (erythrocytes) to change shape and bend without rupturing, which is crucial for their efficient movement through narrow blood vessels. This deformability is influenced by several factors including the cell membrane structure, hemoglobin concentration, and intracellular viscosity. A decrease in erythrocyte deformability can negatively impact blood flow and oxygen delivery to tissues, potentially contributing to various pathological conditions such as sickle cell disease, diabetes, and cardiovascular diseases.
Hematocrit is a medical term that refers to the percentage of total blood volume that is made up of red blood cells. It is typically measured as part of a complete blood count (CBC) test. A high hematocrit may indicate conditions such as dehydration, polycythemia, or living at high altitudes, while a low hematocrit may be a sign of anemia, bleeding, or overhydration. It is important to note that hematocrit values can vary depending on factors such as age, gender, and pregnancy status.
An exchange transfusion of whole blood is a medical procedure in which a patient's blood is gradually replaced with donor whole blood. This procedure is typically performed in newborns or infants who have severe jaundice caused by excessive levels of bilirubin, a yellowish pigment that forms when hemoglobin from red blood cells breaks down.
During an exchange transfusion, the baby's blood is removed through a vein or artery and replaced with donor whole blood through another vein or artery. The process is repeated several times until a significant portion of the baby's blood has been exchanged with donor blood. This helps to reduce the levels of bilirubin in the baby's blood, which can help prevent or treat brain damage caused by excessive bilirubin.
Exchange transfusions are typically performed in a neonatal intensive care unit (NICU) and require close monitoring by a team of healthcare professionals. The procedure carries some risks, including infection, bleeding, and changes in blood pressure or heart rate. However, it can be a lifesaving treatment for newborns with severe jaundice who are at risk of developing serious complications.
Hemoglobin C is a type of hemoglobin variant, which is the oxygen-carrying protein in red blood cells. Hemoglobin C is caused by a specific genetic mutation that results in the substitution of lysine for glutamic acid at position 6 on the beta globin chain of the hemoglobin molecule.
This variant is often associated with a benign condition known as hemoglobin C trait, where an individual inherits one copy of the mutated gene from one parent and one normal gene from the other parent. People with this trait usually have no symptoms or only mild anemia, if any. However, if an individual inherits two copies of the Hemoglobin C gene (one from each parent), they will have a more severe form of hemoglobin disorder called Hemoglobin CC disease, which can cause mild to moderate hemolytic anemia and other complications.
It's important to note that Hemoglobin C is most commonly found in people of West African descent, but it can also occur in other populations with African ancestry.
A leg ulcer is a chronic wound that occurs on the lower extremities, typically on the inner or outer ankle. It's often caused by poor circulation, venous insufficiency, or diabetes. Leg ulcers can also result from injury, infection, or inflammatory diseases such as rheumatoid arthritis or lupus. These ulcers can be painful, and they may take a long time to heal, making them prone to infection. Proper diagnosis, treatment, and wound care are essential for healing leg ulcers and preventing complications.
Globins are a group of proteins that contain a heme prosthetic group, which binds and transports oxygen in the blood. The most well-known globin is hemoglobin, which is found in red blood cells and is responsible for carrying oxygen from the lungs to the body's tissues. Other members of the globin family include myoglobin, which is found in muscle tissue and stores oxygen, and neuroglobin and cytoglobin, which are found in the brain and other organs and may have roles in protecting against oxidative stress and hypoxia (low oxygen levels). Globins share a similar structure, with a folded protein surrounding a central heme group. Mutations in globin genes can lead to various diseases, such as sickle cell anemia and thalassemia.
Hematologic pregnancy complications refer to disorders related to the blood and blood-forming tissues that occur during pregnancy. These complications can have serious consequences for both the mother and the fetus if not properly managed. Some common hematologic pregnancy complications include:
1. Anemia: A condition characterized by a decrease in the number of red blood cells or hemoglobin in the blood, which can lead to fatigue, weakness, and shortness of breath. Iron-deficiency anemia is the most common type of anemia during pregnancy.
2. Thrombocytopenia: A condition characterized by a decrease in the number of platelets (cells that help blood clot) in the blood. Mild thrombocytopenia is relatively common during pregnancy, but severe thrombocytopenia can increase the risk of bleeding during delivery.
3. Gestational thrombotic thrombocytopenic purpura (GTTP): A rare but serious disorder that can cause blood clots to form in small blood vessels throughout the body, leading to a decrease in the number of platelets and red blood cells. GTTP can cause serious complications such as stroke, kidney failure, and even death if not promptly diagnosed and treated.
4. Disseminated intravascular coagulation (DIC): A condition characterized by abnormal clotting and bleeding throughout the body. DIC can be triggered by various conditions such as severe infections, pregnancy complications, or cancer.
5. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: A serious complication of pregnancy that can cause damage to the liver and lead to bleeding. HELLP syndrome is often associated with preeclampsia, a condition characterized by high blood pressure and damage to organs such as the liver and kidneys.
It's important for pregnant women to receive regular prenatal care to monitor for these and other potential complications, and to seek prompt medical attention if any concerning symptoms arise.
Erythrocyte aging, also known as red cell aging, is the natural process of changes and senescence that occur in red blood cells (erythrocytes) over time. In humans, mature erythrocytes are devoid of nuclei and organelles, and have a lifespan of approximately 120 days.
During aging, several biochemical and structural modifications take place in the erythrocyte, including:
1. Loss of membrane phospholipids and proteins, leading to increased rigidity and decreased deformability.
2. Oxidative damage to hemoglobin, resulting in the formation of methemoglobin and heinz bodies.
3. Accumulation of denatured proteins and aggregates, which can impair cellular functions.
4. Changes in the cytoskeleton, affecting the shape and stability of the erythrocyte.
5. Increased expression of surface markers, such as Band 3 and CD47, that signal the spleen to remove aged erythrocytes from circulation.
The spleen plays a crucial role in removing senescent erythrocytes by recognizing and phagocytosing those with altered membrane composition or increased expression of surface markers. This process helps maintain the overall health and functionality of the circulatory system.
Erythropoietin (EPO) is a hormone that is primarily produced by the kidneys and plays a crucial role in the production of red blood cells in the body. It works by stimulating the bone marrow to produce more red blood cells, which are essential for carrying oxygen to various tissues and organs.
EPO is a glycoprotein that is released into the bloodstream in response to low oxygen levels in the body. When the kidneys detect low oxygen levels, they release EPO, which then travels to the bone marrow and binds to specific receptors on immature red blood cells called erythroblasts. This binding triggers a series of events that promote the maturation and proliferation of erythroblasts, leading to an increase in the production of red blood cells.
In addition to its role in regulating red blood cell production, EPO has also been shown to have neuroprotective effects and may play a role in modulating the immune system. Abnormal levels of EPO have been associated with various medical conditions, including anemia, kidney disease, and certain types of cancer.
EPO is also used as a therapeutic agent for the treatment of anemia caused by chronic kidney disease, chemotherapy, or other conditions that affect red blood cell production. Recombinant human EPO (rhEPO) is a synthetic form of the hormone that is produced using genetic engineering techniques and is commonly used in clinical practice to treat anemia. However, misuse of rhEPO for performance enhancement in sports has been a subject of concern due to its potential to enhance oxygen-carrying capacity and improve endurance.
In the context of medicine, iron is an essential micromineral and key component of various proteins and enzymes. It plays a crucial role in oxygen transport, DNA synthesis, and energy production within the body. Iron exists in two main forms: heme and non-heme. Heme iron is derived from hemoglobin and myoglobin in animal products, while non-heme iron comes from plant sources and supplements.
The recommended daily allowance (RDA) for iron varies depending on age, sex, and life stage:
* For men aged 19-50 years, the RDA is 8 mg/day
* For women aged 19-50 years, the RDA is 18 mg/day
* During pregnancy, the RDA increases to 27 mg/day
* During lactation, the RDA for breastfeeding mothers is 9 mg/day
Iron deficiency can lead to anemia, characterized by fatigue, weakness, and shortness of breath. Excessive iron intake may result in iron overload, causing damage to organs such as the liver and heart. Balanced iron levels are essential for maintaining optimal health.
An erythrocyte transfusion, also known as a red blood cell (RBC) transfusion, is the process of transferring compatible red blood cells from a donor to a recipient. This procedure is typically performed to increase the recipient's oxygen-carrying capacity, usually in situations where there is significant blood loss, anemia, or impaired red blood cell production.
During the transfusion, the donor's red blood cells are collected, typed, and tested for compatibility with the recipient's blood to minimize the risk of a transfusion reaction. Once compatible units are identified, they are infused into the recipient's circulation through a sterile intravenous (IV) line. The recipient's body will eventually eliminate the donated red blood cells within 100-120 days as part of its normal turnover process.
Erythrocyte transfusions can be lifesaving in various clinical scenarios, such as trauma, surgery, severe anemia due to chronic diseases, and hematologic disorders. However, they should only be used when necessary, as there are potential risks associated with the procedure, including allergic reactions, transmission of infectious diseases, transfusion-related acute lung injury (TRALI), and iron overload in cases of multiple transfusions.
Equine infectious anemia (EIA) is a viral disease that affects horses and other equine animals. It is caused by the Equine Infectious Anemia Virus (EIAV), which is transmitted through the bloodstream of infected animals, often through biting insects such as horseflies and deerflies.
The symptoms of EIA can vary widely, but often include fever, weakness, weight loss, anemia, and edema. In severe cases, the disease can cause death. There is no cure for EIA, and infected animals must be isolated to prevent the spread of the virus.
EIA is diagnosed through blood tests that detect the presence of antibodies to the virus. Horses that test positive for EIA are typically euthanized or permanently quarantined. Prevention measures include testing horses before they are bought, sold, or moved, as well as controlling insect populations and using insect repellents. Vaccines are not available for EIA in most countries.
Reticulocytes are immature red blood cells that still contain remnants of organelles, such as ribosomes and mitochondria, which are typically found in developing cells. These organelles are involved in the process of protein synthesis and energy production, respectively. Reticulocytes are released from the bone marrow into the bloodstream, where they continue to mature into fully developed red blood cells called erythrocytes.
Reticulocytes can be identified under a microscope by their staining characteristics, which reveal a network of fine filaments or granules known as the reticular apparatus. This apparatus is composed of residual ribosomal RNA and other proteins that have not yet been completely eliminated during the maturation process.
The percentage of reticulocytes in the blood can be used as a measure of bone marrow function and erythropoiesis, or red blood cell production. An increased reticulocyte count may indicate an appropriate response to blood loss, hemolysis, or other conditions that cause anemia, while a decreased count may suggest impaired bone marrow function or a deficiency in erythropoietin, the hormone responsible for stimulating red blood cell production.
Pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage. It is a complex phenomenon that can result from various stimuli, such as thermal, mechanical, or chemical irritation, and it can be acute or chronic. The perception of pain involves the activation of specialized nerve cells called nociceptors, which transmit signals to the brain via the spinal cord. These signals are then processed in different regions of the brain, leading to the conscious experience of pain. It's important to note that pain is a highly individual and subjective experience, and its perception can vary widely among individuals.
Splenic infarction is the death of splenic tissue due to blockage of its arterial supply or, less commonly, its venous drainage. This results in ischemia and necrosis of the affected portion of the spleen. The most common cause is embolism from a distant source such as atrial fibrillation, infective endocarditis, or malignancy. Other causes include splenic artery thrombosis, sickle cell disease, hematologic disorders, and trauma. Clinical presentation can vary widely, ranging from being asymptomatic to acute abdominal pain, nausea, vomiting, and fever. Diagnosis is often made with imaging studies such as ultrasound or CT scan. Treatment depends on the underlying cause and severity of symptoms, but may include anticoagulation, antibiotics, or surgical intervention in severe cases.
An erythrocyte, also known as a red blood cell, is a type of cell that circulates in the blood and is responsible for transporting oxygen throughout the body. The erythrocyte membrane refers to the thin, flexible barrier that surrounds the erythrocyte and helps to maintain its shape and stability.
The erythrocyte membrane is composed of a lipid bilayer, which contains various proteins and carbohydrates. These components help to regulate the movement of molecules into and out of the erythrocyte, as well as provide structural support and protection for the cell.
The main lipids found in the erythrocyte membrane are phospholipids and cholesterol, which are arranged in a bilayer structure with the hydrophilic (water-loving) heads facing outward and the hydrophobic (water-fearing) tails facing inward. This arrangement helps to maintain the integrity of the membrane and prevent the leakage of cellular components.
The proteins found in the erythrocyte membrane include integral proteins, which span the entire width of the membrane, and peripheral proteins, which are attached to the inner or outer surface of the membrane. These proteins play a variety of roles, such as transporting molecules across the membrane, maintaining the shape of the erythrocyte, and interacting with other cells and proteins in the body.
The carbohydrates found in the erythrocyte membrane are attached to the outer surface of the membrane and help to identify the cell as part of the body's own immune system. They also play a role in cell-cell recognition and adhesion.
Overall, the erythrocyte membrane is a complex and dynamic structure that plays a critical role in maintaining the function and integrity of red blood cells.
Hemoglobinometry is a method used to measure the amount or concentration of hemoglobin (Hb) in blood. Hemoglobin is a protein in red blood cells that carries oxygen throughout the body. Hemoglobinometry is typically performed on a sample of whole blood and can be done using various methods, including spectrophotometry, colorimetry, or automated analyzers.
The results of hemoglobinometry are reported in units of grams per deciliter (g/dL) or grams per liter (g/L). Normal values for hemoglobin concentration vary depending on factors such as age, sex, and altitude, but in general, a healthy adult male should have a hemoglobin level between 13.5 and 17.5 g/dL, while a healthy adult female should have a level between 12.0 and 15.5 g/dL.
Hemoglobinometry is an important diagnostic tool in the evaluation of various medical conditions, including anemia, polycythemia, and respiratory disorders. It can help identify the cause of symptoms such as fatigue, shortness of breath, or dizziness and guide treatment decisions.
Chicken anemia virus (CAV) is a small, non-enveloped DNA virus that belongs to the family *Circoviridae* and genus *Gyrovirus*. It primarily infects chickens and causes a variety of clinical signs, including severe anemia, immunosuppression, and runting in young birds.
The virus is highly contagious and can be spread through horizontal transmission via feces, contaminated equipment, or vertically from infected breeder hens to their offspring. CAV infection can lead to significant economic losses in the poultry industry due to decreased growth rates, increased mortality, and reduced egg production.
In addition to its impact on the poultry industry, CAV has also been used as a vector for gene delivery in biomedical research. Its small genome size and ability to infect a wide range of avian species make it an attractive candidate for vaccine development and gene therapy applications.
Dyserythropoietic anemia, congenital is a rare type of inherited anemia characterized by ineffective red blood cell production (erythropoiesis) in the bone marrow. This means that the body has difficulty producing healthy and fully mature red blood cells. The condition is caused by mutations in genes responsible for the development and maturation of red blood cells, leading to the production of abnormally shaped and dysfunctional red blood cells.
There are two main types of congenital dyserythropoietic anemia (CDA), type I and type II, each caused by different genetic mutations:
1. CDA Type I (HEMPAS): This form is caused by a mutation in the SEC23B gene. It typically presents in early childhood with mild to moderate anemia, jaundice, and splenomegaly (enlarged spleen). The severity of the condition can vary widely among affected individuals.
2. CDA Type II (HIEM): This form is caused by a mutation in the KIF23 gene or, less commonly, the TCIRG1 gene. It typically presents in infancy with moderate to severe anemia, hepatomegaly (enlarged liver), and splenomegaly. The condition can lead to iron overload due to repeated blood transfusions, which may require chelation therapy to manage.
Both types of congenital dyserythropoietic anemia are characterized by ineffective erythropoiesis, abnormal red blood cell morphology, and increased destruction of red blood cells (hemolysis). Treatment typically involves supportive care, such as blood transfusions to manage anemia, and occasionally chelation therapy to address iron overload. In some cases, bone marrow transplantation may be considered as a curative option.
Erythropoiesis is the process of forming and developing red blood cells (erythrocytes) in the body. It occurs in the bone marrow and is regulated by the hormone erythropoietin (EPO), which is produced by the kidneys. Erythropoiesis involves the differentiation and maturation of immature red blood cell precursors called erythroblasts into mature red blood cells, which are responsible for carrying oxygen to the body's tissues. Disorders that affect erythropoiesis can lead to anemia or other blood-related conditions.
Diamond-Blackfan anemia is a rare, congenital bone marrow failure disorder characterized by a decreased production of red blood cells (erythroblasts) in the bone marrow. This results in a reduced number of circulating red blood cells, leading to anemia and related symptoms such as fatigue, weakness, and pallor. The disorder is typically diagnosed in infancy or early childhood and can also be associated with physical abnormalities.
The exact cause of Diamond-Blackfan anemia is not fully understood, but it is believed to involve genetic mutations that affect the development and function of the bone marrow. In many cases, the disorder is inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the mutated gene from an affected parent. However, some cases may arise spontaneously due to new genetic mutations.
Treatment for Diamond-Blackfan anemia typically involves regular blood transfusions to maintain adequate red blood cell levels and alleviate symptoms. Corticosteroid therapy may also be used to stimulate red blood cell production in some cases. In severe or refractory cases, stem cell transplantation may be considered as a curative treatment option.
Vascular diseases are medical conditions that affect the circulatory system, specifically the blood vessels (arteries, veins, and capillaries). These diseases can include conditions such as:
1. Atherosclerosis: The buildup of fats, cholesterol, and other substances in and on the walls of the arteries, which can restrict blood flow.
2. Peripheral Artery Disease (PAD): A condition caused by atherosclerosis where there is narrowing or blockage of the peripheral arteries, most commonly in the legs. This can lead to pain, numbness, and cramping.
3. Coronary Artery Disease (CAD): Atherosclerosis of the coronary arteries that supply blood to the heart muscle. This can lead to chest pain, shortness of breath, or a heart attack.
4. Carotid Artery Disease: Atherosclerosis of the carotid arteries in the neck that supply blood to the brain. This can increase the risk of stroke.
5. Cerebrovascular Disease: Conditions that affect blood flow to the brain, including stroke and transient ischemic attack (TIA or "mini-stroke").
6. Aneurysm: A weakened area in the wall of a blood vessel that causes it to bulge outward and potentially rupture.
7. Deep Vein Thrombosis (DVT): A blood clot that forms in the deep veins, usually in the legs, which can cause pain, swelling, and increased risk of pulmonary embolism if the clot travels to the lungs.
8. Varicose Veins: Swollen, twisted, and often painful veins that have filled with an abnormal collection of blood, usually appearing in the legs.
9. Vasculitis: Inflammation of the blood vessels, which can cause damage and narrowing, leading to reduced blood flow.
10. Raynaud's Phenomenon: A condition where the small arteries that supply blood to the skin become narrowed, causing decreased blood flow, typically in response to cold temperatures or stress.
These are just a few examples of vascular conditions that fall under the umbrella term "cerebrovascular disease." Early diagnosis and treatment can significantly improve outcomes for many of these conditions.
Blood viscosity is a measure of the thickness or flow resistance of blood. It is defined as the ratio of shear stress to shear rate within the flowing blood, which reflects the internal friction or resistance to flow. Blood viscosity is primarily determined by the concentration and size of red blood cells (hematocrit), plasma proteins, and other blood constituents. An increase in any of these components can raise blood viscosity, leading to impaired blood flow, reduced oxygen delivery to tissues, and potential cardiovascular complications if not managed appropriately.
A homozygote is an individual who has inherited the same allele (version of a gene) from both parents and therefore possesses two identical copies of that allele at a specific genetic locus. This can result in either having two dominant alleles (homozygous dominant) or two recessive alleles (homozygous recessive). In contrast, a heterozygote has inherited different alleles from each parent for a particular gene.
The term "homozygote" is used in genetics to describe the genetic makeup of an individual at a specific locus on their chromosomes. Homozygosity can play a significant role in determining an individual's phenotype (observable traits), as having two identical alleles can strengthen the expression of certain characteristics compared to having just one dominant and one recessive allele.
Fanconi anemia (FA) is a genetic disorder characterized by various developmental abnormalities, bone marrow failure, and increased risk of malignancies. It is caused by mutations in genes involved in the FA complementation group, which are responsible for repairing damaged DNA.
The FA complementation group proteins include FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1, FANCL, FANCM, and FAAP100. These proteins work together to form the FA core complex, which is responsible for monoubiquitinating FANCD2 and FANCI in response to DNA damage. This modification allows for the recruitment of downstream effectors that facilitate DNA repair and maintain genomic stability.
Defects in any of these FA complementation group proteins can lead to Fanconi anemia, with varying clinical manifestations depending on the specific gene involved and the severity of the mutation.
Iron overload is a condition characterized by an excessive accumulation of iron in the body's tissues and organs, particularly in the liver, heart, and pancreas. This occurs when the body absorbs more iron than it can use or eliminate, leading to iron levels that are higher than normal.
Iron overload can result from various factors, including hereditary hemochromatosis, a genetic disorder that affects how the body absorbs iron from food; frequent blood transfusions, which can cause iron buildup in people with certain chronic diseases such as sickle cell anemia or thalassemia; and excessive consumption of iron supplements or iron-rich foods.
Symptoms of iron overload may include fatigue, joint pain, abdominal discomfort, irregular heartbeat, and liver dysfunction. If left untreated, it can lead to serious complications such as cirrhosis, liver failure, diabetes, heart problems, and even certain types of cancer. Treatment typically involves regular phlebotomy (removal of blood) to reduce iron levels in the body, along with dietary modifications and monitoring by a healthcare professional.
Ferritin is a protein in iron-metabolizing cells that stores iron in a water-soluble form. It is found inside the cells (intracellular) and is released into the bloodstream when the cells break down or die. Measuring the level of ferritin in the blood can help determine the amount of iron stored in the body. High levels of ferritin may indicate hemochromatosis, inflammation, liver disease, or other conditions. Low levels of ferritin may indicate anemia, iron deficiency, or other conditions.
Oxygen is a colorless, odorless, tasteless gas that constitutes about 21% of the earth's atmosphere. It is a crucial element for human and most living organisms as it is vital for respiration. Inhaled oxygen enters the lungs and binds to hemoglobin in red blood cells, which carries it to tissues throughout the body where it is used to convert nutrients into energy and carbon dioxide, a waste product that is exhaled.
Medically, supplemental oxygen therapy may be provided to patients with conditions such as chronic obstructive pulmonary disease (COPD), pneumonia, heart failure, or other medical conditions that impair the body's ability to extract sufficient oxygen from the air. Oxygen can be administered through various devices, including nasal cannulas, face masks, and ventilators.
Erythrocyte aggregation, also known as rouleaux formation, is the clumping together of red blood cells (erythrocytes) in a way that resembles a stack of coins. This phenomenon is typically observed under low-shear conditions, such as those found in small blood vessels and capillaries.
The aggregation of erythrocytes is influenced by several factors, including the concentration of plasma proteins, the charge and shape of the red blood cells, and the flow characteristics of the blood. One of the most important proteins involved in this process is fibrinogen, a large plasma protein that can bridge between adjacent red blood cells and cause them to stick together.
Erythrocyte aggregation can have significant effects on blood flow and rheology (the study of how blood flows), particularly in diseases such as diabetes, sickle cell disease, and certain types of anemia. Increased erythrocyte aggregation can lead to reduced oxygen delivery to tissues, increased blood viscosity, and impaired microcirculatory flow, all of which can contribute to tissue damage and organ dysfunction.
Gamma-globulins are a type of globulin, which are proteins found in the blood plasma. More specifically, gamma-globulins are a class of immunoglobulins, also known as antibodies, that play a crucial role in the immune system's response to foreign substances and infectious agents.
Immunoglobulins are divided into several classes based on their structure and function. Gamma-globulins include IgG, IgA, and IgD isotypes of immunoglobulins. Among these, IgG is the most abundant type found in the blood and other body fluids, responsible for providing protection against bacterial and viral infections.
Gamma-globulins are produced by B cells, a type of white blood cell involved in the immune response. They can be measured in the blood as part of a complete blood count (CBC) or specific protein electrophoresis tests to assess immune system function or diagnose various medical conditions such as infections, inflammation, and autoimmune disorders.
I'm sorry for any confusion, but "Nigeria" is not a medical term. It is a country located in West Africa, and it is the most populous country in Africa. If you have any questions about medical conditions or terms, I would be happy to help clarify those for you.
A reticulocyte count is a laboratory test that measures the percentage of reticulocytes in the peripheral blood. Reticulocytes are immature red blood cells produced in the bone marrow and released into the bloodstream. They contain residual ribosomal RNA, which gives them a reticular or net-like appearance under a microscope when stained with certain dyes.
The reticulocyte count is often used as an indicator of the rate of red blood cell production in the bone marrow. A higher than normal reticulocyte count may indicate an increased production of red blood cells, which can be seen in conditions such as hemolysis, blood loss, or response to treatment of anemia. A lower than normal reticulocyte count may suggest a decreased production of red blood cells, which can be seen in conditions such as bone marrow suppression, aplastic anemia, or vitamin deficiencies.
The reticulocyte count is usually expressed as a percentage of the total number of red blood cells, but it can also be reported as an absolute reticulocyte count (the actual number of reticulocytes per microliter of blood). The normal range for the reticulocyte count varies depending on the laboratory and the population studied.
Oxyhemoglobin is the form of hemoglobin that is combined with oxygen in red blood cells. It's created when oxygen molecules bind to the iron-containing heme groups of the hemoglobin protein inside the lungs, allowing for the transportation of oxygen from the lungs to body tissues. The affinity of hemoglobin for oxygen is influenced by factors such as pH, carbon dioxide concentration, and temperature, which can affect the release of oxygen from oxyhemoglobin in different parts of the body based on their specific needs.
Hemorheology is the study of the flow properties of blood and its components, including red blood cells, white blood cells, platelets, and plasma. Specifically, it examines how these components interact with each other and with the walls of blood vessels to affect the flow characteristics of blood under different conditions. Hemorheological factors can influence blood viscosity, which is a major determinant of peripheral vascular resistance and cardiac workload. Abnormalities in hemorheology have been implicated in various diseases such as atherosclerosis, hypertension, diabetes, and sickle cell disease.
Beta-globins are the type of globin proteins that make up the beta-chain of hemoglobin, the oxygen-carrying protein in red blood cells. Hemoglobin is composed of four polypeptide chains, two alpha-globin and two beta-globin chains, arranged in a specific structure. The beta-globin gene is located on chromosome 11, and mutations in this gene can lead to various forms of hemoglobin disorders such as sickle cell anemia and beta-thalassemia.
Dimethyl adipimidate is a chemical compound that is used as a cross-linking agent in various biochemical and medical applications. It is an imidate ester of adipic acid, which contains two reactive dimethylamino groups. These groups can react with amino groups on proteins or other molecules to form covalent bonds, creating a cross-linked network.
In the context of medical research and diagnostics, dimethyl adipimidate is sometimes used to modify proteins in order to study their structure and function. For example, it can be used to create stable, cross-linked complexes between different proteins or protein domains, which can then be analyzed using various biochemical techniques.
It's important to note that dimethyl adipimidate is not a drug or therapeutic agent itself, but rather a tool used in laboratory research and diagnostics. As with any chemical compound, it should be handled with care and used only by trained professionals in a controlled environment.
Neonatal anemia is a condition characterized by a lower-than-normal number of red blood cells or lower-than-normal levels of hemoglobin in the blood of a newborn infant. Hemoglobin is the protein in red blood cells that carries oxygen to the body's tissues.
There are several types and causes of neonatal anemia, including:
1. Anemia of prematurity: This is the most common type of anemia in newborns, especially those born before 34 weeks of gestation. It occurs due to a decrease in red blood cell production and a shorter lifespan of red blood cells in premature infants.
2. Hemolytic anemia: This type of anemia is caused by the destruction of red blood cells at a faster rate than they can be produced. It can result from various factors, such as incompatibility between the mother's and baby's blood types, genetic disorders like G6PD deficiency, or infections.
3. Fetomaternal hemorrhage: This condition occurs when there is a significant transfer of fetal blood into the maternal circulation during pregnancy or childbirth, leading to anemia in the newborn.
4. Iron-deficiency anemia: Although rare in newborns, iron-deficiency anemia can occur if the mother has low iron levels during pregnancy, and the infant does not receive adequate iron supplementation after birth.
5. Anemia due to nutritional deficiencies: Rarely, neonatal anemia may result from a lack of essential vitamins or minerals like folate, vitamin B12, or copper in the newborn's diet.
Symptoms of neonatal anemia can vary but may include pallor, lethargy, poor feeding, rapid heartbeat, and difficulty breathing. Diagnosis typically involves a complete blood count (CBC) to measure red blood cell count, hemoglobin levels, and other parameters. Treatment depends on the underlying cause of anemia and may include iron supplementation, transfusions, or management of any underlying conditions.
Prevalence, in medical terms, refers to the total number of people in a given population who have a particular disease or condition at a specific point in time, or over a specified period. It is typically expressed as a percentage or a ratio of the number of cases to the size of the population. Prevalence differs from incidence, which measures the number of new cases that develop during a certain period.
Splenic diseases refer to a range of medical conditions that affect the structure, function, or health of the spleen. The spleen is an organ located in the upper left quadrant of the abdomen, which plays a vital role in filtering the blood and fighting infections. Some common splenic diseases include:
1. Splenomegaly: Enlargement of the spleen due to various causes such as infections, liver disease, blood disorders, or cancer.
2. Hypersplenism: Overactivity of the spleen leading to excessive removal of blood cells from circulation, causing anemia, leukopenia, or thrombocytopenia.
3. Splenic infarction: Partial or complete blockage of the splenic artery or its branches, resulting in tissue death and potential organ dysfunction.
4. Splenic rupture: Traumatic or spontaneous tearing of the spleen capsule, causing internal bleeding and potentially life-threatening conditions.
5. Infections: Bacterial (e.g., sepsis, tuberculosis), viral (e.g., mononucleosis, cytomegalovirus), fungal (e.g., histoplasmosis), or parasitic (e.g., malaria) infections can affect the spleen and cause various symptoms.
6. Hematologic disorders: Conditions such as sickle cell disease, thalassemia, hemolytic anemias, lymphomas, leukemias, or myeloproliferative neoplasms can involve the spleen and lead to its enlargement or dysfunction.
7. Autoimmune diseases: Conditions like rheumatoid arthritis, systemic lupus erythematosus, or vasculitis can affect the spleen and cause various symptoms.
8. Cancers: Primary (e.g., splenic tumors) or secondary (e.g., metastatic cancer from other organs) malignancies can involve the spleen and lead to its enlargement, dysfunction, or rupture.
9. Vascular abnormalities: Conditions such as portal hypertension, Budd-Chiari syndrome, or splenic vein thrombosis can affect the spleen and cause various symptoms.
10. Trauma: Accidental or intentional injuries to the spleen can lead to bleeding, infection, or organ dysfunction.
The Lutheran blood group system is a relatively less known and rare blood group system, discovered by Dr. Karl Landsteiner and Dr. Wiener in 1940. It is named after the Lutheran Church in Brooklyn where the serum that led to its discovery was obtained. The Lutheran blood group system consists of four main antigens: Lu^a, Lu^b, Lu^a/b, and In(Lu). These antigens are found on the surface of red blood cells (RBCs) and can cause an immune response when foreign antigens are introduced into the body.
The Lutheran system is inherited in an autosomal dominant manner, which means that a person needs only one copy of the gene to express the antigen. Approximately 98% of the population expresses the Lu(a-b-) phenotype, which lacks both Lu^a and Lu^b antigens. The other common phenotypes include Lu(a+b-) and Lu(a-b+), while the rarest is Lu(a+b+).
Individuals with the Lu(a-b-) phenotype can produce antibodies against both Lu^a and Lu^b antigens, which can cause transfusion reactions or hemolytic disease of the newborn (HDN) if they receive blood from a donor with either Lu^a or Lu^b antigens. Therefore, it is essential to consider the Lutheran blood group system during blood transfusions and pregnancy to ensure compatibility and prevent adverse effects.
In summary, the Lutheran blood group system consists of four main antigens (Lu^a, Lu^b, Lu^a/b, and In(Lu)) found on RBCs, which can cause an immune response in some individuals. Proper identification and matching of these antigens are crucial to prevent transfusion reactions and HDN.
Hematinics are a class of medications and dietary supplements that are used to enhance the production of red blood cells or hemoglobin in the body. They typically contain iron, vitamin B12, folic acid, or other nutrients that are essential for the synthesis of hemoglobin and the formation of red blood cells.
Iron is a critical component of hematinics because it plays a central role in the production of hemoglobin, which is the protein in red blood cells that carries oxygen throughout the body. Vitamin B12 and folic acid are also important nutrients for red blood cell production, as they help to regulate the growth and division of red blood cells in the bone marrow.
Hematinics are often prescribed to treat anemia, which is a condition characterized by a low red blood cell count or abnormally low levels of hemoglobin in the blood. Anemia can be caused by a variety of factors, including nutritional deficiencies, chronic diseases, and inherited genetic disorders.
Examples of hematinics include ferrous sulfate (an iron supplement), cyanocobalamin (vitamin B12), and folic acid. These medications are available in various forms, such as tablets, capsules, and liquids, and can be taken orally or by injection. It is important to follow the dosage instructions carefully and to inform your healthcare provider of any other medications you are taking, as hematinics can interact with certain drugs and may cause side effects.
Refractory anemia with excess blasts is a type of blood disorder that is characterized by the presence of increased numbers of immature blood cells, or "blasts," in the bone marrow and peripheral blood. This condition is considered a subtype of myelodysplastic syndrome (MDS), which is a group of disorders caused by abnormalities in the production of blood cells in the bone marrow.
In refractory anemia with excess blasts, the bone marrow fails to produce sufficient numbers of healthy red blood cells, white blood cells, and platelets. This results in anemia (low red blood cell count), neutropenia (low white blood cell count), and thrombocytopenia (low platelet count). Additionally, there is an increased number of blasts in the bone marrow and peripheral blood, which can indicate the development of acute myeloid leukemia (AML), a more aggressive form of blood cancer.
Refractory anemia with excess blasts is considered "refractory" because it does not respond well to treatment, including chemotherapy and stem cell transplantation. The prognosis for this condition varies depending on the severity of the disease and other individual factors, but it is generally poor, with many patients progressing to AML within a few years.
Kidney papillary necrosis is a medical condition characterized by the death (necrosis) of the renal papillae, which are the small conical projections at the ends of the renal tubules in the kidneys. This condition typically occurs due to reduced blood flow to the kidneys or as a result of toxic injury from certain medications, chronic infections, diabetes, sickle cell disease, and systemic vasculitides.
The necrosis of the papillae can lead to the formation of small stones or debris that can obstruct the flow of urine, causing further damage to the kidneys. Symptoms of kidney papillary necrosis may include fever, flank pain, nausea, vomiting, and bloody or foul-smelling urine. The diagnosis is typically made through imaging studies such as CT scans or MRI, and treatment may involve addressing the underlying cause, administering antibiotics to prevent infection, and providing supportive care to maintain kidney function.
Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency is a genetic disorder that affects the normal functioning of an enzyme called G6PD. This enzyme is found in red blood cells and plays a crucial role in protecting them from damage.
In people with G6PD deficiency, the enzyme's activity is reduced or absent, making their red blood cells more susceptible to damage and destruction, particularly when they are exposed to certain triggers such as certain medications, infections, or foods. This can lead to a condition called hemolysis, where the red blood cells break down prematurely, leading to anemia, jaundice, and in severe cases, kidney failure.
G6PD deficiency is typically inherited from one's parents in an X-linked recessive pattern, meaning that males are more likely to be affected than females. While there is no cure for G6PD deficiency, avoiding triggers and managing symptoms can help prevent complications.
Priapism is defined as a persistent and painful erection of the penis that lasts for more than four hours and occurs without sexual stimulation. It's a serious medical condition that requires immediate attention, as it can lead to permanent damage to the penis if left untreated.
Priapism can be classified into two types: ischemic (or low-flow) priapism and nonischemic (or high-flow) priapism. Ischemic priapism is the more common form, and it occurs when blood flow to the penis is obstructed, leading to the accumulation of deoxygenated blood in the corpora cavernosa. Nonischemic priapism, on the other hand, is usually caused by unregulated arterial blood flow into the corpora cavernosa, often as a result of trauma or surgery.
The causes of priapism can vary, but some common underlying conditions include sickle cell disease, leukemia, spinal cord injuries, and certain medications such as antidepressants and drugs used to treat erectile dysfunction. Treatment for priapism depends on the type and cause of the condition, and may involve medication, aspiration of blood from the penis, or surgical intervention.
A "Blood Cell Count" is a medical laboratory test that measures the number of red blood cells (RBCs), white blood cells (WBCs), and platelets in a sample of blood. This test is often used as a part of a routine check-up or to help diagnose various medical conditions, such as anemia, infection, inflammation, and many others.
The RBC count measures the number of oxygen-carrying cells in the blood, while the WBC count measures the number of immune cells that help fight infections. The platelet count measures the number of cells involved in clotting. Abnormal results in any of these counts may indicate an underlying medical condition and further testing may be required for diagnosis and treatment.
Erythrocyte inclusions refer to the presence of abnormal structures or substances within red blood cells (erythrocytes). These inclusions can be composed of various materials such as proteins, pigments, or foreign bodies. They may be seen in a variety of medical conditions and can provide important diagnostic clues.
Some examples of erythrocyte inclusions include:
1. Howell-Jolly bodies: small remnants of nuclear material left behind after the red blood cell matures. They are typically seen in individuals with an absent or nonfunctional spleen.
2. Heinz bodies: denatured hemoglobin that forms clumps within the red blood cells. They can be seen in conditions such as hemolytic anemia, G6PD deficiency, and exposure to certain drugs or toxins.
3. Pappenheimer bodies: aggregates of iron-containing proteins called ferritin or hemosiderin. They are typically seen in conditions associated with increased red blood cell destruction, such as thalassemia or lead poisoning.
4. Basophilic stippling: small, basophilic (blue-staining) granules within the red blood cells. They can be seen in various conditions, including lead poisoning, megaloblastic anemias, and certain inherited disorders.
5. Parasites: organisms such as malaria or babesia that infect and multiply within the red blood cells.
The detection of erythrocyte inclusions typically requires specialized testing, such as peripheral blood smears stained with specific dyes to highlight the abnormal structures. The presence and type of inclusions can help diagnose certain medical conditions and guide appropriate treatment.
Osmotic fragility is a term used in medicine, specifically in the field of hematology. It refers to the susceptibility or tendency of red blood cells (RBCs) to undergo lysis (rupture or breaking open) when exposed to hypotonic solutions (solutions with lower osmotic pressure than the RBCs). This test is often used to diagnose and monitor hereditary spherocytosis, a genetic disorder that affects the structure and stability of red blood cells.
In this condition, the RBC membrane proteins are defective, leading to abnormally shaped and fragile cells. When these abnormal RBCs come into contact with hypotonic solutions, they rupture more easily than normal RBCs due to their decreased osmotic resistance. The degree of osmotic fragility can be measured through a laboratory test called the "osmotic fragility test," which evaluates the stability and structural integrity of RBCs in response to varying osmotic pressures.
In summary, osmotic fragility is a medical term that describes the increased susceptibility of red blood cells to lysis when exposed to hypotonic solutions, often associated with hereditary spherocytosis or other conditions affecting RBC membrane stability.
2,3-Diphosphoglycerate (2,3-DPG) is a molecule found in red blood cells that plays a crucial role in regulating the affinity of hemoglobin for oxygen. It is a byproduct of the glycolytic pathway, which is a series of biochemical reactions that convert glucose into energy.
In the tissues where oxygen demand is high, such as muscles and organs, 2,3-DPG concentrations are typically elevated. This molecule binds to deoxygenated hemoglobin at specific sites on the beta chains, reducing its affinity for oxygen and promoting the release of oxygen to the tissues.
Conversely, in the lungs where oxygen is abundant, 2,3-DPG concentrations are lower, allowing hemoglobin to bind more readily to oxygen and load up with oxygen for delivery to the tissues. Therefore, 2,3-DPG helps optimize the matching of oxygen supply and demand in the body.
Neonatal screening is a medical procedure in which specific tests are performed on newborn babies within the first few days of life to detect certain congenital or inherited disorders that are not otherwise clinically apparent at birth. These conditions, if left untreated, can lead to serious health problems, developmental delays, or even death.
The primary goal of neonatal screening is to identify affected infants early so that appropriate treatment and management can be initiated as soon as possible, thereby improving their overall prognosis and quality of life. Commonly screened conditions include phenylketonuria (PKU), congenital hypothyroidism, galactosemia, maple syrup urine disease, sickle cell disease, cystic fibrosis, and hearing loss, among others.
Neonatal screening typically involves collecting a small blood sample from the infant's heel (heel stick) or through a dried blood spot card, which is then analyzed using various biochemical, enzymatic, or genetic tests. In some cases, additional tests such as hearing screenings and pulse oximetry for critical congenital heart disease may also be performed.
It's important to note that neonatal screening is not a diagnostic tool but rather an initial step in identifying infants who may be at risk of certain conditions. Positive screening results should always be confirmed with additional diagnostic tests before any treatment decisions are made.
Fanconi anemia complementation group C protein, also known as FANCC protein, is a component of the Fanconi anemia (FA) DNA repair pathway. This protein plays a critical role in protecting cells from oxidative stress and maintaining genomic stability. Mutations in the FANCC gene can lead to Fanconi anemia, a rare genetic disorder characterized by bone marrow failure, congenital abnormalities, and increased risk of cancer.
FANCC protein functions as part of a complex that includes other FA proteins, which work together to repair DNA damage caused by interstrand crosslinks (ICLs) - a type of DNA lesion that can lead to genomic instability and cancer. When the FA pathway is activated in response to ICLs, FANCC protein undergoes monoubiquitination, which allows it to interact with other proteins involved in DNA repair and chromatin remodeling.
Defects in the FANCC protein can result in impaired DNA repair and increased sensitivity to DNA-damaging agents, leading to the characteristic features of Fanconi anemia. Additionally, mutations in the FANCC gene have been associated with an increased risk of developing acute myeloid leukemia (AML) and other cancers.
Fanconi Anemia Complementation Group D2 Protein, also known as FANCD2 protein, is a key player in the Fanconi anemia (FA) pathway, which is a DNA repair pathway that helps to maintain genomic stability. The FA pathway is responsible for the repair of DNA interstrand cross-links (ICLs), which are harmful lesions that can lead to genomic instability and cancer.
FANCD2 protein is part of the E3 ubiquitin ligase complex that monoubiquitinates FANCI protein, forming a heterodimeric complex known as ID2. The monoubiquitination of FANCD2/FANCI is a critical step in the FA pathway and is required for the recruitment of downstream repair factors to the site of DNA damage.
Mutations in the gene that encodes FANCD2 protein can lead to Fanconi anemia, a rare genetic disorder characterized by bone marrow failure, congenital abnormalities, and an increased risk of cancer. The disease is typically inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
Retinal diseases refer to a group of conditions that affect the retina, which is the light-sensitive tissue located at the back of the eye. The retina is responsible for converting light into electrical signals that are sent to the brain and interpreted as visual images. Retinal diseases can cause vision loss or even blindness, depending on their severity and location in the retina.
Some common retinal diseases include:
1. Age-related macular degeneration (AMD): A progressive disease that affects the central part of the retina called the macula, causing blurred or distorted vision.
2. Diabetic retinopathy: A complication of diabetes that can damage the blood vessels in the retina, leading to vision loss.
3. Retinal detachment: A serious condition where the retina becomes separated from its underlying tissue, requiring immediate medical attention.
4. Macular edema: Swelling or thickening of the macula due to fluid accumulation, which can cause blurred vision.
5. Retinitis pigmentosa: A group of inherited eye disorders that affect the retina's ability to respond to light, causing progressive vision loss.
6. Macular hole: A small break in the macula that can cause distorted or blurry vision.
7. Retinal vein occlusion: Blockage of the retinal veins that can lead to bleeding, swelling, and potential vision loss.
Treatment for retinal diseases varies depending on the specific condition and its severity. Some treatments include medication, laser therapy, surgery, or a combination of these options. Regular eye exams are essential for early detection and treatment of retinal diseases.
Fanconi anemia complementation group A protein (FANCA) is a protein encoded by the FANCA gene in humans. It is a part of the Fanconi anemia (FA) pathway, which is a group of proteins that play a critical role in maintaining genomic stability and preventing cancer.
The FA pathway is involved in the repair of DNA interstrand crosslinks (ICLs), which are harmful lesions that can block replication and transcription of DNA. FANCA protein, along with other FA proteins, forms a complex called the "FA core complex" that monoubiquitinates another FA protein called FANCD2. This monoubiquitination event is essential for the recruitment of downstream repair factors to damaged DNA and restoration of normal DNA structure.
Mutations in the FANCA gene can lead to Fanconi anemia, a rare genetic disorder characterized by congenital abnormalities, bone marrow failure, and increased risk of cancer. The disease is typically inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
Hemoglobin A2 is a type of hemoglobin that is found in human red blood cells. Hemoglobin is the protein in red blood cells that carries oxygen throughout the body. Hemoglobin A2 is made up of two alpha-like globin chains and two delta-globin chains, and it accounts for approximately 1.5 to 3.5% of the total hemoglobin in adult humans.
Hemoglobin A2 is not normally present in significant amounts until after a child has passed through their first year of life. Its level remains relatively constant throughout adulthood, and it is often used as a diagnostic marker for certain types of anemia, such as beta-thalassemia. In people with beta-thalassemia, the production of beta-globin chains is reduced or absent, leading to an increase in the relative proportion of Hemoglobin A2 and Hemoglobin F (fetal hemoglobin) in the red blood cells.
It's important to note that Hemoglobin A2 measurement alone is not enough for a definitive diagnosis of beta-thalassemia, but it can be used as a supportive test along with other investigations such as complete blood count (CBC), hemoglobin electrophoresis and molecular genetic testing.
Hyphema is defined as the presence of blood in the anterior chamber of the eye, which is the space between the cornea and the iris. This condition usually results from trauma or injury to the eye, but it can also occur due to various medical conditions such as severe eye inflammation, retinal surgery, or blood disorders that affect clotting.
The blood in the anterior chamber can vary in amount, ranging from a few drops to a complete fill, which is called an "eight-ball hyphema." Hyphema can be painful and cause sensitivity to light (photophobia), blurred vision, or even loss of vision if not treated promptly.
Immediate medical attention is necessary for hyphema to prevent complications such as increased intraocular pressure, corneal blood staining, glaucoma, or cataracts. Treatment options may include bed rest, eye drops to reduce inflammation and control intraocular pressure, and sometimes surgery to remove the blood from the anterior chamber.
A splenectomy is a surgical procedure in which the spleen is removed from the body. The spleen is an organ located in the upper left quadrant of the abdomen, near the stomach and behind the ribs. It plays several important roles in the body, including fighting certain types of infections, removing old or damaged red blood cells from the circulation, and storing platelets and white blood cells.
There are several reasons why a splenectomy may be necessary, including:
* Trauma to the spleen that cannot be repaired
* Certain types of cancer, such as Hodgkin's lymphoma or non-Hodgkin's lymphoma
* Sickle cell disease, which can cause the spleen to enlarge and become damaged
* A ruptured spleen, which can be life-threatening if not treated promptly
* Certain blood disorders, such as idiopathic thrombocytopenic purpura (ITP) or hemolytic anemia
A splenectomy is typically performed under general anesthesia and may be done using open surgery or laparoscopically. After the spleen is removed, the incision(s) are closed with sutures or staples. Recovery time varies depending on the individual and the type of surgery performed, but most people are able to return to their normal activities within a few weeks.
It's important to note that following a splenectomy, individuals may be at increased risk for certain types of infections, so it's recommended that they receive vaccinations to help protect against these infections. They should also seek medical attention promptly if they develop fever, chills, or other signs of infection.
A newborn infant is a baby who is within the first 28 days of life. This period is also referred to as the neonatal period. Newborns require specialized care and attention due to their immature bodily systems and increased vulnerability to various health issues. They are closely monitored for signs of well-being, growth, and development during this critical time.
Pulmonary hypertension is a medical condition characterized by increased blood pressure in the pulmonary arteries, which are the blood vessels that carry blood from the right side of the heart to the lungs. This results in higher than normal pressures in the pulmonary circulation and can lead to various symptoms and complications.
Pulmonary hypertension is typically defined as a mean pulmonary artery pressure (mPAP) greater than or equal to 25 mmHg at rest, as measured by right heart catheterization. The World Health Organization (WHO) classifies pulmonary hypertension into five groups based on the underlying cause:
1. Pulmonary arterial hypertension (PAH): This group includes idiopathic PAH, heritable PAH, drug-induced PAH, and associated PAH due to conditions such as connective tissue diseases, HIV infection, portal hypertension, congenital heart disease, and schistosomiasis.
2. Pulmonary hypertension due to left heart disease: This group includes conditions that cause elevated left atrial pressure, such as left ventricular systolic or diastolic dysfunction, valvular heart disease, and congenital cardiovascular shunts.
3. Pulmonary hypertension due to lung diseases and/or hypoxia: This group includes chronic obstructive pulmonary disease (COPD), interstitial lung disease, sleep-disordered breathing, alveolar hypoventilation disorders, and high altitude exposure.
4. Chronic thromboembolic pulmonary hypertension (CTEPH): This group includes persistent obstruction of the pulmonary arteries due to organized thrombi or emboli.
5. Pulmonary hypertension with unclear and/or multifactorial mechanisms: This group includes hematologic disorders, systemic disorders, metabolic disorders, and other conditions that can cause pulmonary hypertension but do not fit into the previous groups.
Symptoms of pulmonary hypertension may include shortness of breath, fatigue, chest pain, lightheadedness, and syncope (fainting). Diagnosis typically involves a combination of medical history, physical examination, imaging studies, and invasive testing such as right heart catheterization. Treatment depends on the underlying cause but may include medications, oxygen therapy, pulmonary rehabilitation, and, in some cases, surgical intervention.
A premarital examination is a medical evaluation typically consisting of screening tests and counseling, performed for individuals who are planning to get married. The purpose of this examination is to identify any potential health issues that may affect the couple's future family plans or overall well-being. These evaluations often include:
1. Medical History Review: Detailed review of past medical history, surgical history, allergies, current medications, and immunization status.
2. Physical Examination: Complete physical examination to identify any existing health conditions.
3. Infectious Disease Screening: Tests for sexually transmitted infections (STIs) such as HIV, syphilis, hepatitis B, and sometimes gonorrhea and chlamydia.
4. Genetic Disorder Screening: Depending on family history or ethnic background, screening for genetic disorders may be recommended.
5. Blood Type Testing: Determination of blood types (A, B, AB, O) and Rh factor (positive or negative).
6. Counseling: Discussion about reproductive health, family planning, birth control methods, and prevention of sexually transmitted infections.
7. Vaccination Status Check: Ensuring up-to-date vaccinations for both partners.
8. Other Tests: Depending on specific circumstances, other tests like tuberculosis screening or cancer screenings might be advised.
It's important to note that laws regarding premarital examinations vary by country and state. Some places require certain tests by law while others do not.
Heinz bodies are small, irregularly shaped inclusions found in the red blood cells (RBCs). They are aggregates of denatured hemoglobin and are typically seen in RBCs that have been exposed to oxidative stress. This can occur due to various factors such as exposure to certain chemicals, drugs, or diseases.
The presence of Heinz bodies can lead to the premature destruction of RBCs, a condition known as hemolysis. This can result in anemia and related symptoms such as fatigue, weakness, and shortness of breath. It's important to note that while Heinz bodies are often associated with certain diseases, they can also be present in otherwise healthy individuals who have been exposed to oxidative stress.
It's worth mentioning that the term "Heinz bodies" comes from the name of the scientist Robert Heinz, who first described them in 1890.
Splenomegaly is a medical term that refers to an enlargement or expansion of the spleen beyond its normal size. The spleen is a vital organ located in the upper left quadrant of the abdomen, behind the stomach and below the diaphragm. It plays a crucial role in filtering the blood, fighting infections, and storing red and white blood cells and platelets.
Splenomegaly can occur due to various underlying medical conditions, including infections, liver diseases, blood disorders, cancer, and inflammatory diseases. The enlarged spleen may put pressure on surrounding organs, causing discomfort or pain in the abdomen, and it may also lead to a decrease in red and white blood cells and platelets, increasing the risk of anemia, infections, and bleeding.
The diagnosis of splenomegaly typically involves a physical examination, medical history, and imaging tests such as ultrasound, CT scan, or MRI. Treatment depends on the underlying cause and may include medications, surgery, or other interventions to manage the underlying condition.
Medical Definition:
"Risk factors" are any attribute, characteristic or exposure of an individual that increases the likelihood of developing a disease or injury. They can be divided into modifiable and non-modifiable risk factors. Modifiable risk factors are those that can be changed through lifestyle choices or medical treatment, while non-modifiable risk factors are inherent traits such as age, gender, or genetic predisposition. Examples of modifiable risk factors include smoking, alcohol consumption, physical inactivity, and unhealthy diet, while non-modifiable risk factors include age, sex, and family history. It is important to note that having a risk factor does not guarantee that a person will develop the disease, but rather indicates an increased susceptibility.
Methemoglobin is a form of hemoglobin in which the iron within the heme group is in the ferric (Fe3+) state instead of the ferrous (Fe2+) state. This oxidation reduces its ability to bind and transport oxygen effectively, leading to methemoglobinemia when methemoglobin levels become too high. Methemoglobin has a limited capacity to release oxygen to tissues, which can result in hypoxia (reduced oxygen supply) and cyanosis (bluish discoloration of the skin and mucous membranes).
Methemoglobin is normally present in small amounts in the blood, but certain factors such as exposure to oxidizing agents, genetic predisposition, or certain medications can increase its levels. Elevated methemoglobin levels can be treated with methylene blue, which helps restore the iron within hemoglobin back to its ferrous state and improves oxygen transport capacity.
Diphosphoglycerates (also known as 2,3-diphosphoglycerates or 2,3-DPG) are organic molecules found in red blood cells. They play a crucial role in regulating the affinity of hemoglobin for oxygen. Hemoglobin is the protein in red blood cells that carries oxygen from the lungs to the body's tissues.
When the concentration of diphosphoglycerates in red blood cells increases, it reduces the ability of hemoglobin to bind with oxygen, which allows more oxygen to be released into the tissues. This is particularly important in conditions where there is low oxygen availability, such as at high altitudes or in diseases that cause poor oxygen delivery to the tissues, like heart failure and chronic obstructive pulmonary disease (COPD).
In summary, diphosphoglycerates are essential molecules that help regulate hemoglobin's affinity for oxygen, ensuring optimal oxygen delivery to the body's tissues.
Enuresis is a medical term that refers to the involuntary or unconscious release of urine, especially at night during sleep, in children who are at least 5 years old. It's commonly known as bedwetting. Enuresis can be classified into two types: primary and secondary. Primary enuresis occurs when a child has never achieved consistent dryness during sleep, while secondary enuresis happens when a child starts wetting the bed again after having been dry for at least six months.
Enuresis can have various causes, including developmental delays, small bladder capacity, urinary tract infections, constipation, sleep disorders, and emotional stress. In some cases, it may also be associated with genetic factors. Treatment options depend on the underlying cause and may include behavioral interventions, bladder training, alarm systems, medication, or a combination of these approaches.
I'm sorry for any confusion, but "Mediterranean Islands" is not a term that has a specific medical definition. The Mediterranean Sea is surrounded by many countries and islands, including Cyprus, Sicily, Sardinia, Corsica, Crete, and numerous Greek islands, among others. If you're looking for medical information related to these areas, I would need more specificity. However, if you're interested in general health statistics or demographic data related to these islands, I could try to provide some relevant information.
Hereditary Spherocytosis is a genetic disorder that affects the red blood cells (RBCs) causing them to take on a spherical shape instead of their normal biconcave disc shape. This occurs due to mutations in the genes responsible for the proteins that maintain the structure and flexibility of RBCs, such as ankyrin, band 3, spectrin, and protein 4.2.
The abnormally shaped RBCs are fragile and prone to hemolysis (premature destruction), which can lead to anemia, jaundice, and gallstones. Symptoms can vary from mild to severe and may include fatigue, weakness, shortness of breath, and a yellowing of the skin and eyes (jaundice). Diagnosis is typically made through a combination of family history, physical examination, complete blood count (CBC), and specialized tests such as osmotic fragility test, eosin-5'-maleimide binding test, or direct antiglobulin test. Treatment may include monitoring, supplementation with folic acid, and in severe cases, splenectomy (surgical removal of the spleen) to reduce RBC destruction.
Prospective studies, also known as longitudinal studies, are a type of cohort study in which data is collected forward in time, following a group of individuals who share a common characteristic or exposure over a period of time. The researchers clearly define the study population and exposure of interest at the beginning of the study and follow up with the participants to determine the outcomes that develop over time. This type of study design allows for the investigation of causal relationships between exposures and outcomes, as well as the identification of risk factors and the estimation of disease incidence rates. Prospective studies are particularly useful in epidemiology and medical research when studying diseases with long latency periods or rare outcomes.
Hemolytic anemia, congenital nonspherocytic is a rare type of inherited anemia characterized by the premature destruction (hemolysis) of red blood cells. This condition is caused by defects in enzymes or proteins that help maintain the structural integrity and function of red blood cells.
In this form of hemolytic anemia, the red blood cells are not spherical in shape like spherocytes; instead, they may be oval or elongated. The most common types of congenital nonspherocytic hemolytic anemia are caused by deficiencies in enzymes such as glucose-6-phosphate dehydrogenase (G6PD) and pyruvate kinase.
Symptoms of this condition may include fatigue, weakness, pale skin, jaundice, dark urine, and an enlarged spleen. Treatment may involve blood transfusions, medications to manage symptoms, and avoidance of certain triggers that can exacerbate the hemolysis. In some cases, a bone marrow transplant may be considered as a curative treatment option.
Pallor is a medical term that refers to an abnormal pale appearance of the skin, mucous membranes, or nail beds. It can be a sign of various underlying medical conditions such as anemia (a decrease in red blood cells or hemoglobin), blood loss, malnutrition, vitamin deficiencies, or certain diseases that affect circulation or oxygenation of the blood. Pallor can also occur due to emotional distress or fear, leading to a temporary reduction in blood flow to the skin. It is important to note that pallor should be evaluated in conjunction with other symptoms and medical history for an accurate diagnosis.
Retrospective studies, also known as retrospective research or looking back studies, are a type of observational study that examines data from the past to draw conclusions about possible causal relationships between risk factors and outcomes. In these studies, researchers analyze existing records, medical charts, or previously collected data to test a hypothesis or answer a specific research question.
Retrospective studies can be useful for generating hypotheses and identifying trends, but they have limitations compared to prospective studies, which follow participants forward in time from exposure to outcome. Retrospective studies are subject to biases such as recall bias, selection bias, and information bias, which can affect the validity of the results. Therefore, retrospective studies should be interpreted with caution and used primarily to generate hypotheses for further testing in prospective studies.
Hematologic tests, also known as hematology tests, are a group of diagnostic exams that evaluate the health and function of different components of blood, such as red and white blood cells, platelets, and clotting factors. These tests can detect various disorders, including anemia, infection, bleeding problems, and several types of cancer. Common hematologic tests include complete blood count (CBC), coagulation studies, peripheral smear examination, and erythrocyte sedimentation rate (ESR). The specific test or combination of tests ordered will depend on the patient's symptoms, medical history, and physical examination findings.
Hemoglobinuria is a medical condition characterized by the presence of hemoglobin in the urine. Hemoglobin is a protein found in red blood cells that carries oxygen throughout the body. Normally, when red blood cells die, they are broken down and their hemoglobin is recycled. However, in certain conditions such as intravascular hemolysis (the destruction of red blood cells inside blood vessels), hemoglobin can be released into the bloodstream and then filtered by the kidneys into the urine.
Hemoglobinuria can be a symptom of various underlying medical conditions, including hemolytic anemias, disseminated intravascular coagulation (DIC), severe infections, snake bites, and exposure to certain toxins or medications. It is important to identify the underlying cause of hemoglobinuria, as treatment will depend on the specific condition.
In some cases, hemoglobinuria can lead to kidney damage due to the toxic effects of free hemoglobin on the renal tubules. This can result in acute or chronic kidney injury, and in severe cases, it may require dialysis or transplantation.
Treatment outcome is a term used to describe the result or effect of medical treatment on a patient's health status. It can be measured in various ways, such as through symptoms improvement, disease remission, reduced disability, improved quality of life, or survival rates. The treatment outcome helps healthcare providers evaluate the effectiveness of a particular treatment plan and make informed decisions about future care. It is also used in clinical research to compare the efficacy of different treatments and improve patient care.
Tricuspid valve insufficiency, also known as tricuspid regurgitation, is a cardiac condition in which the tricuspid valve located between the right atrium and right ventricle of the heart does not close properly, allowing blood to flow back into the right atrium during contraction of the right ventricle. This results in a portion of the blood being pumped inefficiently, which can lead to volume overload of the right side of the heart and potentially result in symptoms such as fatigue, weakness, shortness of breath, and fluid retention. The condition can be congenital or acquired, with common causes including dilated cardiomyopathy, infective endocarditis, rheumatic heart disease, and trauma.
 Sickle Cell Anemia, a Molecular Disease
Sickle Cell Anemia, a Molecular Disease Sickle Cell Disease | Sickle Cell Anemia | MedlinePlus
Sickle Cell Disease | Sickle Cell Anemia | MedlinePlus Preventing Sickle Cell Anemia Complications in Children | VitalSigns | CDC
Preventing Sickle Cell Anemia Complications in Children | VitalSigns | CDC Sickle Cell Anemia Skeletal Imaging: Practice Essentials, Radiography, Computed Tomography
Sickle Cell Anemia Skeletal Imaging: Practice Essentials, Radiography, Computed Tomography Sickle Cell Anemia: Types, Symptoms, and Treatment
Sickle Cell Anemia: Types, Symptoms, and Treatment Sickle cell anemia in African Americans: Symptoms, causes, and more
Sickle cell anemia in African Americans: Symptoms, causes, and more Sickle cell anemia | health.am
Sickle cell anemia | health.am Gene editing offers hope for curing sickle cell anemia - Xinhua | English.news.cn
Gene editing offers hope for curing sickle cell anemia - Xinhua | English.news.cn The Bad Blood: My Life With Sickle Cell Anaemia
The Bad Blood: My Life With Sickle Cell Anaemia Sickle cell anemia: Could gene therapy cure sickle cell anemia? - "60 Minutes" - CBS News
Sickle cell anemia: Could gene therapy cure sickle cell anemia? - "60 Minutes" - CBS News Sickle Cell Disease in Babies: Sickle Cell Anemia Causes, Symptoms & Treatment
Sickle Cell Disease in Babies: Sickle Cell Anemia Causes, Symptoms & Treatment Support Sickle Cell Anemia Patient in Maharashtra - GlobalGiving
Support Sickle Cell Anemia Patient in Maharashtra - GlobalGiving Porteus awarded grant for work on possible treatment for sickle cell anemia | News Center | Stanford Medicine
Porteus awarded grant for work on possible treatment for sickle cell anemia | News Center | Stanford Medicine Johns Hopkins Researchers Develop Human Stem Cell Line Containing Sickle Cell Anemia Mutation - 05/29/2008
Johns Hopkins Researchers Develop Human Stem Cell Line Containing Sickle Cell Anemia Mutation - 05/29/2008 Sickle Cell Test: Reasons & Test Results: Inexplicable Anemia
Sickle Cell Test: Reasons & Test Results: Inexplicable Anemia The thorny history of sickle cell anemia | Hub
The thorny history of sickle cell anemia | Hub Glossary of Terms | Sickle Cell Anemia | Health & Senior Services
Glossary of Terms | Sickle Cell Anemia | Health & Senior Services Asthma is associated with Increased mortality in individuals with sickle cell anemia
| Haematologica
Asthma is associated with Increased mortality in individuals with sickle cell anemia
| Haematologica
 Genes and Health - Mutation, Genetic Disease, Sickle-Cell Anemia, Gene Therapy
Genes and Health - Mutation, Genetic Disease, Sickle-Cell Anemia, Gene Therapy Sickle Cell Anemia Therapeutics Market Is Booming Worldwide 2021-2028 | Bristol-Myers Squibb, GlycoMimetics, Pfizer,Anthera...
Sickle Cell Anemia Therapeutics Market Is Booming Worldwide 2021-2028 | Bristol-Myers Squibb, GlycoMimetics, Pfizer,Anthera... Pleiotropic and epistatic genes in sickle cell anaemia | HSTalks
Pleiotropic and epistatic genes in sickle cell anaemia | HSTalks A novel deep learning approach for sickle cell anemia detection in human RBCs using an improved wrapper-based feature selection...
A novel deep learning approach for sickle cell anemia detection in human RBCs using an improved wrapper-based feature selection... A Revolutionary New Gene Therapy Treatment for Sickle Cell Anemia Is Imminent | American Council on Science and Health
A Revolutionary New Gene Therapy Treatment for Sickle Cell Anemia Is Imminent | American Council on Science and Health Many Kids With Sickle Cell Anemia Do Not Receive Stroke Screening, Treatment - Real Health
Many Kids With Sickle Cell Anemia Do Not Receive Stroke Screening, Treatment - Real Health 2nd Annual Black Tie Gala Benefiting Children with Sickle Cell Anemia - Cobb Galleria Centre
2nd Annual Black Tie Gala Benefiting Children with Sickle Cell Anemia - Cobb Galleria Centre What are indications, complications of acute blood transfusions in sickle cell anemia? Key Points Additional Reading - Page 2...
What are indications, complications of acute blood transfusions in sickle cell anemia? Key Points Additional Reading - Page 2... Paper: Progressive Loss of Brain Volume in Children with Sickle Cell Anemia: A Report from the Silent Cerebral Infarct...
Paper: Progressive Loss of Brain Volume in Children with Sickle Cell Anemia: A Report from the Silent Cerebral Infarct... Management of Sickle Cell Anemia in Nigeria with Medicinal Plants: Cationic Evaluation of Extracts and Possible Effects on the...
Management of Sickle Cell Anemia in Nigeria with Medicinal Plants: Cationic Evaluation of Extracts and Possible Effects on the... Brooklyn screens for sickle cell anemia.
Brooklyn screens for sickle cell anemia.