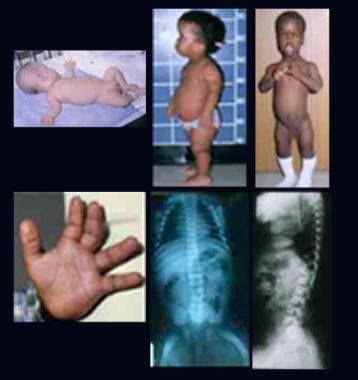
Aspartylglucosaminuria
Aspartylglucosylaminase
Lysosomal Storage Diseases
Encyclopedias as Topic
Fabry Disease
Angiokeratoma
alpha-Galactosidase
alpha-N-Acetylgalactosaminidase
Trihexosylceramides
Enzyme Replacement Therapy
New Zealand
Overgrowth of oral mucosa and facial skin, a novel feature of aspartylglucosaminuria. (1/19)
Aspartylglucosaminuria (AGU) is a lysosomal storage disorder caused by deficiency of aspartylglucosaminidase (AGA). The main symptom is progressive mental retardation. A spectrum of different mutations has been reported in this disease, one missense mutation (Cys163Ser) being responsible for the majority of Finnish cases. We were able to examine 66 Finnish AGU patients for changes in the oral mucosa and 44 of these for changes in facial skin. Biopsy specimens of 16 oral lesions, 12 of them associated with the teeth, plus two facial lesions were studied histologically. Immunohistochemical staining for AGA was performed on 15 oral specimens. Skin was seborrhoeic in adolescent and adult patients, with erythema of the facial skin already common in childhood. Of 44 patients, nine (20%) had facial angiofibromas, tumours primarily occurring in association with tuberous sclerosis. Oedemic buccal mucosa (leucoedema) and gingival overgrowths were more frequent in AGU patients than in controls (p<0.001). Of 16 oral mucosal lesions studied histologically, 15 represented fibroepithelial or epithelial hyperplasias and were reactive in nature. Cytoplasmic vacuolisation was evident in four. Immunohistochemically, expression of AGA in AGU patients' mucosal lesions did not differ from that seen in corresponding lesions of normal subjects. Thus, the high frequency of mucosal overgrowth in AGU patients does not appear to be directly associated with lysosomal storage or with alterations in the level of AGA expression. (+info)Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations. (2/19)
A deficiency of functional aspartylglucosaminidase (AGA) causes a lysosomal storage disease, aspartylglucosaminuria (AGU). The recessively inherited disease is enriched in the Finnish population, where 98% of AGU alleles contain one founder mutation, AGU(Fin). Elsewhere in the world, we and others have described 18 different sporadic AGU mutations. Many of these are predicted to interfere with the complex intracellular maturation and processing of the AGA polypeptide. Proper initial folding of AGA in the endoplasmic reticulum (ER) is dependent on intramolecular disulfide bridge formation and dimerization of two precursor polypeptides. The subsequent activation of AGA occurs autocatalytically in the ER and the protein is transported via the Golgi to the lysosomal compartment using the mannose-6-phosphate receptor pathway. Here we use the three-dimensional structure of AGA to predict structural consequences of AGU mutations, including six novel mutations, and make an effort to characterize every known disease mutation by dissecting the effect of mutations on intracellular stability, maturation, transport and the activity of AGA. Most mutations are substitutions replacing the original amino acid with a bulkier residue. Mutations of the dimer interface prevent dimerization in the ER, whereas active site mutations not only destroy the activity but also affect maturation of the precursor. Depending on their effects on the AGA polypeptide the mutations can be categorized as mild, moderate or severe. These data contribute to the expanding body of knowledge pertaining to molecular pathogenesis of AGU. (+info)Human leukocyte glycosylasparaginase: cell-to-cell transfer and properties in correction of aspartylglycosaminuria. (3/19)
Aspartylglycosaminuria (AGU), a severe lysosomal storage disease, is caused by the deficiency of the lysosomal enzyme, glycosylasparaginase (GA), and accumulation of aspartylglucosamine (GlcNAc-Asn) in tissues. Here we show that human leukocyte glycosylasparaginase can correct the metabolic defect in Epstein-Barr virus (EBV)-transformed AGU lymphocytes rapidly and effectively by mannose-6-phosphate receptor-mediated endocytosis or by contact-mediated cell-to-cell transfer from normal EBV-transformed lymphocytes, and that 2-7% of normal activity is sufficient to correct the GlcNAc-Asn metabolism in the cells. Cell-to-cell contact is obligatory for the transfer of GA since normal transformed lymphocytes do not excrete GA into extracellular medium. The combined evidence indicates that cell-to-cell transfer of GA plays a main role in enzyme replacement therapy of AGU by normal lymphocytes. (+info)Characterization of two glycoasparagines isolated from the urine of patients with aspartylglycosylaminuria (AGU). (4/19)
Two major glycoasparagines (2-acetamido-N-(4'-L-aspartyl)-2-deoxy-beta-D-glycosylamines) were isolated from the urine of patients with aspartylglycosylaminuria (AGU). They were composed of equimolar amounts of sialic acid, galactose, glucosamine, and aspartic acid. They were isomeric with respect to the position of sialic acid attachment, since they produced the same glycoasparagine on incubation with the neuraminidase from Clostridium perfringens. The structure of the resulting sialic acid-free glycoasparagine was determined as beta-Gal-(1 leads to 4)-beta-GlcNAc-Asn based on the following findings. It produced galactose on incubation with beta-galactosidase, and N-acetyllactosamine and aspartic acid on incubation with 4-L-aspartylglycosylamine amindo hydrolase. (+info)Quantitative determination of rare mRNA species by PCR and solid-phase minisequencing. (5/19)
We present a new method for quantification of mRNA, in which the limitations of the current quantitative PCR methods can be overcome. A known amount of a synthetic RNA standard differing from the mRNA to be quantified by a single nucleotide is reverse-transcribed and amplified together with the mRNA template using a biotinylated primer. The biotinylated PCR product is immobilized on a streptavidin-coated solid support and denatured. The ratio between the two amplified sequences is determined by separate "mini-sequencing" reactions, in which a detection step primer annealing immediately adjacent to the site of the variable nucleotide is elongated by a single labeled dNTP complementary to the nucleotide at the variable site. The ratio between the incorporated labels accurately determines the ratio between the two sequences in the original RNA sample. We applied this method to quantify the mRNA of human aspartylglucosaminidase (AGA) in tissues and cultured cells. AGA is a lysosomal enzyme participating in the degradation of glycoproteins. A mutation in the AGA gene abolishes the enzyme activity and leads to aspartylglucosaminuria (AGU), a recessively inherited metabolic disorder. The mRNA quantification revealed that the normal and mutant genes are expressed at similar levels in kidney, liver, and cultured fibroblast, whereas the amount of AGA mRNA in normal placenta and brain is significantly higher than that found in the corresponding samples from AGU patients. The method presented here is generally applicable for PCR-based quantification of rare mRNAs and DNA as well. (+info)Human aspartylglucosaminidase. A biochemical and immunocytochemical characterization of the enzyme in normal and aspartylglucosaminuria fibroblasts. (6/19)
Aspartylglucosaminidase (AGA, EC 3.5.1.26) is an essential enzyme in the degradation of asparagine-linked glycoproteins. In man, deficient activity of this enzyme leads to aspartylglucosaminuria (AGU), a recessively inherited lysosomal storage disease. Here we used affinity-purified polyclonal antibodies against the native AGA and its denatured subunits to establish the molecular structure and intracellular location of the enzyme in normal and AGU fibroblasts. Inactivation of the enzyme was found to coincide with the dissociation of the heterodimeric enzyme complex into subunits. Although the subunits were not linked by covalent forces, the intrapolypeptide disulphide bridges were found to be essential for the normal function of AGA. AGA was localized into lysosomes in control fibroblasts by both immunofluorescence microscopy and immuno-electron microscopy, whereas in AGU cells the location of antigen was different, suggesting that, owing to the mutation, a missing disulphide bridge, most of the enzyme molecules get retarded in the cis-Golgi region and most probably face intracellular degradation. (+info)Massive accumulation of Man2GlcNAc2-Asn in nonneuronal tissues of glycosylasparaginase-deficient mice and its removal by enzyme replacement therapy. (7/19)
Aspartylglycosaminuria (AGU) is caused by deficient enzymatic activity of glycosylasparaginase (GA). The disease is characterized by accumulation of aspartylglucosamine (GlcNAc-Asn) and other glycoasparagines in tissues and body fluids of AGU patients and in an AGU mouse model. In the current study, we characterized a glycoasparagine carrying the tetrasaccharide moiety of alpha-D-Man-(1-->6)-beta-D-Man-(1-->4)-beta-D-GlcNAc-(1-->4)-beta-D-GlcNAc-(1-->N )-Asn (Man2GlcNAc2-Asn) in urine of an AGU patient and also in the tissues of the AGU mouse model. Quantitative analysis demonstrated a massive accumulation of the compound especially in nonneuronal tissues of the AGU mice, in which the levels of Man2GlcNAc2-Asn were typically 30-87% of those of GlcNAc-Asn. The highest level of Man2GlcNAc2-Asn was found in the liver, spleen, and heart tissues of the AGU mice, the respective amounts being 87%, 76%, and 57% of the GlcNAc-Asn levels. In the brain tissue of AGU mice the Man2GlcNAc2-Asn storage was only 9% of that of GlcNAc-Asn. In contrast to GlcNAc-Asn, the storage of Man2GlcNAc2-Asn markedly increased in the liver and spleen tissues of AGU mice as they grew older. Enzyme replacement therapy with glycosylasparaginase for 3.5 weeks reduced the amount of Man2GlcNAc2-Asn by 66-97% in nonneuronal tissues, but only by 13% in the brain tissue of the AGU mice. In conclusion, there is evidence for a role for storage of glycoasparagines other than aspartylglucosamine in the pathogenesis of AGU, and this possibility should be taken into consideration in the treatment of the disease. (+info)Aspartylglucosaminuria: cDNA encoding human aspartylglucosaminidase and the missense mutation causing the disease. (8/19)
We have isolated a 2.1 kb cDNA which encodes human aspartylglucosaminidase (AGA, E.C. 3.5.1.26). The activity of this lysosomal enzyme is deficient in aspartylglucosaminuria (AGU), a recessively inherited lysosomal accumulation disease resulting in severe mental retardation. The polypeptide chain deduced from the AGA cDNA consists of 346 amino acids, has two potential N-glycosylation sites and 11 cysteine residues. Transient expression of this cDNA in COS-1 cells resulted in increased expression of immunoprecipitable AGA protein. Direct sequencing of amplified AGA cDNA from an AGU patient revealed a G----C transition resulting in the substitution of cysteine 163 with serine. This mutation was subsequently found in all the 20 analyzed Finnish AGU patients, in the heterozygous form in all 53 carriers and in none of 67 control individuals, suggesting that it represents the major AGU causing mutation enriched in this isolated population. Since the mutation produces a change in the predicted flexibility of the AGA polypeptide chain and removes an intramolecular S-S bridge, it most probably explains the deficient enzyme activity found in cells and tissues of AGU patients. (+info)Aspartylglucosaminuria (AGU) is a rare inherited metabolic disorder caused by a deficiency of the enzyme Aspartylglucosaminidase (AGA). This enzyme is responsible for breaking down complex sugars called glycoproteins in the body. When AGA is deficient, glycoproteins accumulate in various tissues and organs, leading to progressive damage.
The symptoms of AGU usually become apparent during early childhood, around 2-6 years of age. These may include developmental delays, intellectual disability, coarse facial features, recurrent respiratory infections, and skeletal abnormalities. Over time, individuals with AGU may experience worsening neurological symptoms, such as seizures, ataxia (loss of muscle coordination), and spasticity (stiff or rigid muscles).
AGU is an autosomal recessive disorder, which means that an individual must inherit two copies of the defective gene, one from each parent, to develop the condition. If both parents are carriers of the AGU gene mutation, they have a 25% chance of having a child with the disease in each pregnancy.
Currently, there is no cure for AGU, and treatment is focused on managing symptoms and improving quality of life. Regular follow-up with a healthcare team experienced in treating metabolic disorders is essential to monitor disease progression and adjust treatment plans as needed.
Aspartylglucosaminuria (AGA) is a rare inherited metabolic disorder caused by a deficiency in the enzyme Aspartylglucosaminidase (AGAse). This enzyme is responsible for breaking down complex sugars called glycoproteins in the body. Without adequate levels of this enzyme, glycoproteins accumulate in various tissues and organs, leading to a range of symptoms including developmental delays, intellectual disability, coarse facial features, skeletal abnormalities, and skin issues.
AGA is an autosomal recessive disorder, which means that an individual must inherit two copies of the defective gene (one from each parent) in order to develop the condition. If only one copy of the gene is inherited, the person will not develop the disorder but will be a carrier and may pass the gene on to their offspring.
There is currently no cure for Aspartylglucosaminuria, and treatment is focused on managing symptoms and improving quality of life. Regular monitoring and early intervention can help to address any complications that arise as a result of the disorder.
Lysosomal storage diseases (LSDs) are a group of rare inherited metabolic disorders caused by defects in lysosomal function. Lysosomes are membrane-bound organelles within cells that contain enzymes responsible for breaking down and recycling various biomolecules, such as proteins, lipids, and carbohydrates. In LSDs, the absence or deficiency of specific lysosomal enzymes leads to the accumulation of undigested substrates within the lysosomes, resulting in cellular dysfunction and organ damage.
These disorders can affect various organs and systems in the body, including the brain, nervous system, bones, skin, and visceral organs. Symptoms may include developmental delays, neurological impairment, motor dysfunction, bone abnormalities, coarse facial features, hepatosplenomegaly (enlarged liver and spleen), and recurrent infections.
Examples of LSDs include Gaucher disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, Pompe disease, and mucopolysaccharidoses (MPS). Treatment options for LSDs may include enzyme replacement therapy, substrate reduction therapy, or bone marrow transplantation. Early diagnosis and intervention can help improve the prognosis and quality of life for affected individuals.
Acetylglucosamine is a type of sugar that is commonly found in the body and plays a crucial role in various biological processes. It is a key component of glycoproteins and proteoglycans, which are complex molecules made up of protein and carbohydrate components.
More specifically, acetylglucosamine is an amino sugar that is formed by the addition of an acetyl group to glucosamine. It can be further modified in the body through a process called acetylation, which involves the addition of additional acetyl groups.
Acetylglucosamine is important for maintaining the structure and function of various tissues in the body, including cartilage, tendons, and ligaments. It also plays a role in the immune system and has been studied as a potential therapeutic target for various diseases, including cancer and inflammatory conditions.
In summary, acetylglucosamine is a type of sugar that is involved in many important biological processes in the body, and has potential therapeutic applications in various diseases.
An encyclopedia is a comprehensive reference work containing articles on various topics, usually arranged in alphabetical order. In the context of medicine, a medical encyclopedia is a collection of articles that provide information about a wide range of medical topics, including diseases and conditions, treatments, tests, procedures, and anatomy and physiology. Medical encyclopedias may be published in print or electronic formats and are often used as a starting point for researching medical topics. They can provide reliable and accurate information on medical subjects, making them useful resources for healthcare professionals, students, and patients alike. Some well-known examples of medical encyclopedias include the Merck Manual and the Stedman's Medical Dictionary.
Fabry disease is a rare X-linked inherited lysosomal storage disorder caused by mutations in the GLA gene, which encodes the enzyme alpha-galactosidase A. This enzyme deficiency leads to the accumulation of glycosphingolipids, particularly globotriaosylceramide (Gb3 or GL-3), in various tissues and organs throughout the body. The accumulation of these lipids results in progressive damage to multiple organ systems, including the heart, kidneys, nerves, and skin.
The symptoms of Fabry disease can vary widely among affected individuals, but common manifestations include:
1. Pain: Acroparesthesias (burning or tingling sensations) in the hands and feet, episodic pain crises, chronic pain, and neuropathy.
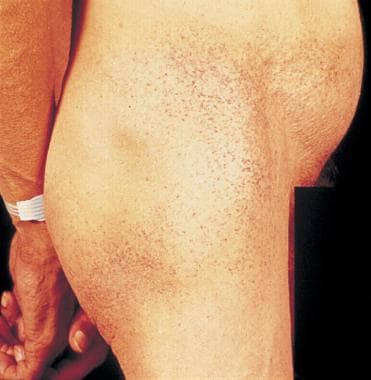
2. Skin: Angiokeratomas (small, red, rough bumps on the skin), hypohidrosis (decreased sweating), and anhydrosis (absent sweating).
3. Gastrointestinal: Abdominal pain, diarrhea, constipation, nausea, and vomiting.
4. Cardiovascular: Left ventricular hypertrophy (enlargement of the heart muscle), cardiomyopathy, ischemic heart disease, arrhythmias, and valvular abnormalities.
5. Renal: Proteinuria (protein in the urine), hematuria (blood in the urine), chronic kidney disease, and end-stage renal disease.
6. Nervous system: Hearing loss, tinnitus, vertigo, stroke, and cognitive decline.
7. Ocular: Corneal opacities, cataracts, and retinal vessel abnormalities.
8. Pulmonary: Chronic cough, bronchial hyperresponsiveness, and restrictive lung disease.
9. Reproductive system: Erectile dysfunction in males and menstrual irregularities in females.
Fabry disease affects both males and females, but the severity of symptoms is generally more pronounced in males due to the X-linked inheritance pattern. Early diagnosis and treatment with enzyme replacement therapy (ERT) or chaperone therapy can help manage the progression of the disease and improve quality of life.
Angiokeratoma is a cutaneous condition characterized by the presence of small, dilated blood vessels (capillaries) in the upper dermis, which are covered by thickened epidermis. These lesions appear as dark red to black papules or plaques on the skin surface. They can occur spontaneously or as a result of an underlying medical condition such as Fabry disease. Angiokeratomas are typically asymptomatic but may occasionally cause mild discomfort or itching. They most commonly affect the lower extremities, particularly the buttocks and genital region, but can also appear on other parts of the body.
Alpha-galactosidase is an enzyme that breaks down complex carbohydrates, specifically those containing alpha-galactose molecules. This enzyme is found in humans, animals, and microorganisms. In humans, a deficiency of this enzyme can lead to a genetic disorder known as Fabry disease, which is characterized by the accumulation of these complex carbohydrates in various tissues and organs, leading to progressive damage. Alpha-galactosidase is also used as a medication for the treatment of Fabry disease, where it is administered intravenously to help break down the accumulated carbohydrates and alleviate symptoms.
Alpha-N-Acetylgalactosaminidase (also known as alpha-GalNAcase) is an enzyme that belongs to the class of glycoside hydrolases. Its systematic name is N-acetyl-alpha-galactosaminide galactosaminohydrolase. This enzyme is responsible for catalyzing the hydrolysis of the terminal, non-reducing N-acetyl-D-galactosamine residues in gangliosides and glycoproteins.
Gangliosides are sialic acid-containing glycosphingolipids found in animal tissues, especially in the nervous system. Glycoproteins are proteins that contain oligosaccharide chains (glycans) covalently attached to their polypeptide backbone.
Deficiency or dysfunction of alpha-N-Acetylgalactosaminidase can lead to various genetic disorders, such as Schindler and Kanzaki diseases, which are characterized by the accumulation of gangliosides and glycoproteins in lysosomes, leading to progressive neurological deterioration.
Trihexosylceramides are a type of glycosphingolipids, which are complex lipids found in animal tissues. They consist of a ceramide molecule (a sphingosine and fatty acid) with three hexose sugars attached to it in a specific sequence, typically glucose-galactose-galactose.
Trihexosylceramides are further classified into two types based on the type of ceramide they contain: lactosylceramide (Gal-Glc-Cer) and isoglobotrihexosylceramide (GalNAcβ1-4Galβ1-4Glc-Cer).
These lipids are important components of the cell membrane and play a role in various biological processes, including cell recognition, signal transduction, and cell adhesion. Abnormal accumulation of trihexosylceramides has been implicated in certain diseases, such as Gaucher disease and Tay-Sachs disease, which are caused by deficiencies in enzymes involved in their breakdown.
Enzyme Replacement Therapy (ERT) is a medical treatment approach in which functional copies of a missing or deficient enzyme are introduced into the body to compensate for the lack of enzymatic activity caused by a genetic disorder. This therapy is primarily used to manage lysosomal storage diseases, such as Gaucher disease, Fabry disease, Pompe disease, and Mucopolysaccharidoses (MPS), among others.
In ERT, the required enzyme is produced recombinantly in a laboratory using biotechnological methods. The purified enzyme is then administered to the patient intravenously at regular intervals. Once inside the body, the exogenous enzyme is taken up by cells, particularly those affected by the disorder, and helps restore normal cellular functions by participating in essential metabolic pathways.
ERT aims to alleviate disease symptoms, slow down disease progression, improve quality of life, and increase survival rates for patients with lysosomal storage disorders. However, it does not cure the underlying genetic defect responsible for the enzyme deficiency.
I'm sorry for any confusion, but "New Zealand" is not a medical term or concept. It is a country located in the southwestern Pacific Ocean, known for its stunning landscapes, unique wildlife, and as the filming location for the "Lord of the Rings" films. If you have any questions related to medicine or health, I'd be happy to try and help answer those for you!
 Aspartylglucosaminuria
Aspartylglucosaminuria
List of OMIM disorder codes
Aspartylglucosamine
Aspartylglucosaminidase
Leukodystrophy
Fucosidosis
Finnish heritage disease
Galactosialidosis
Brachycephaly
Glycoproteinosis
AGU
Congenital disorders of amino acid metabolism
Lysosomal storage disease
List of conditions treated with hematopoietic stem cell transplantation
List of diseases (A)
Coarse facial features
Aspartylglucosaminuria - Wikipedia
 Aspartylglucosaminuria: MedlinePlus Genetics
Aspartylglucosaminuria: MedlinePlus Genetics
 Aspartylglycosaminuria - Supra-Regional Assay Service
Aspartylglycosaminuria - Supra-Regional Assay Service
 What causes Aspartylglucosaminuria? - Idsemergencymanagement.com
What causes Aspartylglucosaminuria? - Idsemergencymanagement.com
 Background - AGU Rare Disease: Aspartylglucosaminuria
Background - AGU Rare Disease: Aspartylglucosaminuria
Susceptibility-Weighted Imaging Findings in Aspartylglucosaminuria<...
 A capillary electrophoresis procedure for the screening of oligosaccharidoses and related diseases
A capillary electrophoresis procedure for the screening of oligosaccharidoses and related diseases
 WikiGenes
WikiGenes
Arvio P[au] - Search Results - PubMed
Genetic Brain Disorders | MedlinePlus
 Lana Lee Harder, PhD, ABPP - Pediatric Neuropsychologist - Children's Health
Lana Lee Harder, PhD, ABPP - Pediatric Neuropsychologist - Children's Health
 Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US
 Krabbe disease | Radiology Reference Article | Radiopaedia.org
Krabbe disease | Radiology Reference Article | Radiopaedia.org
Fucosidosis | Radiology Reference Article | Radiopaedia.org
 Developmental Insult and Intervention - Murdoch Children's Research Institute
Developmental Insult and Intervention - Murdoch Children's Research Institute
 Fabry disease | DermNet
Fabry disease | DermNet
 Lysosomal Storage Disease: Overview, Classification of Lysosomal Storage Diseases, Glycogen Storage Disease Type II
Lysosomal Storage Disease: Overview, Classification of Lysosomal Storage Diseases, Glycogen Storage Disease Type II
 How To Buy Methocarbamol Generic Pharmacy Canada, Purchase methocarbamol purchase toronto manitoba º Centre for Research on the...
How To Buy Methocarbamol Generic Pharmacy Canada, Purchase methocarbamol purchase toronto manitoba º Centre for Research on the...
 Search | Preprints.org
Search | Preprints.org
Inborn Errors of Metabolism: Practice Essentials, Pathophysiology, Etiology
 Baby’s Pregnancy Calendar
Baby’s Pregnancy Calendar
 Nationwide Children's Hospital - Overview, News & Competitors | ZoomInfo.com
Nationwide Children's Hospital - Overview, News & Competitors | ZoomInfo.com
 Vancouver mother of girl with rare, fatal illness now raising money for clinical trial | CTV News
Vancouver mother of girl with rare, fatal illness now raising money for clinical trial | CTV News
 Download Multihacks | Mods, Semi-Rage, Bunny Hop - Alcha Shop
Download Multihacks | Mods, Semi-Rage, Bunny Hop - Alcha Shop
 Namespace
Namespace
Lysosomal Storage Diseases | Profiles RNS
GlyCosmos Portal
 Katalog - Sjelden
Katalog - Sjelden
 Gpt2 Mouse Gene Details | glutamic pyruvate transaminase (alanine aminotransferase) 2 | International Mouse Phenotyping...
Gpt2 Mouse Gene Details | glutamic pyruvate transaminase (alanine aminotransferase) 2 | International Mouse Phenotyping...Symptoms of aspartylglucosaminuria2
- At birth, there is no sign that a child will develop symptoms of aspartylglucosaminuria. (wikipedia.org)
- loss of these cells causes many of the signs and symptoms of aspartylglucosaminuria. (medlineplus.gov)
Autosomal recessive2
- Aspartylglucosaminuria is an autosomal recessive genetic condition that is inherited from both parents. (wikipedia.org)
- Aspartylglucosaminuria (AGU) is a severe autosomal recessive lysosomal storage disorder that involves the central nervous system and causes skeletal abnormalities as well as connective tissue lesions. (nih.gov)
Inherited disease1
- Aspartylglucosaminuria (AGU) is an inherited disease that is characterized by a decline in mental functioning, accompanied by an increase in skin, bone and joint issues. (wikipedia.org)
Lysosomal diseases1
- Aspartylglucosaminuria (AGU) is one of the rare lysosomal diseases, very little understood, for the time being a cure still remains to be developed and administered to humans. (raretraitswiss.ch)
Urine1
- Only urine from patients with aspartylglucosaminuria and Schindler disease displayed normal results. (nih.gov)
Mutation2
- I, Ikonen S, Riekkinen P, Ginns EI, Peltonen L. Mice with an aspartylglucosaminuria mutation similar to humans replicate the pathophysiology in patients. (umassmed.edu)
- Localization of the disulfide bond involved in post-translational processing of glycosylasparaginase and apex legends scripting engine by a mutation in the Finnish-type aspartylglycosaminuria. (spitswimclub.org)
Patients4
- Dysmorphic facial features in aspartylglucosaminuria patients and carriers. (medlineplus.gov)
- the charity of patients with Aspartylglucosaminuria (AGU), information for families of patients and healthcare professionals who want to know more about (AGU). (raretraitswiss.ch)
- Impaired oral health in patients with aspartylglucosaminuria. (nih.gov)
- Reduction in head size in patients with aspartylglucosaminuria. (nih.gov)
Progressive2
- Aspartylglucosaminuria is a condition that causes a progressive decline in mental functioning. (idsemergencymanagement.com)
- Progressive nature of aspartylglucosaminuria. (nih.gov)
Finland3
- In Finland, it is estimated that 1 to 3 individuals are born with aspartylglucosaminuria each year. (medlineplus.gov)
- How many people are affected by aspartylglucosaminuria in Finland? (idsemergencymanagement.com)
- Aspartylglucosaminuria is estimated to affect 1 in 18,500 people in Finland. (idsemergencymanagement.com)
Typically1
- Infants with aspartylglucosaminuria appear healthy at birth, and development is typically normal throughout early childhood. (medlineplus.gov)
Facial1
- Overgrowth of oral mucosa and facial skin, a novel feature of aspartylglucosaminuria. (nih.gov)
Treatment4
- No treatment is available to cure or slow down the progression of aspartylglucosaminuria. (wikipedia.org)
- Which is the best treatment for aspartylglycosaminuria? (idsemergencymanagement.com)
- Treatment of Aspartylglycosaminuria is symptomatic and supportive. (idsemergencymanagement.com)
- COLUMBUS, Ohio--(BUSINESS WIRE)--Battelle, Andelyn Biosciences, Inc., and AmplifyBio, have been awarded a new task order from the National Institute of Neurological Disorders and Stroke (NINDS) to manufacture suspension process gene therapy for the transformative treatment of Aspartylglucosaminuria (AGU). (zoominfo.com)
People1
- Less severe symptoms include: enlargement of the spleen and liver diarrhea People with aspartylglucosaminuria may have lower than average height, because they tend to go through puberty earlier. (wikipedia.org)
Children2
- Since ear infections and respiratory infections are common for children diagnosed with aspartylglucosaminuria, it is best to have regular checkups for both the ears and the respiratory tract. (wikipedia.org)
- Your donation will help fund research, drug and gene therapy, clinical trials and more to find a cure for all children affected by Aspartylglucosaminuria. (raretrait.com)
Develop2
- In order to develop aspartylglucosaminuria, the individual must inherit changes in both of his AGU genes (autonomic recessive inheritance). (wikipedia.org)
- Adults with aspartylglucosaminuria often have psychological disorders and may develop seizures. (medlineplus.gov)
Features1
- Optical coherence tomography features in brothers with aspartylglucosaminuria. (childrens.com)
Review1
- Arvio M, Mononen I. Aspartylglycosaminuria: a review. (medlineplus.gov)
Condition1
- Aspartylglucosaminuria is a condition that primarily affects mental functioning and movement. (medlineplus.gov)
Child3
- When families have a child who has already been diagnosed with AGU, they have the option to observe the enzyme's activity that codes for AGU in future pregnancy, to help determine if the next child will also have a positive diagnosis for aspartylglucosaminuria. (wikipedia.org)
- When does a child show signs of aspartylglucosaminuria? (idsemergencymanagement.com)
- Rare Trait Swiss is a Swiss non-profit association, founded in March 2021 by the parents of a child affected by this rare lysosomal storage disease, aspartylglucosaminuria (AGU). (raretraitswiss.ch)
Rare1
- Makeda has a rare neurodegenerative disease called aspartylglucosaminuria, or AGU. (ctvnews.ca)
Specific1
- The activity of the deficient enzyme in aspartylglycosaminuria, aspartylglycosylaminase, was assayed with a specific gas-chromatographic method. (wikigenes.org)
Clinical Presentation and Potential Therapies1
- Aspartylglucosaminuria: Clinical Presentation and Potential Therapies. (medlineplus.gov)
Aspartylglucosaminidase1
- Aspartylglucosaminuria (AGU) is a lysosomal storage disorder caused by insufficient aspartylglucosaminidase (AGA) activity leading to chronic neurodegeneration. (bvsalud.org)
Lysosomal2
- Aspartylglucosaminuria itself is characterized as a lysosomal disease because it does deal with inadequate activity in an enzyme's function. (wikipedia.org)
- The possible presence of disturbed sleep-wake behaviour in the lysosomal storage disorder aspartylglucosaminuria, has not been previously studied, however. (omeka.net)
Genetic2
- Aspartylglucosaminuria is an autosomal recessive genetic condition that is inherited from both parents. (wikipedia.org)
- Carriers of the aspartylglucosaminuria genetic mutation and chronic arthritis. (nih.gov)
Children diagnosed2
- Since ear infections and respiratory infections are common for children diagnosed with aspartylglucosaminuria, it is best to have regular checkups for both the ears and the respiratory tract. (wikipedia.org)
- A family with two children diagnosed with aspartylglucosaminuria-case report and literature review]. (nih.gov)
Glycosylasparaginase1
- A fluorometric assay for glycosylasparaginase counter strike skin changer cheat and detection of aspartylglycosaminuria. (divinasdeuzas.com.br)
Seizures1
- Adults with aspartylglucosaminuria often have psychological disorders and may develop seizures. (medlineplus.gov)
Gene2
- Variants (also known as mutations) in the AGA gene cause aspartylglucosaminuria. (medlineplus.gov)
- COLUMBUS, Ohio-(BUSINESS WIRE)-Battelle, Andelyn Biosciences, Inc. , and AmplifyBio, have been awarded a new task order from the National Institute of Neurological Disorders and Stroke (NINDS) to manufacture suspension process gene therapy for the transformative treatment of Aspartylglucosaminuria (AGU). (com.ng)