Epidermolysis Bullosa
Epidermolysis Bullosa Dystrophica
Epidermolysis Bullosa Simplex
Epidermolysis Bullosa, Junctional
Epidermolysis Bullosa Acquisita
Collagen Type VII
Plectin
Keratin-14
Skin
Pylorus
Non-Fibrillar Collagens
Integrin beta4
Hemidesmosomes
Keratin-5
Keratins
Skin Diseases, Vesiculobullous
Keratinocytes
Fibril-Associated Collagens
Pedigree
Desmosomes
Frameshift Mutation
Basement Membrane
Codon, Nonsense
Intermediate Filament Proteins
Mutation
Skin Diseases, Genetic
Collagen
Skin, Artificial
Intestinal Atresia
Autoantigens
Codon, Terminator
Pemphigoid, Bullous
Intermediate Filaments
Reticulin
Phenotype
Prurigo
Turbinates
Laminin
Integrin alpha6beta4
Genes, Dominant
Point Mutation
Mosaicism
Fatal Outcome
Microbial Collagenase
Exons
Integrin alpha6
Heterozygote
Epidermis
Autoantibodies
Molecular Sequence Data
Cell Adhesion Molecules
Acantholysis
Genetic Therapy
Rare Diseases
Base Sequence
Microscopy, Electron
Mutation, Missense
Mouth Diseases
Fibroblasts
Pruritus
Prenatal Diagnosis
Hyperkeratosis, Epidermolytic
Family Health
Ichthyosis Bullosa of Siemens
Fluorescent Antibody Technique
Revertant mosaicism: partial correction of a germ-line mutation in COL17A1 by a frame-restoring mutation. (1/53)
Generalized atrophic benign epidermolysis bullosa is an autosomal recessive subepidermal blistering disease typified by null mutations in COL17A1. In 1 large kindred, affected individuals were homozygous for a 2-bp deletion in COL17A1, 4003delTC, which resulted in a downstream premature termination codon, nonsense-mediated mRNA decay, and abrogation of type XVII collagen synthesis. Interestingly, 1 of these patients, although phenotypically identical to her affected siblings, showed focal expression of type XVII collagen in epidermal basement membrane in a pattern suggestive of revertant mosaicism. When studies of randomly obtained epidermal, oromucosal, and peripheral blood cells failed to identify the genetic basis of this apparent mosaicism, microscopic subpopulations of potentially revertant epidermal cells (i.e., those overlying basement membrane containing type XVII collagen) were selectively isolated using laser capture microdissection. Analysis of DNA and RNA from these cells revealed a second mutation, 4080insGG, on 1 allele of COL17A1. This 2-bp insertion corrected the reading frame just proximal to the premature termination codon, countered nonsense-mediated mRNA decay, and allowed protein production by patient keratinocytes in vivo and in vitro. These studies elucidate the molecular basis of a novel form of revertant mosaicism in humans. (+info)BP180 gene delivery in junctional epidermolysis bullosa. (2/53)
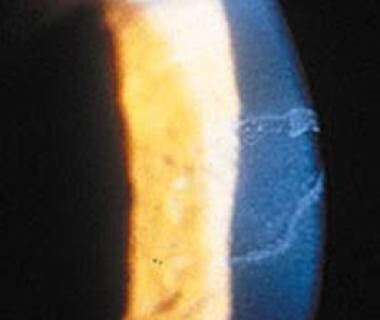
Epidermolysis bullosa (EB) comprises a family of inherited blistering skin diseases for which current therapy is only palliative. Junctional EB (JEB) involves dissociation of the dermal-epidermal junction and results from mutations in a number of genes that encode vital structural proteins, including BP180 (type XVII collagen/BPAG2). In order to develop a model of corrective gene delivery for JEB, we produced a retroviral expression vector for wild-type human BP180 and used it to restore BP180 protein expression to primary keratinocytes from BP180-negative patients with generalized atrophic JEB. Restoration of full-length BP180 protein expression was associated with adhesion parameter normalization of primary JEB keratinocytes in vitro. These cells were then used to regenerate human skin on immune-deficient mice. BP180 gene-transduced tissue demonstrated restoration of BP180 gene expression at the dermal-epidermal junction in vivo while untransduced regenerated JEB skin entirely lacked BP180 expression. These findings provide a basis for future efforts to achieve gene delivery in human EB skin tissue. (+info)The eye in epidermolysis bullosa. (3/53)
AIMS: To describe the ophthalmic findings in a large cohort of epidermolysis bullosa (EB) patients managed in one large specialist centre. METHODS: A case note review of consecutive patients seen at Great Ormond Street Children's Hospital. Data on the dermatological disease, ophthalmic history, and examination were collected and coded onto a data sheet. RESULTS: 181 patients: 50 (28%) simplex EB; 15 (8%) junctional EB; 28 (15%) autosomal dominant dystrophic EB; 72 (40%) autosomal recessive dystrophic EB; nine patients (5%) with dystrophic EB whose inheritance could not be ascertained; and seven cases (4%) of EB that could not be classified. Ocular problems were found in 12% (n = 6) of simplex patients and 40% (n = 6) of those with junctional disease. One patient (of 28) in the autosomal dominant dystrophic group had ocular involvement and 51% (37/72) of patients in the autosomal recessive dystrophic group had ophthalmic complications: corneal (25/72), lid ectropions (3/72), lid blisters (5/72), and symblepharon (3/72). CONCLUSION: Ophthalmic complications are common in EB overall but the incidence varies widely with subtype. Ophthalmic complications are the most severe in the dystrophic recessive and junctional subtypes where there is a need for extra vigilance. The major treatment modality was use of ocular lubricants. (+info)Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. (4/53)
Laminin 5 regulates anchorage and motility of epithelial cells through integrins alpha6beta4 and alpha3beta1, respectively. We used targeted disruption of the LAMA3 gene, which encodes the alpha3 subunit of laminin 5 and other isoforms, to examine developmental functions that are regulated by adhesion to the basement membrane (BM). In homozygous null animals, profound epithelial abnormalities were detected that resulted in neonatal lethality, consistent with removal of all alpha3-laminin isoforms from epithelial BMs. Alterations in three different cellular functions were identified. First, using a novel tissue adhesion assay, we found that the mutant BM could not induce stable adhesion by integrin alpha6beta4, consistent with the presence of junctional blisters and abnormal hemidesmosomes. In the absence of laminin 5 function, we were able to detect a new ligand for integrin alpha3beta1 in the epidermal BM, suggesting that basal keratinocytes can utilize integrin alpha3beta1 to interact with an alternative ligand. Second, we identified a survival defect in mutant epithelial cells that could be rescued by exogenous laminin 5, collagen, or an antibody against integrin alpha6beta4, suggesting that signaling through beta1 or beta4 integrins is sufficient for survival. Third, we detected abnormalities in ameloblast differentiation in developing mutant incisors indicating that events downstream of adhesion are affected in mutant animals. These results indicate that laminin 5 has an important role in regulating tissue organization, gene expression, and survival of epithelium. (+info)Moderation of phenotypic severity in dystrophic and junctional forms of epidermolysis bullosa through in-frame skipping of exons containing non-sense or frameshift mutations. (5/53)
Non-sense mutations on both alleles of either the type VII collagen gene (COL7A1) or the genes encoding laminin 5 (LAMA3, LAMB3, or LAMC2) usually result in clinically severe forms of recessive dystrophic or junctional epidermolysis bullosa, respectively. In this study we assessed two unrelated families whose mutations in genomic DNA predicted severe recessive dystrophic epidermolysis bullosa or junctional epidermolysis bullosa phenotypes but in whom the manifestations were milder than expected. The recessive dystrophic epidermolysis bullosa patients had a homozygous single base-pair frameshift mutation in exon 19 of COL7A1 (2470insG). Clinically, there was generalized blistering but only mild scarring. Skin biopsy revealed positive type VII collagen immunoreactivity and recognizable anchoring fibrils. The junctional epidermolysis bullosa patients were compound heterozygotes for a frameshift/non-sense combination of mutations in exons 3 and 17 of LAMB3 (29insC/Q834X). These patients did not have the lethal form of junctional epidermolysis bullosa but, as adults, displayed the milder generalized atrophic benign epidermolysis bullosa variant. There was undetectable laminin 5 staining at the dermal-epidermal junction using an antibody to the beta3 chain, but faintly positive alpha3 and gamma2 chain labeling, and there was variable hypoplasia of hemidesmosomes. To explain the milder recessive dystrophic epidermolysis bullosa and junctional epidermolysis bullosa phenotypes in these families, reverse transcription-polymerase chain reaction, using RNA extracted from frozen skin, was able to provide evidence for some rescue of mutant mRNA transcripts with restoration of the open- reading frame. In the recessive dystrophic epidermolysis bullosa patients, transcripts containing in-frame skipping of exon 19 of COL7A1 in the cDNA were detected, and in the junctional epidermolysis bullosa patients transcripts with in-frame skipping of exon 17 of LAMB3 were identified. The truncated proteins encoded by these transcripts are expected to lack certain critical domains involved in cell-matrix attachment, but may still be able to contribute to adhesion thereby moderating the severity of the skin blistering. This study shows the limitations in predicting phenotype in epidermolysis bullosa solely based on mutation analysis of genomic DNA and emphasizes the importance of immunohistochemistry, electron microscopy, and mRNA assessment as parallel investigations. (+info)Splicing modulation of integrin beta4 pre-mRNA carrying a branch point mutation underlies epidermolysis bullosa with pyloric atresia undergoing spontaneous amelioration with ageing. (6/53)
A general improvement with ageing has been reported in a few cases of epidermolysis bullosa with pyloric atresia (PA-JEB), an autosomal recessive skin disease characterized by extensive disadhesion of epithelia. In a patient who improved from severe to mild PA-JEB, a search for mutations in the integrin beta4 gene (IGTB4) detected heterozygosity for a novel base substitution 3986-19T-->A in the putative branchpoint sequence of intron 31, and a point mutation 3802+1G-->A in the donor splice site of intron 30 previously associated with severe PA-JEB. Analysis of mRNA showed that the intronic mutation prevents legitimate splicing of the beta4 pre-mRNA. Functional splicing can be restored in vitro by seeding the proband's keratinocytes on feeders of irradiated fibroblasts. Study of mRNA in wild-type keratinocytes transfected with IGTB4 minigenes containing intron 31 with or without mutation 3986-19T-->A, confirmed the causative role of the intronic mutation in PA-JEB, and highlighted the influence of feeders on the maturation process of the mutated beta4 pre-mRNA. Our results show that in a context of overall reduction of the beta4 mRNA levels, activation of the legitimate splice site in the aberrant beta4 pre-mRNA underlies the transient severity of the condition. The results also point to the relevance which the interaction between epithelial and stromal cells may have in modulating expression of integrin receptors. (+info)Digenic junctional epidermolysis bullosa: mutations in COL17A1 and LAMB3 genes. (7/53)
Junctional epidermolysis bullosa (JEB), a genetically heterogeneous group of blistering skin diseases, can be caused by mutations in the genes encoding laminin 5 or collagen XVII, which are components of the hemidesmosome-anchoring filament complex in the skin. Here, a family with severe nonlethal JEB and with mutations in genes for both proteins was identified. The index patient was compound heterozygous for the COL17A1 mutations L855X and R1226X and was heterozygous for the LAMB3 mutation R635X. As a consequence, two functionally related proteins were affected. Absence of collagen XVII and attenuated laminin 5 expression resulted in rudimentary hemidesmosome structure and separation of the epidermis from the basement membrane, with severe skin blistering as the clinical manifestation. In contrast, single heterozygotes carrying either (1) one or the other of the COL17A1 null alleles or (2) a double heterozygote for a COL17A1 and a LAMB3 null allele did not have a pathological skin phenotype. These observations indicate that the known allelic heterogeneity in JEB is further complicated by interactions between unlinked mutations. They also demonstrate that identification of one mutation in one gene is not sufficient for determination of the genetic basis of JEB in a given family. (+info)Compound heterozygosity for novel splice site mutations in the BPAG2/COL17A1 gene underlies generalized atrophic benign epidermolysis bullosa. (8/53)
Generalized atrophic benign epidermolysis bullosa, GABEB (OMIM# 226650), is a nonlethal variant of epidermolysis bullosa with autosomal recessive inheritance pattern. The pathogenesis of this disorder can be caused by mutations affecting two different gene/protein systems. Most of the mutations have been identified in the BPAG2/COL17A1 gene encoding a hemidesmosomal transmembrane protein, the 180 kDa bullous pemphigoid antigen (BP180), also known as type XVII collagen. The minority of the mutations are localized in the LAMB3 gene encoding the beta3 polypeptide of laminin 5. In In this study we describe a GABEB patient who showed absent expression of BP180 in the cultured keratinocytes as well as in the skin. The patient was a compound heterozygote for two different splice site mutations, 3053-1G-->C and 3871+1G-->C, affecting the extra-cellular domain of the protein. These mutations resulted in multiple aberrant splice variants, three of them causing premature termination codons for translation. This case, dealing with out-of-frame splice site mutations in BPAG2/COL17A1, attests to the molecular heterogeneity of GABEB. (+info)Epidermolysis Bullosa (EB) is a group of rare inherited skin disorders that are characterized by the development of blisters, erosions, and scarring following minor trauma or friction. The condition results from a genetic defect that affects the structural proteins responsible for anchoring the epidermis (outer layer of the skin) to the dermis (inner layer of the skin).
There are several types of EB, which vary in severity and clinical presentation. These include:
1. Epidermolysis Bullosa Simplex (EBS): This is the most common form of EB, and it typically affects the skin's superficial layers. Blistering tends to occur after minor trauma or friction, and healing usually occurs without scarring. There are several subtypes of EBS, which vary in severity.
2. Junctional Epidermolysis Bullosa (JEB): This form of EB affects the deeper layers of the skin, and blistering can occur spontaneously or following minor trauma. Healing often results in scarring, and affected individuals may also experience nail loss, dental abnormalities, and fragile mucous membranes.
3. Dystrophic Epidermolysis Bullosa (DEB): DEB affects the deeper layers of the skin, and blistering can lead to significant scarring, contractures, and fusion of fingers and toes. There are two main subtypes of DEB: recessive DEB (RDEB), which is more severe and associated with a higher risk of skin cancer, and dominant DEB (DDEB), which tends to be milder.
4. Kindler Syndrome: This is a rare form of EB that affects both the epidermis and dermis. Blistering can occur spontaneously or following minor trauma, and affected individuals may experience photosensitivity, poikiloderma (a mottled skin appearance), and oral and gastrointestinal abnormalities.
Treatment for EB typically focuses on managing symptoms, preventing blister formation and infection, and promoting wound healing. There is currently no cure for EB, but research is ongoing to develop new therapies and treatments.
Epidermolysis Bullosa Dystrophica (EBD) is a type of inherited skin disorder that belongs to the group of conditions known as Epidermolysis Bullosa. This condition is characterized by the development of fragile, blistering skin that can be caused by minor trauma or friction.
In EBD, the blisters form in the upper layer of the skin (epidermis) and the underlying layer (dermis), leading to scarring and tissue damage. The symptoms of EBD can range from mild to severe and may include:
* Blistering of the skin that can be triggered by friction, heat, or other factors
* Formation of scars, particularly on the hands and feet
* Thickening of the skin (hyperkeratosis)
* Nail abnormalities, such as ridged or brittle nails
* Mouth sores and blisters
* Dental problems, including tooth decay and gum disease
EBD is caused by mutations in the genes that provide instructions for making proteins that help to anchor the skin's layers together. As a result, the skin becomes fragile and prone to blistering.
There are several subtypes of EBD, each with its own specific genetic cause and symptoms. Treatment typically involves wound care, prevention of infection, and management of pain. In severe cases, surgery may be necessary to treat complications such as scarring or contractures.
Epidermolysis Bullosa Simplex (EBS) is a group of genetic skin disorders characterized by the development of blisters and erosions on the skin following minor trauma or friction. It is caused by mutations in genes that encode proteins responsible for anchoring the epidermis (outer layer of the skin) to the dermis (inner layer of the skin).
There are several subtypes of EBS, which vary in severity and clinical presentation. The most common form is called "Dowling-Meara" EBS, which is characterized by blistering at or near birth, widespread blistering, and scarring. Other forms of EBS include "Weber-Cockayne" EBS, which is characterized by localized blistering and healing with minimal scarring, and "Kobner" EBS, which is characterized by blistering in response to heat or physical trauma.
Treatment for EBS typically involves wound care, prevention of infection, and pain management. In some cases, protein therapy or bone marrow transplantation may be considered as a treatment option. It's important to note that the prognosis for individuals with EBS varies depending on the severity and subtype of the disorder.
Junctional Epidermolysis Bullosa (JEB) is a rare genetic skin disorder characterized by the presence of blisters and erosions on the skin and mucous membranes. It results from a defect in one of the proteins that anchors the epidermis (the outermost layer of the skin) to the dermis (the underlying layer of connective tissue). This defect causes the layers to separate easily, leading to blistering with minor friction or trauma.
JEB is usually apparent at birth or within the first few months of life. The severity of the condition can vary widely, even among members of the same family. There are several subtypes of JEB, each caused by mutations in different genes. These include:
1. Herlitz JEB: This is the most severe form, often lethal in infancy. It's characterized by widespread blistering over the entire body, including the mucous membranes, and severe growth retardation.
2. Non-Herlitz JEB: Less severe than Herlitz JEB, this form can still cause significant disability. Blistering tends to be localized to specific areas of the body, such as the hands, feet, and knees.
3. JEB with Pyloric Atresia: This subtype includes gastrointestinal abnormalities like pyloric atresia (a blockage in the lower part of the stomach), in addition to skin fragility.
Treatment for JEB typically focuses on managing symptoms and preventing complications. This may involve wound care, prevention of infection, pain management, nutritional support, and physical therapy. There is currently no cure for JEB.
Epidermolysis Bullosa Acquisita (EBA) is a rare autoimmune blistering disorder characterized by the production of autoantibodies against type VII collagen, a protein that plays a crucial role in anchoring the epidermis to the dermis. This results in the formation of blisters and erosions on the skin and mucous membranes, particularly in areas subjected to friction or trauma.
EBA can be classified into two main forms: the mechanobullous form and the inflammatory form. The mechanobullous form is characterized by spontaneous blistering and mechanical fragility of the skin, while the inflammatory form presents with inflammation and erosions in the mucous membranes.
The onset of EBA can occur at any age, but it is more common in adults, particularly those over 40 years old. The diagnosis of EBA is based on clinical presentation, direct immunofluorescence (DIF) studies, and detection of autoantibodies against type VII collagen.
Treatment of EBA typically involves a combination of wound care, prevention of infection, and immunosuppressive therapy to control the production of autoantibodies. The prognosis of EBA varies depending on the severity and extent of skin and mucous membrane involvement, as well as the response to treatment.
Collagen type VII is a type of collagen that is a major component of the anchoring fibrils, which are structures that help to attach the epidermis (the outermost layer of the skin) to the dermis (the layer of skin directly below the epidermis). Collagen type VII is composed of three identical chains that are encoded by the COL7A1 gene. Mutations in this gene can lead to a group of inherited blistering disorders known as autosomal recessive dystrophic epidermolysis bullosa, which is characterized by fragile skin and mucous membranes that blister and tear easily, often from minor trauma or friction.
A blister is a small fluid-filled bubble that forms on the skin due to friction, burns, or contact with certain chemicals or irritants. Blisters are typically filled with a clear fluid called serum, which is a component of blood. They can also be filled with blood (known as blood blisters) if the blister is caused by a more severe injury.
Blisters act as a natural protective barrier for the underlying skin and tissues, preventing infection and promoting healing. It's generally recommended to leave blisters intact and avoid breaking them, as doing so can increase the risk of infection and delay healing. If a blister is particularly large or painful, medical attention may be necessary to prevent complications.
Plectin is a large cytolinker protein that plays a crucial role in the structural organization and stability of the cell. It has the ability to interact with various components of the cytoskeleton, including intermediate filaments, microtubules, and actin filaments, thereby providing a critical link between these structures. Plectin is widely expressed in many tissues and is involved in maintaining the integrity and functionality of cells under both physiological and pathological conditions. Mutations in the gene encoding plectin have been associated with several human diseases, including epidermolysis bullosa, muscular dystrophy, and neuropathies.
Keratin-14 is a type of keratin protein that is specifically expressed in the suprabasal layers of stratified epithelia, including the epidermis. It is a component of the intermediate filament cytoskeleton and plays an important role in maintaining the structural integrity and stability of epithelial cells. Mutations in the gene encoding keratin-14 have been associated with several genetic skin disorders, such as epidermolysis bullosa simplex and white sponge nevus.
In medical terms, the skin is the largest organ of the human body. It consists of two main layers: the epidermis (outer layer) and dermis (inner layer), as well as accessory structures like hair follicles, sweat glands, and oil glands. The skin plays a crucial role in protecting us from external factors such as bacteria, viruses, and environmental hazards, while also regulating body temperature and enabling the sense of touch.
The pylorus is the lower, narrow part of the stomach that connects to the first part of the small intestine (duodenum). It consists of the pyloric canal, which is a short muscular tube, and the pyloric sphincter, a circular muscle that controls the passage of food from the stomach into the duodenum. The pylorus regulates the entry of chyme (partially digested food) into the small intestine by adjusting the size and frequency of the muscular contractions that push the chyme through the pyloric sphincter. This process helps in further digestion and absorption of nutrients in the small intestine.
Non-fibrillar collagens are a type of collagen that do not form fibrous structures, unlike the more common fibrillar collagens. They are a group of structurally diverse collagens that play important roles in various biological processes such as cell adhesion, migration, and differentiation. Non-fibrillar collagens include types IV, VI, VIII, X, XII, XIV, XVI, XIX, XXI, and XXVIII. They are often found in basement membranes and other specialized extracellular matrix structures.
Type IV collagen is a major component of the basement membrane and forms a network-like structure that provides a scaffold for other matrix components. Type VI collagen has a beaded filament structure and is involved in the organization of the extracellular matrix. Type VIII collagen is found in the eyes and helps to maintain the structural integrity of the eye. Type X collagen is associated with cartilage development and bone formation. Type XII and XIV collagens are fibril-associated collagens that help to regulate the organization and diameter of fibrillar collagens. The other non-fibrillar collagens have various functions, including cell adhesion, migration, and differentiation.
Overall, non-fibrillar collagens are important structural components of the extracellular matrix and play critical roles in various biological processes.
Integrin beta4, also known as ITGB4 or CD104, is a type of integrin subunit that forms part of the integrin receptor along with an alpha subunit. Integrins are transmembrane proteins involved in cell-cell and cell-extracellular matrix (ECM) adhesion, signal transduction, and regulation of various cellular processes such as proliferation, differentiation, and migration.
Integrin beta4 is unique among the integrin subunits because it has a large cytoplasmic domain that can interact with several intracellular signaling molecules, making it an important regulator of cell behavior. Integrin beta4 is widely expressed in various tissues, including epithelial cells, endothelial cells, and hematopoietic cells.
Integrin beta4 forms heterodimers with integrin alpha6 to form the receptor for laminins, which are major components of the basement membrane. This receptor is involved in maintaining the integrity of epithelial tissues and regulating cell migration during development, tissue repair, and cancer progression. Mutations in ITGB4 have been associated with several human diseases, including epidermolysis bullosa, a group of inherited skin disorders characterized by fragile skin and blistering.
Hemidesmosomes are specialized structures found in the cell membranes of epithelial cells that help to anchor them to the underlying basement membrane. They are composed of several proteins, including integrins and collagen type XVII, which interact with both intracellular keratin filaments and extracellular matrix components such as laminin-332. Hemidesmosomes play a crucial role in maintaining the integrity and stability of epithelial tissues by providing strong adhesive bonds between the epithelial cells and the underlying basement membrane, which is essential for normal tissue function and homeostasis. Mutations in genes encoding hemidesmosomal proteins can lead to various inherited skin blistering disorders, such as epidermolysis bullosa.
Keratin 5 is a type of keratin protein that is primarily expressed in the basal layer of epithelial tissues, including the skin, hair follicles, and nails. It forms heterodimers with keratin 14 and plays a crucial role in maintaining the structural integrity and stability of these tissues. Mutations in the gene that encodes keratin 5 (KRT5) can lead to several genetic disorders, such as epidermolysis bullosa simplex, which is characterized by blistering of the skin and mucous membranes.
Keratins are a type of fibrous structural proteins that constitute the main component of the integumentary system, which includes the hair, nails, and skin of vertebrates. They are also found in other tissues such as horns, hooves, feathers, and reptilian scales. Keratins are insoluble proteins that provide strength, rigidity, and protection to these structures.
Keratins are classified into two types: soft keratins (Type I) and hard keratins (Type II). Soft keratins are found in the skin and simple epithelial tissues, while hard keratins are present in structures like hair, nails, horns, and hooves.
Keratin proteins have a complex structure consisting of several domains, including an alpha-helical domain, beta-pleated sheet domain, and a non-repetitive domain. These domains provide keratin with its unique properties, such as resistance to heat, chemicals, and mechanical stress.
In summary, keratins are fibrous structural proteins that play a crucial role in providing strength, rigidity, and protection to various tissues in the body.
Recessive genes refer to the alleles (versions of a gene) that will only be expressed when an individual has two copies of that particular allele, one inherited from each parent. If an individual inherits one recessive allele and one dominant allele for a particular gene, the dominant allele will be expressed and the recessive allele will have no effect on the individual's phenotype (observable traits).
Recessive genes can still play a role in determining an individual's genetic makeup and can be passed down through generations even if they are not expressed. If two carriers of a recessive gene have children, there is a 25% chance that their offspring will inherit two copies of the recessive allele and exhibit the associated recessive trait.
Examples of genetic disorders caused by recessive genes include cystic fibrosis, sickle cell anemia, and albinism.
Vesiculobullous skin diseases are a group of disorders characterized by the formation of blisters (vesicles) and bullae (larger blisters) on the skin. These blisters form when there is a separation between the epidermis (outer layer of the skin) and the dermis (layer beneath the epidermis) due to damage in the area where they join, known as the dermo-epidermal junction.
There are several types of vesiculobullous diseases, each with its own specific causes and symptoms. Some of the most common types include:
1. Pemphigus vulgaris: an autoimmune disorder where the immune system mistakenly attacks proteins that help to hold the skin together, causing blisters to form.
2. Bullous pemphigoid: another autoimmune disorder, but in this case, the immune system attacks a different set of proteins, leading to large blisters and inflammation.
3. Dermatitis herpetiformis: a skin condition associated with celiac disease, where gluten ingestion triggers an immune response that leads to the formation of itchy blisters.
4. Pemphigoid gestationis: a rare autoimmune disorder that occurs during pregnancy and causes blisters on the abdomen and other parts of the body.
5. Epidermolysis bullosa: a group of inherited disorders where there is a fragile skin structure, leading to blistering and wound formation after minor trauma or friction.
Treatment for vesiculobullous diseases depends on the specific diagnosis and may include topical or systemic medications, such as corticosteroids, immunosuppressants, or antibiotics, as well as wound care and prevention of infection.
Keratinocytes are the predominant type of cells found in the epidermis, which is the outermost layer of the skin. These cells are responsible for producing keratin, a tough protein that provides structural support and protection to the skin. Keratinocytes undergo constant turnover, with new cells produced in the basal layer of the epidermis and older cells moving upward and eventually becoming flattened and filled with keratin as they reach the surface of the skin, where they are then shed. They also play a role in the immune response and can release cytokines and other signaling molecules to help protect the body from infection and injury.
Fibril-Associated Collagens (also known as FACIT collagens) are a group of collagen proteins that are characterized by their association with the surface of collagen fibrils. They play a role in the organization, stability, and diameter regulation of collagen fibrils. These collagens include types XII, XIV, XVI, XIX, XXI, and XXII.
Type XII collagen is found in various tissues such as tendons, ligaments, skin, and cornea. It has a triple-helical domain that interacts with the surface of collagen fibrils and a non-collagenous domain that can bind to other extracellular matrix proteins.
Type XIV collagen is also found in various tissues and has a similar structure to type XII collagen, but it has a larger non-collagenous domain. It plays a role in regulating the diameter of collagen fibrils.
Type XVI collagen is primarily found in cartilage and has a unique structure with multiple interruptions in its triple-helical domain. It is involved in the regulation of collagen fibrillogenesis and may also have roles in cell adhesion and signaling.
Types XIX and XXI collagens are similar to each other and are found in various tissues, including skin, tendons, and blood vessels. They have a short triple-helical domain and large non-collagenous domains that contain multiple binding sites for other extracellular matrix proteins.
Type XXII collagen is primarily found in the cornea and has a similar structure to type XIX collagen. It plays a role in regulating the diameter of collagen fibrils and may also have roles in cell adhesion and signaling.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Desmosomes are specialized intercellular junctions that provide strong adhesion between adjacent epithelial cells and help maintain the structural integrity and stability of tissues. They are composed of several proteins, including desmoplakin, plakoglobin, and cadherins, which form complex structures that anchor intermediate filaments (such as keratin) to the cell membrane. This creates a network of interconnected cells that can withstand mechanical stresses. Desmosomes are particularly abundant in tissues subjected to high levels of tension, such as the skin and heart.
A frameshift mutation is a type of genetic mutation that occurs when the addition or deletion of nucleotides in a DNA sequence is not divisible by three. Since DNA is read in groups of three nucleotides (codons), which each specify an amino acid, this can shift the "reading frame," leading to the insertion or deletion of one or more amino acids in the resulting protein. This can cause a protein to be significantly different from the normal protein, often resulting in a nonfunctional protein and potentially causing disease. Frameshift mutations are typically caused by insertions or deletions of nucleotides, but they can also result from more complex genetic rearrangements.
The basement membrane is a thin, specialized layer of extracellular matrix that provides structural support and separates epithelial cells (which line the outer surfaces of organs and blood vessels) from connective tissue. It is composed of two main layers: the basal lamina, which is produced by the epithelial cells, and the reticular lamina, which is produced by the connective tissue. The basement membrane plays important roles in cell adhesion, migration, differentiation, and survival.
The basal lamina is composed mainly of type IV collagen, laminins, nidogens, and proteoglycans, while the reticular lamina contains type III collagen, fibronectin, and other matrix proteins. The basement membrane also contains a variety of growth factors and cytokines that can influence cell behavior.
Defects in the composition or organization of the basement membrane can lead to various diseases, including kidney disease, eye disease, and skin blistering disorders.
The digestive system is a complex series of organs and glands that process food. Abnormalities in the digestive system can refer to a wide range of conditions that affect any part of the system, including the esophagus, stomach, small intestine, large intestine, liver, pancreas, and gallbladder. These abnormalities can be present at birth (congenital) or acquired later in life due to various factors such as infection, inflammation, injury, or disease.
Some examples of digestive system abnormalities include:
1. Gastroesophageal Reflux Disease (GERD): A condition where the stomach acid flows back into the esophagus, causing heartburn and damage to the esophageal lining.
2. Peptic Ulcers: Open sores that develop on the lining of the stomach or duodenum, often caused by bacterial infections or long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs).
3. Inflammatory Bowel Disease (IBD): A group of chronic inflammatory conditions of the intestine, including Crohn's disease and ulcerative colitis.
4. Irritable Bowel Syndrome (IBS): A functional gastrointestinal disorder characterized by abdominal pain, bloating, and altered bowel habits.
5. Celiac Disease: An autoimmune disorder where the ingestion of gluten leads to damage in the small intestine.
6. Diverticulosis: The presence of small pouches or sacs that form on the lining of the intestine, which can become inflamed or infected (diverticulitis).
7. Hiatal Hernia: A condition where a portion of the stomach protrudes through the diaphragm into the chest cavity.
8. Hepatitis: Inflammation of the liver, often caused by viral infections or toxins.
9. Cirrhosis: A chronic liver disease characterized by scarring and loss of liver function, often due to long-term alcohol abuse or hepatitis.
10. Gallstones: Small, hard deposits that form in the gallbladder and can cause pain and inflammation.
These are just a few examples of gastrointestinal disorders, and there are many others. If you are experiencing symptoms such as abdominal pain, bloating, diarrhea, constipation, or difficulty swallowing, it is important to speak with your healthcare provider to determine the cause and develop an appropriate treatment plan.
A nonsense codon is a sequence of three nucleotides in DNA or RNA that does not code for an amino acid. Instead, it signals the end of the protein-coding region of a gene and triggers the termination of translation, the process by which the genetic code is translated into a protein.
In DNA, the nonsense codons are UAA, UAG, and UGA, which are also known as "stop codons." When these codons are encountered during translation, they cause the release of the newly synthesized polypeptide chain from the ribosome, bringing the process of protein synthesis to a halt.
Nonsense mutations are changes in the DNA sequence that result in the appearance of a nonsense codon where an amino acid-coding codon used to be. These types of mutations can lead to premature termination of translation and the production of truncated, nonfunctional proteins, which can cause genetic diseases or contribute to cancer development.
Intermediate filament proteins (IFPs) are a type of cytoskeletal protein that form the intermediate filaments (IFs), which are one of the three major components of the cytoskeleton in eukaryotic cells, along with microtubules and microfilaments. These proteins have a unique structure, characterized by an alpha-helical rod domain flanked by non-helical head and tail domains.
Intermediate filament proteins are classified into six major types based on their amino acid sequence: Type I (acidic) and Type II (basic) keratins, Type III (desmin, vimentin, glial fibrillary acidic protein, and peripherin), Type IV (neurofilaments), Type V (lamins), and Type VI (nestin). Each type of IFP has a distinct pattern of expression in different tissues and cell types.
Intermediate filament proteins play important roles in maintaining the structural integrity and mechanical strength of cells, providing resilience to mechanical stress, and regulating various cellular processes such as cell division, migration, and signal transduction. Mutations in IFP genes have been associated with several human diseases, including cancer, neurodegenerative disorders, and genetic skin fragility disorders.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Genetic skin diseases are a group of disorders caused by mutations or alterations in the genetic material (DNA), which can be inherited from one or both parents. These mutations affect the structure, function, or development of the skin and can lead to various conditions with different symptoms, severity, and prognosis.
Some examples of genetic skin diseases include:
1. Epidermolysis Bullosa (EB): A group of disorders characterized by fragile skin and mucous membranes that blister and tear easily, leading to painful sores and wounds. There are several types of EB, each caused by mutations in different genes involved in anchoring the epidermis to the dermis.
2. Ichthyosis: A family of genetic disorders characterized by dry, thickened, scaly, or rough skin. The severity and symptoms can vary widely, depending on the specific type and underlying genetic cause.
3. Neurofibromatosis: A group of conditions caused by mutations in the NF1 gene, which regulates cell growth and division. The most common types, NF1 and NF2, are characterized by the development of benign tumors called neurofibromas on the skin and nerves, as well as other symptoms affecting various organs and systems.
4. Tuberous Sclerosis Complex (TSC): A genetic disorder caused by mutations in the TSC1 or TSC2 genes, which control cell growth and division. TSC is characterized by the development of benign tumors in multiple organs, including the skin, brain, heart, kidneys, and lungs.
5. Xeroderma Pigmentosum (XP): A rare genetic disorder caused by mutations in genes responsible for repairing DNA damage from ultraviolet (UV) radiation. People with XP are extremely sensitive to sunlight and have a high risk of developing skin cancer and other complications.
6. Incontinentia Pigmenti (IP): A genetic disorder that affects the development and growth of skin, hair, nails, teeth, and eyes. IP is caused by mutations in the IKBKG gene and primarily affects females.
7. Darier's Disease: An inherited skin disorder characterized by greasy, crusted, keratotic papules and plaques, usually located on the trunk, scalp, and seborrheic areas of the body. Darier's disease is caused by mutations in the ATP2A2 gene.
These are just a few examples of genetic skin disorders. There are many more, each with its unique set of symptoms, causes, and treatments. If you or someone you know has a genetic skin disorder, it is essential to consult with a dermatologist or other healthcare professional for proper diagnosis and treatment.
Collagen is the most abundant protein in the human body, and it is a major component of connective tissues such as tendons, ligaments, skin, and bones. Collagen provides structure and strength to these tissues and helps them to withstand stretching and tension. It is made up of long chains of amino acids, primarily glycine, proline, and hydroxyproline, which are arranged in a triple helix structure. There are at least 16 different types of collagen found in the body, each with slightly different structures and functions. Collagen is important for maintaining the integrity and health of tissues throughout the body, and it has been studied for its potential therapeutic uses in various medical conditions.
Artificial Skin is a synthetic substitute or equivalent that is used to replace, support, or enhance the function of damaged or absent skin. It can be made from various materials such as biopolymers, composites, or biosynthetic materials. The main purpose of artificial skin is to provide a temporary or permanent covering for wounds, burns, or ulcers that cannot be healed with conventional treatments. Additionally, it may serve as a platform for the delivery of medications or as a matrix for the growth of cells and tissues during skin grafting procedures. Artificial skin must possess properties such as biocompatibility, durability, flexibility, and permeability to air and water vapor in order to promote optimal healing and minimize scarring.
Intestinal atresia is a congenital condition characterized by the absence or complete closure of a portion of the intestine, preventing the passage of digested food from the stomach to the remaining part of the intestines. This results in a blockage in the digestive system, which can be life-threatening if not treated promptly after birth. The condition can occur anywhere along the small or large intestine and may affect either a single segment or multiple segments of the intestine.
There are several types of intestinal atresia, including:
1. Jejunal atresia: A closure or absence in the jejunum, a part of the small intestine located between the duodenum and ileum.
2. Ileal atresia: A closure or absence in the ileum, the lower portion of the small intestine that connects to the large intestine (cecum).
3. Colonic atresia: A closure or absence in the colon, a part of the large intestine responsible for storing and eliminating waste.
4. Duodenal atresia: A closure or absence in the duodenum, the uppermost portion of the small intestine that receives chyme (partially digested food) from the stomach.
5. Multiple atresias: When more than one segment of the intestines is affected by atresia.
The exact cause of intestinal atresia remains unclear, but it is believed to be related to disruptions in fetal development during pregnancy. Treatment typically involves surgical correction to reconnect the affected segments of the intestine and restore normal digestive function. The prognosis for infants with intestinal atresia depends on the severity and location of the atresia, as well as any associated conditions or complications.
Autoantigens are substances that are typically found in an individual's own body, but can stimulate an immune response because they are recognized as foreign by the body's own immune system. In autoimmune diseases, the immune system mistakenly attacks and damages healthy tissues and organs because it recognizes some of their components as autoantigens. These autoantigens can be proteins, DNA, or other molecules that are normally present in the body but have become altered or exposed due to various factors such as infection, genetics, or environmental triggers. The immune system then produces antibodies and activates immune cells to attack these autoantigens, leading to tissue damage and inflammation.
A codon is a sequence of three adjacent nucleotides in DNA or RNA that specifies a particular amino acid during the process of protein synthesis, or codes for the termination of translation. In DNA, these triplets are read in a 5' to 3' direction, while in mRNA, they are read in a 5' to 3' direction as well. There are 64 possible codons (4^3) in the genetic code, and 61 of them specify amino acids. The remaining three codons, UAA, UAG, and UGA, are terminator or stop codons that signal the end of protein synthesis.
Terminator codons, also known as nonsense codons, do not code for any amino acids. Instead, they cause the release of the newly synthesized polypeptide chain from the ribosome, which is the complex machinery responsible for translating the genetic code into a protein. This process is called termination or translation termination.
In prokaryotic cells, termination occurs when a release factor recognizes and binds to the stop codon in the A site of the ribosome. This triggers the hydrolysis of the peptidyl-tRNA bond, releasing the completed polypeptide chain from the tRNA and the ribosome. In eukaryotic cells, a similar process occurs, but it involves different release factors and additional steps to ensure accurate termination.
In summary, a codon is a sequence of three adjacent nucleotides in DNA or RNA that specifies an amino acid or signals the end of protein synthesis. Terminator codons are specific codons that do not code for any amino acids and instead signal the end of translation, leading to the release of the newly synthesized polypeptide chain from the ribosome.
According to the American Academy of Ophthalmology and the National Organization for Rare Disorders, bullous pemphigoid is an autoimmune blistering disorder characterized by the formation of large, fluid-filled blisters (bullae) on the skin and mucous membranes. This condition primarily affects older adults, with most cases occurring in individuals over 60 years of age.
In bullous pemphigoid, the immune system mistakenly produces antibodies against proteins called BP230 and BP180, which are found in the basement membrane zone – a layer that separates the epidermis (outer skin layer) from the dermis (inner skin layer). This autoimmune response leads to the formation of blisters, causing significant discomfort and potential complications if left untreated.
The symptoms of bullous pemphigoid typically include:
1. Large, fluid-filled blisters on the skin, often appearing on the trunk, arms, or legs. These blisters may be itchy or painful.
2. Blisters that rupture easily, leading to raw, open sores.
3. Mucous membrane involvement, such as blisters in the mouth, nose, eyes, or genital area.
4. Skin redness and irritation.
5. Fluid-filled bumps (papules) or pus-filled bumps (pustules).
6. Scarring and skin discoloration after blisters heal.
Treatment for bullous pemphigoid usually involves a combination of medications to control the immune response, reduce inflammation, and promote healing. These may include corticosteroids, immunosuppressants, or other targeted therapies. In some cases, antibiotics may also be prescribed to help manage secondary infections that can occur due to blister formation.
It is essential to consult with a healthcare professional for an accurate diagnosis and treatment plan if you suspect you have bullous pemphigoid or are experiencing related symptoms.
Intermediate filaments (IFs) are a type of cytoskeletal filament found in the cytoplasm of eukaryotic cells, including animal cells. They are called "intermediate" because they are smaller in diameter than microfilaments and larger than microtubules, two other types of cytoskeletal structures.
Intermediate filaments are composed of fibrous proteins that form long, unbranched, and flexible filaments. These filaments provide structural support to the cell and help maintain its shape. They also play a role in cell-to-cell adhesion, intracellular transport, and protection against mechanical stress.
Intermediate filaments are classified into six types based on their protein composition: Type I (acidic keratins), Type II (neutral/basic keratins), Type III (vimentin, desmin, peripherin), Type IV (neurofilaments), Type V (lamins), and Type VI (nestin). Each type of intermediate filament has a specific function and is expressed in different cell types. For example, Type I and II keratins are found in epithelial cells, while vimentin is expressed in mesenchymal cells.
Overall, intermediate filaments play an essential role in maintaining the structural integrity of cells and tissues, and their dysfunction has been implicated in various human diseases, including cancer, neurodegenerative disorders, and genetic disorders.
Reticulin is a type of protein fiber that forms part of the extracellular matrix in various connective tissues in the body. It is composed of collagenous and non-collagenous proteins, and it has a reticular or network-like structure when viewed under a microscope. In histology (the study of the microscopic structure of tissues), reticulin fibers are often stained to help identify certain types of cells or structures.
In particular, reticulin fibers are often found in close association with certain types of cells, such as hematopoietic stem cells and neurons. They provide structural support and help regulate the function of these cells. In addition, reticulin fibers play a role in the immune response, wound healing, and tissue repair.
Abnormal accumulations of reticulin fibers can be seen in various disease states, such as fibrosis (excessive scarring) and certain types of cancer. For example, increased reticulin fibers are often found in the liver in patients with cirrhosis, a condition characterized by extensive scarring and damage to the liver. Similarly, abnormal reticulin fiber deposition is seen in some forms of lymphoma, a type of cancer that affects the lymphatic system.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Prurigo is a dermatological condition characterized by the development of persistent, itchy papules (small, solid, raised bumps) on the skin. These lesions often result in scratching or rubbing, which can further exacerbate the itching and lead to the formation of new papules. The exact cause of prurigo is not well understood, but it may be associated with various underlying conditions such as atopic dermatitis, diabetes, HIV infection, or chronic renal failure.
There are two main types of prurigo: acute and chronic. Acute prurigo typically lasts for less than six months and is often triggered by an insect bite, drug reaction, or other short-term factors. Chronic prurigo, on the other hand, can persist for years and may be more resistant to treatment.
Prurigo can significantly affect a person's quality of life due to constant itching, discomfort, and potential sleep disturbances. Dermatological evaluation, identification of underlying causes, and appropriate management strategies are essential in addressing this condition effectively.
In medical terms, turbinates refer to the curled bone shelves that are present inside the nasal passages. They are covered by a mucous membrane and are responsible for warming, humidifying, and filtering the air that we breathe in through our nose. There are three pairs of turbinates in each nasal passage: inferior, middle, and superior turbinates. The inferior turbinate is the largest and most significant contributor to nasal airflow resistance. Inflammation or enlargement of the turbinates can lead to nasal congestion and difficulty breathing through the nose.
Laminin is a family of proteins that are an essential component of the basement membrane, which is a specialized type of extracellular matrix. Laminins are large trimeric molecules composed of three different chains: α, β, and γ. There are five different α chains, three different β chains, and three different γ chains that can combine to form at least 15 different laminin isoforms.
Laminins play a crucial role in maintaining the structure and integrity of basement membranes by interacting with other components of the extracellular matrix, such as collagen IV, and cell surface receptors, such as integrins. They are involved in various biological processes, including cell adhesion, differentiation, migration, and survival.
Laminin dysfunction has been implicated in several human diseases, including cancer, diabetic nephropathy, and muscular dystrophy.
Integrin α6β4 is a type of cell surface receptor that is composed of two subunits, α6 and β4. It is also known as CD49f/CD104. This integrin is primarily expressed in epithelial cells and plays important roles in cell adhesion, migration, and signal transduction.
Integrin α6β4 specifically binds to laminin-332 (also known as laminin-5), a component of the basement membrane, and forms a stable anchorage complex that links the cytoskeleton to the extracellular matrix. This interaction is critical for maintaining the integrity of epithelial tissues and regulating cell behavior during processes such as wound healing and tissue regeneration.
Mutations in the genes encoding integrin α6β4 have been associated with various human diseases, including epidermolysis bullosa, a group of inherited skin disorders characterized by fragile skin and blistering. Additionally, integrin α6β4 has been implicated in cancer progression and metastasis, as its expression is often upregulated in tumor cells and contributes to their invasive behavior.
Dominant genes refer to the alleles (versions of a gene) that are fully expressed in an individual's phenotype, even if only one copy of the gene is present. In dominant inheritance patterns, an individual needs only to receive one dominant allele from either parent to express the associated trait. This is in contrast to recessive genes, where both copies of the gene must be the recessive allele for the trait to be expressed. Dominant genes are represented by uppercase letters (e.g., 'A') and recessive genes by lowercase letters (e.g., 'a'). If an individual inherits one dominant allele (A) from either parent, they will express the dominant trait (A).
A point mutation is a type of genetic mutation where a single nucleotide base (A, T, C, or G) in DNA is altered, deleted, or substituted with another nucleotide. Point mutations can have various effects on the organism, depending on the location of the mutation and whether it affects the function of any genes. Some point mutations may not have any noticeable effect, while others might lead to changes in the amino acids that make up proteins, potentially causing diseases or altering traits. Point mutations can occur spontaneously due to errors during DNA replication or be inherited from parents.
Mosaicism, in the context of genetics and medicine, refers to the presence of two or more cell lines with different genetic compositions in an individual who has developed from a single fertilized egg. This means that some cells have one genetic makeup, while others have a different genetic makeup. This condition can occur due to various reasons such as errors during cell division after fertilization.
Mosaicism can involve chromosomes (where whole or parts of chromosomes are present in some cells but not in others) or it can involve single genes (where a particular gene is present in one form in some cells and a different form in others). The symptoms and severity of mosaicism can vary widely, depending on the type and location of the genetic difference and the proportion of cells that are affected. Some individuals with mosaicism may not experience any noticeable effects, while others may have significant health problems.
A fatal outcome is a term used in medical context to describe a situation where a disease, injury, or illness results in the death of an individual. It is the most severe and unfortunate possible outcome of any medical condition, and is often used as a measure of the severity and prognosis of various diseases and injuries. In clinical trials and research, fatal outcome may be used as an endpoint to evaluate the effectiveness and safety of different treatments or interventions.
Microbial collagenase is not a medical term per se, but it does refer to an enzyme that is used in various medical and research contexts. Collagenases are a group of enzymes that break down collagen, a structural protein found in connective tissues such as skin, tendons, and ligaments. Microbial collagenase is a type of collagenase that is produced by certain bacteria, such as Clostridium histolyticum.
In medical terms, microbial collagenase is used in various therapeutic and research applications, including:
1. Wound healing: Microbial collagenase can be used to break down and remove necrotic tissue from wounds, which can help promote healing and prevent infection.
2. Dental applications: Collagenases have been used in periodontal therapy to remove calculus and improve the effectiveness of root planing and scaling procedures.
3. Research: Microbial collagenase is a valuable tool for researchers studying the structure and function of collagen and other extracellular matrix proteins. It can be used to digest tissue samples, allowing scientists to study the individual components of the extracellular matrix.
It's important to note that while microbial collagenase has many useful applications, it must be used with care, as excessive or improper use can damage healthy tissues and cause adverse effects.
Exons are the coding regions of DNA that remain in the mature, processed mRNA after the removal of non-coding intronic sequences during RNA splicing. These exons contain the information necessary to encode proteins, as they specify the sequence of amino acids within a polypeptide chain. The arrangement and order of exons can vary between different genes and even between different versions of the same gene (alternative splicing), allowing for the generation of multiple protein isoforms from a single gene. This complexity in exon structure and usage significantly contributes to the diversity and functionality of the proteome.
Integrin α6 (also known as CD49f) is a type of integrin, which is a heterodimeric transmembrane receptor that mediates cell-cell and cell-extracellular matrix (ECM) interactions. Integrins play crucial roles in various biological processes such as cell adhesion, migration, proliferation, differentiation, and survival.
Integrin α6 is a 130 kDa glycoprotein that pairs with integrin β1, β4 or β5 to form three distinct heterodimeric complexes: α6β1, α6β4, and α6β5. Among these, the α6β4 integrin is the most extensively studied. It specifically binds to laminins in the basement membrane and plays essential roles in maintaining epithelial tissue architecture and function.
The α6β4 integrin has a unique structure with an extended cytoplasmic domain of β4 that can interact with intracellular signaling molecules, cytoskeletal proteins, and other adhesion receptors. This interaction allows the formation of stable adhesion complexes called hemidesmosomes, which anchor epithelial cells to the basement membrane and provide mechanical stability to tissues.
Mutations in integrin α6 or its partners can lead to various human diseases, including epidermolysis bullosa, a group of inherited skin disorders characterized by fragile skin and mucous membranes that blister and tear easily.
A skin cream is not a medical term per se, but it generally refers to a topical emollient preparation intended for application to the skin. It contains a mixture of water, oil, and active ingredients, which are formulated to provide various benefits such as moisturizing, protecting, soothing, or treating specific skin conditions. The exact definition and composition may vary depending on the product's intended use and formulation.
Examples of active ingredients in skin creams include:
1. Moisturizers (e.g., glycerin, hyaluronic acid) - help to retain water in the skin, making it feel softer and smoother.
2. Emollients (e.g., shea butter, coconut oil, petrolatum) - provide a protective barrier that helps prevent moisture loss and soften the skin.
3. Humectants (e.g., urea, lactic acid, alpha-hydroxy acids) - attract water from the environment or deeper layers of the skin to hydrate the surface.
4. Anti-inflammatory agents (e.g., hydrocortisone, aloe vera) - help reduce redness, swelling, and itching associated with various skin conditions.
5. Antioxidants (e.g., vitamin C, vitamin E, green tea extract) - protect the skin from free radical damage and environmental stressors that can lead to premature aging.
6. Sunscreen agents (e.g., zinc oxide, titanium dioxide, chemical filters) - provide broad-spectrum protection against UVA and UVB rays.
7. Skin lighteners (e.g., hydroquinone, kojic acid, arbutin) - help reduce the appearance of hyperpigmentation and even out skin tone.
8. Acne treatments (e.g., benzoyl peroxide, salicylic acid, retinoids) - target acne-causing bacteria, unclog pores, and regulate cell turnover to prevent breakouts.
It is essential to choose a skin cream based on your specific skin type and concerns, as well as any medical conditions or allergies you may have. Always consult with a dermatologist or healthcare provider before starting a new skincare regimen.
A heterozygote is an individual who has inherited two different alleles (versions) of a particular gene, one from each parent. This means that the individual's genotype for that gene contains both a dominant and a recessive allele. The dominant allele will be expressed phenotypically (outwardly visible), while the recessive allele may or may not have any effect on the individual's observable traits, depending on the specific gene and its function. Heterozygotes are often represented as 'Aa', where 'A' is the dominant allele and 'a' is the recessive allele.
The epidermis is the outermost layer of the skin, composed mainly of stratified squamous epithelium. It forms a protective barrier that prevents water loss and inhibits the entry of microorganisms. The epidermis contains no blood vessels, and its cells are nourished by diffusion from the underlying dermis. The bottom-most layer of the epidermis, called the stratum basale, is responsible for generating new skin cells that eventually move up to replace dead cells on the surface. This process of cell turnover takes about 28 days in adults.
The most superficial part of the epidermis consists of dead cells called squames, which are constantly shed and replaced. The exact rate at which this happens varies depending on location; for example, it's faster on the palms and soles than elsewhere. Melanocytes, the pigment-producing cells, are also located in the epidermis, specifically within the stratum basale layer.
In summary, the epidermis is a vital part of our integumentary system, providing not only physical protection but also playing a crucial role in immunity and sensory perception through touch receptors called Pacinian corpuscles.
Autoantibodies are defined as antibodies that are produced by the immune system and target the body's own cells, tissues, or organs. These antibodies mistakenly identify certain proteins or molecules in the body as foreign invaders and attack them, leading to an autoimmune response. Autoantibodies can be found in various autoimmune diseases such as rheumatoid arthritis, lupus, and thyroiditis. The presence of autoantibodies can also be used as a diagnostic marker for certain conditions.
DNA Mutational Analysis is a laboratory test used to identify genetic variations or changes (mutations) in the DNA sequence of a gene. This type of analysis can be used to diagnose genetic disorders, predict the risk of developing certain diseases, determine the most effective treatment for cancer, or assess the likelihood of passing on an inherited condition to offspring.
The test involves extracting DNA from a patient's sample (such as blood, saliva, or tissue), amplifying specific regions of interest using polymerase chain reaction (PCR), and then sequencing those regions to determine the precise order of nucleotide bases in the DNA molecule. The resulting sequence is then compared to reference sequences to identify any variations or mutations that may be present.
DNA Mutational Analysis can detect a wide range of genetic changes, including single-nucleotide polymorphisms (SNPs), insertions, deletions, duplications, and rearrangements. The test is often used in conjunction with other diagnostic tests and clinical evaluations to provide a comprehensive assessment of a patient's genetic profile.
It is important to note that not all mutations are pathogenic or associated with disease, and the interpretation of DNA Mutational Analysis results requires careful consideration of the patient's medical history, family history, and other relevant factors.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
A homozygote is an individual who has inherited the same allele (version of a gene) from both parents and therefore possesses two identical copies of that allele at a specific genetic locus. This can result in either having two dominant alleles (homozygous dominant) or two recessive alleles (homozygous recessive). In contrast, a heterozygote has inherited different alleles from each parent for a particular gene.
The term "homozygote" is used in genetics to describe the genetic makeup of an individual at a specific locus on their chromosomes. Homozygosity can play a significant role in determining an individual's phenotype (observable traits), as having two identical alleles can strengthen the expression of certain characteristics compared to having just one dominant and one recessive allele.
Cell adhesion molecules (CAMs) are a type of protein found on the surface of cells that mediate the attachment or adhesion of cells to either other cells or to the extracellular matrix (ECM), which is the network of proteins and carbohydrates that provides structural and biochemical support to surrounding cells.
CAMs play crucial roles in various biological processes, including tissue development, differentiation, repair, and maintenance of tissue architecture and function. They are also involved in cell signaling, migration, and regulation of the immune response.
There are several types of CAMs, classified based on their structure and function, such as immunoglobulin-like CAMs (IgCAMs), cadherins, integrins, and selectins. Dysregulation of CAMs has been implicated in various diseases, including cancer, inflammation, and neurological disorders.
Acantholysis is a medical term that refers to the separation of the cells in the upper layer of the skin (the epidermis), specifically between the pickle cell layer (stratum spinosum) and the granular cell layer (stratum granulosum). This separation results in the formation of distinct, round, or oval cells called acantholytic cells, which are typically seen in certain skin conditions.
Acantholysis is a characteristic feature of several skin disorders, including:
1. Pemphigus vulgaris: A rare autoimmune blistering disorder where the immune system produces antibodies against desmoglein-1 and -3 proteins, leading to acantholysis and formation of flaccid blisters.
2. Pemphigus foliaceus: Another autoimmune blistering disorder that specifically targets desmoglein-1 protein, causing superficial blisters and erosions on the skin.
3. Hailey-Hailey disease (familial benign chronic pemphigus): An autosomal dominant genetic disorder affecting ATP2C1 gene, leading to defective calcium transport and abnormal keratinocyte adhesion, resulting in acantholysis and recurrent skin eruptions.
4. Darier's disease (keratosis follicularis): An autosomal dominant genetic disorder affecting ATP2A2 gene, causing dysfunction of calcium transport and abnormal keratinocyte adhesion, resulting in acantholysis and characteristic papular or keratotic skin lesions.
5. Grover's disease (transient acantholytic dermatosis): An acquired skin disorder of unknown cause, characterized by the development of pruritic, red, and scaly papules and vesicles due to acantholysis.
The presence of acantholysis in these conditions can be confirmed through histopathological examination of skin biopsies.
Genetic therapy, also known as gene therapy, is a medical intervention that involves the use of genetic material, such as DNA or RNA, to treat or prevent diseases. It works by introducing functional genes into cells to replace missing or faulty ones caused by genetic disorders or mutations. The introduced gene is incorporated into the recipient's genome, allowing for the production of a therapeutic protein that can help manage the disease symptoms or even cure the condition.
There are several approaches to genetic therapy, including:
1. Replacing a faulty gene with a healthy one
2. Inactivating or "silencing" a dysfunctional gene causing a disease
3. Introducing a new gene into the body to help fight off a disease, such as cancer
Genetic therapy holds great promise for treating various genetic disorders, including cystic fibrosis, muscular dystrophy, hemophilia, and certain types of cancer. However, it is still an evolving field with many challenges, such as efficient gene delivery, potential immune responses, and ensuring the safety and long-term effectiveness of the therapy.
A rare disease, also known as an orphan disease, is a health condition that affects fewer than 200,000 people in the United States or fewer than 1 in 2,000 people in Europe. There are over 7,000 rare diseases identified, and many of them are severe, chronic, and often life-threatening. The causes of rare diseases can be genetic, infectious, environmental, or degenerative. Due to their rarity, research on rare diseases is often underfunded, and treatments may not be available or well-studied. Additionally, the diagnosis of rare diseases can be challenging due to a lack of awareness and understanding among healthcare professionals.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
Electron microscopy (EM) is a type of microscopy that uses a beam of electrons to create an image of the sample being examined, resulting in much higher magnification and resolution than light microscopy. There are several types of electron microscopy, including transmission electron microscopy (TEM), scanning electron microscopy (SEM), and reflection electron microscopy (REM).
In TEM, a beam of electrons is transmitted through a thin slice of the sample, and the electrons that pass through the sample are focused to form an image. This technique can provide detailed information about the internal structure of cells, viruses, and other biological specimens, as well as the composition and structure of materials at the atomic level.
In SEM, a beam of electrons is scanned across the surface of the sample, and the electrons that are scattered back from the surface are detected to create an image. This technique can provide information about the topography and composition of surfaces, as well as the structure of materials at the microscopic level.
REM is a variation of SEM in which the beam of electrons is reflected off the surface of the sample, rather than scattered back from it. This technique can provide information about the surface chemistry and composition of materials.
Electron microscopy has a wide range of applications in biology, medicine, and materials science, including the study of cellular structure and function, disease diagnosis, and the development of new materials and technologies.
A missense mutation is a type of point mutation in which a single nucleotide change results in the substitution of a different amino acid in the protein that is encoded by the affected gene. This occurs when the altered codon (a sequence of three nucleotides that corresponds to a specific amino acid) specifies a different amino acid than the original one. The function and/or stability of the resulting protein may be affected, depending on the type and location of the missense mutation. Missense mutations can have various effects, ranging from benign to severe, depending on the importance of the changed amino acid for the protein's structure or function.
Mouth diseases refer to a variety of conditions that affect the oral cavity, including the lips, gums, teeth, tongue, palate, and lining of the mouth. These diseases can be caused by bacteria, viruses, fungi, or other organisms. They can also result from injuries, chronic illnesses, or genetic factors.
Some common examples of mouth diseases include dental caries (cavities), periodontal disease (gum disease), oral herpes, candidiasis (thrush), lichen planus, and oral cancer. Symptoms may include pain, swelling, redness, bleeding, bad breath, difficulty swallowing or speaking, and changes in the appearance of the mouth or teeth. Treatment depends on the specific diagnosis and may involve medications, dental procedures, or lifestyle changes.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
Pruritus is a medical term derived from Latin, in which "prurire" means "to itch." It refers to an unpleasant sensation on the skin that provokes the desire or reflex to scratch. This can be caused by various factors, such as skin conditions (e.g., dryness, eczema, psoriasis), systemic diseases (e.g., liver disease, kidney failure), nerve disorders, psychological conditions, or reactions to certain medications.
Pruritus can significantly affect a person's quality of life, leading to sleep disturbances, anxiety, and depression. Proper identification and management of the underlying cause are essential for effective treatment.
Prenatal diagnosis is the medical testing of fetuses, embryos, or pregnant women to detect the presence or absence of certain genetic disorders or birth defects. These tests can be performed through various methods such as chorionic villus sampling (CVS), amniocentesis, or ultrasound. The goal of prenatal diagnosis is to provide early information about the health of the fetus so that parents and healthcare providers can make informed decisions about pregnancy management and newborn care. It allows for early intervention, treatment, or planning for the child's needs after birth.
A newborn infant is a baby who is within the first 28 days of life. This period is also referred to as the neonatal period. Newborns require specialized care and attention due to their immature bodily systems and increased vulnerability to various health issues. They are closely monitored for signs of well-being, growth, and development during this critical time.
Epidermolytic hyperkeratosis (EH) is a rare genetic skin disorder characterized by the abnormal growth and accumulation of keratin, a protein found in the outermost layer of the skin (epidermis). This condition results in widespread blistering and peeling of the skin, particularly in areas prone to friction such as the hands, feet, knees, and elbows.
EH is caused by mutations in the KRT1 or KRT10 genes, which provide instructions for making keratin proteins that are essential for maintaining the structure and integrity of the epidermis. When these genes are mutated, the keratin proteins become unstable and form clumps, leading to the formation of blisters and areas of thickened, scaly skin (hyperkeratosis).
EH is typically present at birth or appears in early childhood, and it can range from mild to severe. In addition to the skin symptoms, individuals with EH may also experience nail abnormalities, hair loss, and an increased risk of skin infections. Treatment for EH is focused on managing symptoms and preventing complications, and may include topical creams or ointments, wound care, and protection from friction and injury.
"Family Health" is not a term that has a single, widely accepted medical definition. However, in the context of healthcare and public health, "family health" often refers to the physical, mental, and social well-being of all members of a family unit. It includes the assessment, promotion, and prevention of health conditions that affect individual family members as well as the family as a whole.
Family health may also encompass interventions and programs that aim to strengthen family relationships, communication, and functioning, as these factors can have a significant impact on overall health outcomes. Additionally, family health may involve addressing social determinants of health, such as poverty, housing, and access to healthcare, which can affect the health of families and communities.
Overall, family health is a holistic approach to healthcare that recognizes the importance of considering the needs and experiences of all family members in promoting and maintaining good health.
Ichthyosis Bullosa of Siemens (IBS) is a rare genetic skin disorder that is characterized by the presence of blisters and erosions on the skin. It is caused by mutations in the KRT5 or KRT14 gene, which provide instructions for making keratin proteins that are essential for the structural integrity of the skin.
In IBS, the mutated keratin proteins are fragile and can form clumps, leading to the formation of blisters in response to minor trauma or friction. These blisters typically occur on the palms of the hands, soles of the feet, and other areas of the body that are subjected to frequent rubbing or pressure.
IBS is usually present at birth or develops during infancy, and it can vary in severity from mild to severe. In addition to blistering, individuals with IBS may also experience skin thickening, hyperpigmentation, and scaling. The condition can be associated with other medical issues, such as nail abnormalities, hair loss, and dental problems.
There is no cure for IBS, but treatment is focused on managing symptoms and preventing complications. This may include the use of topical creams or ointments to protect the skin, antibiotics to treat infections, and pain management strategies. In severe cases, systemic medications such as retinoids may be used to help reduce blistering and promote skin healing.
The Fluorescent Antibody Technique (FAT) is a type of immunofluorescence assay used in laboratory medicine and pathology for the detection and localization of specific antigens or antibodies in tissues, cells, or microorganisms. In this technique, a fluorescein-labeled antibody is used to selectively bind to the target antigen or antibody, forming an immune complex. When excited by light of a specific wavelength, the fluorescein label emits light at a longer wavelength, typically visualized as green fluorescence under a fluorescence microscope.
The FAT is widely used in diagnostic microbiology for the identification and characterization of various bacteria, viruses, fungi, and parasites. It has also been applied in the diagnosis of autoimmune diseases and certain cancers by detecting specific antibodies or antigens in patient samples. The main advantage of FAT is its high sensitivity and specificity, allowing for accurate detection and differentiation of various pathogens and disease markers. However, it requires specialized equipment and trained personnel to perform and interpret the results.
No data available that match "epidermolysis bullosa junctional"
 Junctional epidermolysis bullosa - Wikipedia
Junctional epidermolysis bullosa - Wikipedia
 Junctional epidermolysis bullosa: MedlinePlus Genetics
Junctional epidermolysis bullosa: MedlinePlus Genetics
 Junctional Epidermolysis Bullosa (Discovered in the Australian Shepherd)
Junctional Epidermolysis Bullosa (Discovered in the Australian Shepherd)
 Kidney-Urinary Tract Involvement with Intermediate Junctional Epidermolysis Bullosa
Kidney-Urinary Tract Involvement with Intermediate Junctional Epidermolysis Bullosa
 A very mild form of non-Herlitz junctional epidermolysis bullosa: BP180 rescue by outsplicing of mutated exon 30 coding for the...
A very mild form of non-Herlitz junctional epidermolysis bullosa: BP180 rescue by outsplicing of mutated exon 30 coding for the...
 Junctional Epidermolysis Bullosa - GeneReviews® - NCBI Bookshelf
Junctional Epidermolysis Bullosa - GeneReviews® - NCBI Bookshelf
 Junctional Epidermolysis Bullosa (JEB)
Junctional Epidermolysis Bullosa (JEB)
 Epidermolysis Bullosa: Practice Essentials, Pathophysiology, Etiology
Epidermolysis Bullosa: Practice Essentials, Pathophysiology, Etiology
Junctional Epidermolysis Bullosa (Discovered in the Australian Cattle Dog Mix)
 Junctional epidermolysis bullosa of the larynx<...
Junctional epidermolysis bullosa of the larynx<...
 Epidermolysis bullosa: Effects, types, and symptoms
Epidermolysis bullosa: Effects, types, and symptoms
 General Data Protection Regulation Compliance - 23andMe Europe
General Data Protection Regulation Compliance - 23andMe Europe
 Protecting Sight #240: What happens when you remove the hippocampus? Horizontal chop technique & junctional epidermolysis...
Protecting Sight #240: What happens when you remove the hippocampus? Horizontal chop technique & junctional epidermolysis...
A de novo COL17A1 splicesite mutation causing a 7-bp deletion in a Taiwanese patient with junctional epidermolysis bullosa<...
Lamc2 MGI Mouse Gene Detail - MGI:99913 - laminin, gamma 2
Epidermolysis Bullosa: Practice Essentials, Pathophysiology, Etiology
 Epidermolysis bullosa - Diagnosis and treatment - Mayo Clinic
Epidermolysis bullosa - Diagnosis and treatment - Mayo Clinic
Heterozygous COL17A1 variants are a frequent cause of amelogenesis imperfecta | Journal of Medical Genetics
 Stanford EB Publications
Stanford EB Publications
Nephrology | Medical News & Expert Insight | HCPLive | Page 5
 palmoplantar keratoderma and congenital alopecia 1 - Ontology Browser - Rat Genome Database
palmoplantar keratoderma and congenital alopecia 1 - Ontology Browser - Rat Genome Database
 SMART: FN3 domain annotation
SMART: FN3 domain annotation
SMART: FN3 domain annotation
 Eloxx Pharmaceuticals Announces FDA Clearance to Begin Single Ascending Dose Study of ZKN-013
Eloxx Pharmaceuticals Announces FDA Clearance to Begin Single Ascending Dose Study of ZKN-013
Birch Bark Derivative Effective for Epidermolysis Bullosa
Treatment of Oral Lesions in Dystrophic Epidermolysis Bullosa: A Case Series of Cord Blood Platelet Gel and Low-level Laser...
 Epidermolysis Bullosa - ENT Clinic Sydney
Epidermolysis Bullosa - ENT Clinic Sydney
 WikiGenes - Fluorescent Antibody Technique
WikiGenes - Fluorescent Antibody Technique
urofacial syndrome - Ontology Browser - Rat Genome Database
 Psychosocial recommendations for the care of children and adults with epidermolysis bullosa and their family: evidence based...
Psychosocial recommendations for the care of children and adults with epidermolysis bullosa and their family: evidence based...
Simplex11
- Epidermolysis bullosa is classified into four major categories: (1) epidermolysis bullosa simplex (intraepidermal skin separation), (2) junctional epidermolysis bullosa (skin separation in lamina lucida or central BMZ), (3) dystrophic epidermolysis bullosa (sublamina densa BMZ separation, as in the images below), and (4) Kindler syndrome (extremely rare, blistering at any level). (medscape.com)
- Epidermolysis bullosa simplex: Blisters occur in the lower part of the epidermis. (nih.gov)
- Epidermolysis bullosa simplex is the most common form of the disease. (nih.gov)
- Epidermolysis bullosa (EB) simplex blisters form in the levels of the epidermis. (msdmanuals.com)
- Epidermolysis bullosa simplex is the most common and mildest type, occurring in about 80% of cases. (msdmanuals.com)
- In type-specific cases more than 60% of prevalent cases were observed in epidermolysis bullosa simplex, and only 5% were found in junctional epidermolysis bullosa in 2022. (researchandmarkets.com)
- In the more severe subtypes of epidermolysis bullosa simplex (Koebner subtype and Dowling-Maera subtype), the blisters are more widespread and the onset of blistering is first noticed a short period of time after birth. (entclinic.com.au)
- These subtypes cause deeper blisters and erosions than what are seen in epidermolysis bullosa simplex. (entclinic.com.au)
- Paragon founded Castle Creek with the vision to deliver transformative therapies to patients with rare genetic dermatologic diseases, including Epidermolysis Bullosa Simplex," said Paragon Biosciences CE O Jeff Aronin [3]. (pressbooks.pub)
- The three main subtypes of EB are EB simplex (EBS), Junctional EB (JEB) and Dystrophic EB (DEB), where EBS occurs at most. (pressbooks.pub)
- KRT5 and KRT14 Mutations in Epidermolysis Bullosa Simplex with Phenotypic Heterogeneity, and Evidence of Semidominant Inheritance in a Multiplex Family. (jefferson.edu)
Lethal junctional epiderm1
- Muhle C, Jiang QJ, Charlesworth A, Bruckner-Tuderman L, Meneguzzi G, Schneider H. Novel and recurrent mutations in the laminin-5 genes causing lethal junctional epidermolysis bullosa: molecular basis and clinical course of Herlitz disease. (nih.gov)
Subtypes7
- These findings attest to the clinical and molecular heterogeneity of the junctional and hemidesmosomal subtypes of EB. (bmj.com)
- Historically, epidermolysis bullosa subtypes have been classified according to skin morphology. (medscape.com)
- This subtype, similar to other dystrophic and junctional epidermolysis bullosa subtypes, can result in nail dystrophy and loss. (medscape.com)
- Researchers have identified more than 30 subtypes of the disease, which are groupings under the four major types of epidermolysis bullosa. (nih.gov)
- Epidermolysis bullosa is a group of 4 very rare genetic diseases and their subtypes. (msdmanuals.com)
- There are also several subtypes of dystrophic epidermolysis bullosa. (entclinic.com.au)
- The dystrophic epidermolysis bullosa subtypes often produce scarring after the blisters have healed. (entclinic.com.au)
Mutations16
- Junctional epidermolysis bullosa most commonly results from mutations in the LAMA3 , LAMB3 , LAMC2 , and COL17A1 genes. (medlineplus.gov)
- LAMB3 gene mutations are the most common, causing about 70 percent of all cases of junctional epidermolysis bullosa. (medlineplus.gov)
- Very rarely, junctional epidermolysis bullosa is caused by mutations in another gene that provides instructions for making a different protein that helps attach the top layer of skin to underlying layers. (medlineplus.gov)
- More than 100 mutations in the LAMB3 gene have been identified in people with junctional epidermolysis bullosa (JEB). (nih.gov)
- Other LAMB3 gene mutations cause the milder form of junctional epidermolysis bullosa, JEB generalized intermediate. (nih.gov)
- Homozygous LAMB3 mutations cause junctional epidermolysis bullosa (JEB) and enamel defects are seen in JEB cases. (nih.gov)
- Background: Junctional epidermolysis bullosa (JEB) is a group of inherited blistering diseases characterized by epidermal-dermal separation resulting from mutations that affect the function of critical components of the basement membrane zone. (tau.ac.il)
- Epidermolysis bullosa is a family of bullous disorders caused by an absence of basement membrane components due to underlying gene mutations. (medscape.com)
- Epidermolysis bullosa is a group of inherited disorders that involve various genetic mutations. (msdmanuals.com)
- Dystrophic epidermolysis bullosa tends to be due to genetic mutations affecting the collagen in the dermis. (entclinic.com.au)
- The epithelium-specific expression of the gamma 2 chain implied its role as an epithelium attachment molecule, and mutations in this gene have been associated with junctional epidermolysis bullosa, a skin disease characterized by blisters due to disruption of the epidermal-dermal junction. (nih.gov)
- Genome-wide single nucleotide polymorphism-based autozygosity mapping facilitates identification of mutations in consanguineous families with epidermolysis bullosa. (jefferson.edu)
- Mutations in PLOD3, encoding lysyl hydroxylase 3, cause a complex connective tissue disorder including recessive dystrophic epidermolysis bullosa-like blistering phenotype with abnormal anchoring fibrils and type VII collagen deficiency. (jefferson.edu)
- First report of COL7A1 mutations in two patients with recessive dystrophic epidermolysis bullosa from Peru. (jefferson.edu)
- Seven novel COL7A1 mutations identified in patients with recessive dystrophic epidermolysis bullosa from Mexico. (jefferson.edu)
- Ancestral patterns of recessive dystrophic epidermolysis bullosa mutations in Hispanic populations suggest sephardic ancestry. (jefferson.edu)
LAMB31
- LAMB3 missense variant in Australian Shepherd dogs with junctional epidermolysis bullosa. (wisdompanel.com)
Atrophic benign epidermolysis bullosa1
- Finally, in a family with the clinical diagnosis of generalized atrophic benign epidermolysis bullosa, a homozygous non-sense mutation in Col17A1 gene (encoding the BPAG2 antigen) was identified. (tau.ac.il)
Disease12
- Junctional epidermolysis bullosa is a genetic disease. (nih.gov)
- A fifth type of the disease, epidermolysis bullosa acquisita, is a rare autoimmune disorder that causes the body's immune system to attack a certain type of collagen in the person's skin. (nih.gov)
- Epidermolysis bullosa (EB), a rare disease eligible for orphan drug status in various regions, presents abundant opportunities for research, development and investment, fueled by the incentives of market exclusivity and regulatory support. (researchandmarkets.com)
- Epidermolysis bullosa qualifies as a rare disease, making it eligible for orphan drug status in many regions. (researchandmarkets.com)
- Epidermolysis bullosa is a blistering skin disease which is usually first noticed during early childhood. (entclinic.com.au)
- Since the corneal epithelial-stromal separations are in the region identified by electron microscopy as the lamina lucida, the same region affected by the blistering disease junctional epidermolysis bullosa (JEB), we postulated that the molecules that are defective in JEB would be the same ones cleaved by mustard compounds. (nih.gov)
- The evolution of the Epidermolysis bullosa (EB) House Austria in Salzburg has demonstrated from its beginning in 2005 in an exceptional way the establishment of an optimized health care for a hitherto neglected group of patients, suffering from a rare but devastating skin disease: Epidermolysis bullosa. (almclinmed.ru)
- Epidermolysis bullosa is a mucocutaneous disease characterized by the formation of bullae on the skin and mucosa, which grow either spontaneously or from minor traumas. (bvsalud.org)
- This disease is classified as simple, junctional, and dystrophic. (bvsalud.org)
- The case study revolves around a boy with junctional epidermolysis bullosa, a disease that. (webtopnews.com)
- He was born with a genetic disease called junctional epidermolysis bullosa that leaves his skin as fragile as a butterfly's wings. (moscownewsdaily.com)
- Very-Early-Onset Inflammatory Bowel Disease in a Patient With Junctional Epidermolysis Bullosa With a Homozygous Mutation in the a6 Integrin Gene (ITGA6). (jefferson.edu)
Acquisita2
- Related articles include Epidermolysis Bullosa Acquisita and Pediatric Epidermolysis Bullosa . (medscape.com)
- Epidermolysis Bullosa Acquisita Epidermolysis bullosa acquisita is a rare, acquired, chronic condition characterized by subepidermal blistering. (msdmanuals.com)
Basement membrane5
- The type known as Junctional Epidermolysis Bullosa is indicative of the involvement of the epithelial basement membrane. (wisdompanel.com)
- JEB (Junctional Epidermolysis bullosa) is a blistering disorder of the skin and mucous membranes in which tissue separation occurs within the lamina lucida of the basement membrane zone at the dermal-epidermal junction. (labogen.com)
- Based on a better understanding of the basement membrane zone (BMZ) and the genes responsible for its components, newer treatments (eg, gene or protein therapy) may provide solutions to the skin fragility found in patients with epidermolysis bullosa. (medscape.com)
- Junctional epidermolysis bullosa: Blisters occur in the top portion of the basement membrane, due to problems in attachment between the epidermis and basement membrane. (nih.gov)
- Dystrophic epidermolysis bullosa: Blisters occur in the upper dermis due to problems in attachment between the basement membrane and the upper dermis. (nih.gov)
Clinical1
- Please note: It is possible that disorder signs similar to the ones associated with this Junctional Epidermolysis Bullosa variant could develop due to a different genetic or clinical cause. (wisdompanel.com)
Autosomal recessive1
- Form of epidermolysis bullosa having onset at birth or during the neonatal period and transmitted through autosomal recessive inheritance. (nih.gov)
Herlitz1
- Researchers classify junctional epidermolysis bullosa into two main types: JEB generalized severe (formerly known as Herlitz JEB) and JEB generalized intermediate (formerly known as non-Herlitz JEB). (medlineplus.gov)
Genes1
- Proporciona un análisis completo de los genes involucrados en esta enfermedad utilizando secuenciación de próxima generación (NGS) para comprender completamente el espectro de genes relevantes involucrados. (igenomix.com)
Severe3
- Junctional epidermolysis bullosa is usually severe. (nih.gov)
- Extensive mucocutaneous epidermolysis bullosa of any type can cause severe pain. (msdmanuals.com)
- This is the most severe form of junctional epidermolysis bullosa. (entclinic.com.au)
Blisters form2
- Junctional EB blisters form in the lamina lucida. (msdmanuals.com)
- In epidermolysis bullosa, blisters form on the skin following minor skin trauma, such as bumping into objects, sitting on hard surfaces and walking. (entclinic.com.au)
Genetically1
- Comments on: Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. (pasteur.fr)
Dystrophica2
- 10. Management of squamous cell carcinoma in a patient with recessive-type epidermolysis bullosa dystrophica. (nih.gov)
- Epidermolysis bullosa dystrophica. (bvsalud.org)
Disorder1
- JEB (Junctional Epidermolysis bullosa) is a blistering disorder of the skin and mucous membranes. (labogen.com)
Dermis1
- This is one of the rarer types of epidermolysis bullosa, and as the name suggests, it affects the layer of the skin between the epidermis and the dermis. (entclinic.com.au)
Fragility2
- Junctional epidermolysis bullosa (JEB) is characterized by fragility of the skin and mucous membranes, manifest by blistering with little or no trauma. (nih.gov)
- Epidermolysis Bullosa is a group of inherited disorders characterized by skin fragility and multifocal skin blistering and ulcerations. (wisdompanel.com)
Diseases3
- Epidermolysis bullosa (EB), a group of autosomal heritable blistering diseases, is characterised by extensive phenotypic variability with considerable morbidity and mortality. (bmj.com)
- ABCB5+ mesenchymal stromal cells (ABCB5+ MSCs) are noteworthy for their special immunomodulatory and anti-inflammatory properties, making them a promising therapeutic option for various chronic inflammatory diseases, including epidermolysis bullosa. (researchandmarkets.com)
- Transgenic epidermal grafts do so also in genetic skin diseases such as Junctional Epidermolysis Bullosa. (holostem.com)
20221
- In 2022, the market size of epidermolysis bullosa was highest in the US among the 7MM, accounting for approximately USD 1,300 million, which is further expected to increase by 2032. (researchandmarkets.com)
Fragile skin1
- The primary symptom of epidermolysis bullosa is fragile skin that leads to blistering and tearing. (nih.gov)
Patients8
- 1. Morphological and morphometric analysis of cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa: a retrospective study. (nih.gov)
- 2. Immune Disregulation in Cutaneous Squamous Cell Carcinoma of Patients with Recessive Dystrophic Epidermolysis Bullosa: A Single Pilot Study. (nih.gov)
- 5. Squamous Cell Carcinoma in Patients with Inherited Epidermolysis Bullosa: Review of Current Literature. (nih.gov)
- 8. Epidemiology and natural history of cutaneous squamous cell carcinoma in recessive dystrophic epidermolysis bullosa patients: 20 years' experience of a reference centre in Spain. (nih.gov)
- 18. Electrochemotherapy, a local treatment for squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. (nih.gov)
- 19. Expression of mutant p53 gene in squamous carcinoma arising in patients with recessive dystrophic epidermolysis bullosa. (nih.gov)
- Although burn patients are the most common patient population with extensive skin loss, other conditions such as surgical resection of scars, or giant nevus, epidermolysis bullosa, trauma and chronic ulcers from various etiologies, also require replacement therapies. (scirp.org)
- The present article reports on an dental service protocol for patients with recessive generalized dystrophic epidermolysis bullosa, as well as offers some considerations concerning the appropriate dental treatment. (bvsalud.org)
Epidermis1
- In terms of overall cell therapies development Japan is a bit ahead of other countries among the 7MM, and in December 2018, Japan became the first country where the first cell therapy for epidermolysis bullosa got approved as Fujifilm subsidiary Japan Tissue Engineering received government approval to culture and sell Autologous Cultured Epidermis JACE for Epidermolysis Bullosa in Japan. (researchandmarkets.com)
Type2
Molecular1
- Discoveries of the molecular basis of epidermolysis bullosa have resulted in the development of diagnostic tools, including prenatal and preimplantation testing. (medscape.com)
Neonatal1
- A form of junctional epidermolysis bullosa characterized by neonatal onset of localized blistering, and dystrophic or absent nails. (orpha.net)
Symptoms2
Gene therapy1
- Krystal has taken the lead in the race for gene therapy treatments for Epidermolysis Bullosa (EB) by securing FDA approval for VYJUVEK, which is effective for both the recessive and dominant forms of the condition. (researchandmarkets.com)
Occurs1
- Epidermolysis bullosa occurs in 20/million live births. (msdmanuals.com)
Therapies1
- Currently, there are three different therapies approved for Epidermolysis Bullosa (EB) in the United States, European Union, and Japan, respectively. (researchandmarkets.com)
Rare1
- Both types of junctional epidermolysis bullosa are rare, together affecting approximately 3 per million people per year in the United States. (medlineplus.gov)
Mutation2
- Dystrophic Epidermolysis Bullosa: COL7A1 Mutation Landscape in a Multi-Ethnic Cohort of 152 Extended Families with High Degree of Customary Consanguineous Marriages. (jefferson.edu)
- Homozygous ITGA3 Missense Mutation in Adults in a Family with Syndromic Epidermolysis Bullosa (ILNEB) without Pulmonary Involvement. (jefferson.edu)
Form3
- The milder form of junctional epidermolysis bullosa is called JEB generalized intermediate . (medlineplus.gov)
- If you have epidermolysis bullosa, one of these proteins does not form correctly. (nih.gov)
- this form of epidermolysis bullosa affects predominantly the hands and feet. (entclinic.com.au)
Disorders characterized1
- Epidermolysis bullosa (EB) is a group of inherited bullous disorders characterized by blister formation in response to mechanical trauma. (medscape.com)
Diagnosis1
- In total, 265 cases were submitted with the preliminary diagnosis of junctional or hemidesmosomal forms of EB. (bmj.com)
Genetic skin2
- Kindler EB is one of four main types of epidermolysis bullosa (EB) , a painful genetic skin condition causing the skin to tear or blister at the slightest touch. (debra.org.uk)
- Epidermolysis bullosa (EB) is a painful genetic skin blistering condition with no cure. (debra.org.uk)
Types3
- There are four major types of epidermolysis bullosa. (nih.gov)
- All types of epidermolysis bullosa manifest with painful, inappropriate blistering. (msdmanuals.com)
- There are three main types of epidermolysis bullosa which are classified according to the level of the skin which is affected. (entclinic.com.au)
Group1
- I. INTRODUCTION The purpose of this guidance is to assist sponsors with the development of drugs 2 for treatment or prevention of the serious cutaneous manifestations of the heterogeneous group of disorders collectively known as epidermolysis bullosa (EB). (nih.gov)
Treatments1
- however, scientists continue to research possible treatments and cures for epidermolysis bullosa. (nih.gov)