Factor VII Deficiency
Factor VII
Blood Coagulation Disorders, Inherited
Factor VIIa
Factor VII deficiency in a mixed breed dog. (1/64)
Abnormal bleeding following routine orchectomy of a 5-month-old mixed breed was determined to be due to factor VII deficiency. Although pedigree information was unavailable, failure to respond to vitamin K therapy and the absence of a plasma coagulation inhibitor suggested that the factor VII deficiency was likely inherited rather than acquired. (+info)Combined factor VII/protein C deficiency results in intrauterine coagulopathy in mice. (2/64)
To determine whether an additional loss of the coagulation factor VII (FVII) gene influenced the coagulopathy observed in protein C gene-deficient (PC(-/-)) embryos and neonates, we crossed mice doubly heterozygous for the factor VII (FVII(+/-)) and protein C (PC(+/-)) genes to produce offspring possessing the 9 predicted genotypic combinations. FVII(-/-)/PC(-/-) embryos, although present at their expected Mendelian frequency, displayed a phenotype that had not been observed in either the FVII or PC singly deficient embryos. At E12.5 days postcoitum (dpc), FVII(-/-)/PC(-/-) embryos demonstrated an intra- and extravascular coagulopathy that progressed with substantial concomitant hemorrhage and peripheral edema by E17.5dpc, resulting in mortality immediately after birth. FVII(+/-)/PC(-/-) embryos showed a less severe phenotype, suggesting a gene dosage effect. The lack of rescue of PC(-/-) embryos and neonates and augmented coagulopathy resulting from an additional heterozygous or homozygous FVII deficiency are probably due to increased factor Xa and thrombin generation, resulting from loss of FVIIa-dependent tissue factor pathway inhibitor function and the absence of control at the levels of factors Va and VIIIa. The presence of fibrin in embryos in the absence of fetal FVII suggests that significant clot-generating potential exists outside of the embryonic factor VII-dependent pathway. (+info)Modulation of factor VII levels by intron 7 polymorphisms: population and in vitro studies. (3/64)
Previous studies have established that factor VII gene (F7) polymorphisms (5'F7 and R353Q) contribute about one-third of factor VII (FVII) level variation in plasma. However, F7 genotyping in patients with cardiovascular disease has produced conflicting results. Population and expression studies were used to investigate the role of intron 7 (IVS7 ) polymorphisms, including repeat and sequence variations, in controlling activated FVII (FVIIa) and antigen (FVIIag) levels. Genotype-phenotype studies performed in 438 Italian subjects suggested a positive relation between the IVS7 repeat number and FVII levels. The lowest values were associated with the IVS7 + 7G allele. The screening of 52 patients with mild FVII deficiency showed an 8-fold increase in frequency (8%) of this allele, and among heterozygotes for identical mutations, lower FVII levels were observed in the IVS7 + 7G carriers. This frequent genetic component participates in the phenotypic heterogeneity of FVII deficiency. The evaluation of the individual contribution of polymorphisms was assisted by the expression of each IVS7 variant, as a minigene, in eukaryotic cells. The novel quantitative analysis revealed that higher numbers of repeats were associated with higher mRNA expression levels and that the IVS7 + 7G allele, previously defined as a functionally silent polymorphism, was responsible for the lowest relative mRNA expression. Taken together, these findings indicate that the IVS7 polymorphisms contribute to the plasmatic variance of FVII levels via differential efficiency of mRNA splicing. These studies provide further elements to understand the control of FVII levels, which could be of importance to ensure the hemostatic balance under pathologic conditions. (+info)A new mutation in the HNF4 binding region of the factor VII promoter in a patient with severe factor VII deficiency. (4/64)
Investigation of the molecular basis of a severe factor VII (fVII) deficiency revealed compound heterozygosity in the fVII gene. On the paternal allele the patient had 3 structural gene abnormalities frequently associated with fVII deficiency. A new mutation, a C to T transition at position -55 relative to the translational start site, was found on the maternal allele. The study demonstrates that this mutation partially impeded binding of the transcriptional activator, hepatic nuclear factor 4, to the fVII promoter while greatly reducing reporter gene expression in hepatic cells. (Blood. 2000;96:4370-4372) (+info)Abnormal secretion and function of recombinant human factor VII as the result of modification to a calcium binding site caused by a 15-base pair insertion in the F7 gene. (5/64)
A case of a novel mutation in the F7 gene that results in factor VII coagulant activity (VII:c) of less than 1% and VII antigen (VII:Ag) levels of 10% is presented. DNA analysis revealed a homozygous 15-base pair (bp) in-frame insertion-type mutation at nucleotide 10554. This insertion consisted of a duplication of residues leucine (L)213 to aspartic acid (D)217 (leucine, serine, glutamic acid, histidine, and aspartic acid), probably arising by slipped mispairing between 2 copies of a direct repeat (GCGAGCACGAC) separated by 4 bp. Molecular graphic analyses showed that the insertion is located at the surface of the catalytic domain in an exposed loop stabilized by extensive salt-bridge and hydrogen bond formation at which the calcium binding site is located. The mutation probably interferes with protein folding during VII biosynthesis and/or diminishes functional activity through the loss of calcium binding. In vitro expression studies demonstrated that the levels of VII:Ag in lysates of cells transfected with wild type VII (VIIWT) were equivalent to those with mutant type VII (VIIMT), but the level of secreted VIIMT was 5% to 10% that of VIIWT. Pulse chase studies demonstrated that VIIMT did not accumulate intracellularly, and studies with inhibitors of protein degradation showed that recombinant VIIMT was partially degraded in the pre-Golgi compartment. Accordingly, only small amounts of VIIMT with undetectable procoagulant activity were secreted into conditioned media. These results demonstrate that a combination of secretion and functional defects is the mechanism whereby this insertion causes VII deficiency. (+info)Analysis of the genotypes and phenotypes of 37 unrelated patients with inherited factor VII deficiency. (6/64)
Severe inherited factor VII (FVII) deficiency is a rare autosomal recessive disorder with a poor relationship between FVII coagulant activity and bleeding tendency. Both clinical expression and mutational spectrum are highly variable. We have screened for mutations the FVII gene of 37 unrelated patients with a FVII coagulant activity less than 5% of normal pooled plasmas. The nine exons with boundaries and the 5' flanking region of the FVII gene were explored using a combination of denaturing gradient gel electrophoresis and direct DNA sequencing. This strategy allowed us to characterise 68 out of the 74 predicted FVII mutated alleles. They corresponded to a large panel of 40 different mutations. Among these, 18 were not already reported. Genotypes of the severely affected patients comprised, on both alleles, deleterious mutations which appeared to be related to a total absence of activated FVII. We suggest that this absence of functional FVII can explain the severe clinical expression. Whether a small release of FVII is sufficient to initiate the coagulation cascade and to prevent the expression of a severe phenotype, requires further investigations. (+info)Pharmacokinetic evaluation of recombinant, activated factor VII in patients with inherited factor VII deficiency. (7/64)
BACKGROUND AND OBJECTIVES: Recombinant factor VIIa (rFVIIa) has been widely used in the treatment of bleedings occurring in hemophiliacs with inhibitors. Very few reports exist on the use of rFVIIa in patients with inherited FVII deficiency. Pharmacokinetic studies on rFVIIa have been performed exclusively in hemophiliacs, patients with cirrhosis or volunteers pretreated with acenocoumarol. The aim of this study was to evaluate the kinetics of rFVIIa in patients naturally deficient of FVII. DESIGN AND METHODS: A single dose kinetic study with rFVIIa was performed in 5 patients affected by severe congenital deficiency of factor VII in order to evaluate the true kinetic parameters of rFVIIa without the interference of FVII. Two dosages, 15 and 30 microg/kg, were used in a crossover schedule. FVII:C and FVIIa concentration/time curves were analyzed by a model-independent method. Antithrombin (AT), prothombin fragment 1+2 (F1+2) and tissue factor pathway inhibitor (TFPI) were assayed. RESULTS: No differences emerged between the dosages with respect to dose-independent parameters [total body clearance (CL), volume of distribution area (VdArea), mean residence time (MRT)]. No significant changes of AT, TFPI, and F1+2 were observed. Comparing the results with those of other studies performed in adult hemophiliacs, in patients affected by cirrhosis or in volunteers on oral anticoagulant therapy (OAT), CL and VdArea of rFVIIa were definitely higher and in vivo recovery was lower. INTERPRETATION AND CONCLUSIONS: These findings suggest that the kinetics of rFVIIa are not dose-dependent. In the absence of FVII, the changes of VdArea and CL may be in agreement with a mechanism of competition between FVII and rFVIIa for tissue factor binding. (+info)Residual factor VII activity and different hemorrhagic phenotypes in CRM(+) factor VII deficiencies (Gly331Ser and Gly283Ser). (8/64)
Two cross-reacting material-positive (CRM(+)) factor VII (FVII) mutations, associated with similar reductions in coagulant activity (2.5%) but with mild to asymptomatic (Gly331Ser, c184 [in chymotrypsin numbering]) or severe (Gly283Ser, c140) hemorrhagic phenotypes, were investigated. The affected glycines belong to structurally conserved regions in the c184 through c193 and c140s activation domain loops, respectively. The natural mutants 331Ser-FVII and 283Ser-FVII were expressed, and in addition 331Ala-FVII and 283Ala-FVII were expressed because 3 functional serine-proteases bear alanine at these positions. The 331Ser-FVII, present in several asymptomatic subjects, showed detectable factor Xa generation activity in patient plasma (0.7% +/- 0.2%) and in reconstituted system with the recombinant molecules (2.7% +/- 1.1%). The reduced activity of recombinant 283Ala-FVII (7.2% +/- 2.2%) indicates that the full function of FVII requires glycine at this position, and the undetectable activity of 283Ser-FVII suggests that the oxydrile group of Ser283 participates in causing severe CRM(+) deficiency. Furthermore, in a plasma system with limiting thromboplastin concentration, 283Ser-FVII inhibited wild-type FVIIa activity in a dose-dependent manner. (+info)Factor VII deficiency is a bleeding disorder that is caused by a deficiency or dysfunction of coagulation factor VII, which is a protein involved in the coagulation cascade and is necessary for the initiation of blood clotting. This condition can lead to prolonged bleeding after injury or surgery, easy bruising, and spontaneous bleeding. The severity of the disorder varies widely, depending on the level of factor VII activity. In severe cases, factor VII activity may be less than 1% of normal, leading to a high risk of bleeding. In milder cases, factor VII activity may be between 5-40% of normal, leading to a lower risk of bleeding. Treatment typically involves replacement therapy with fresh frozen plasma or recombinant factor VIIa to control bleeding episodes and prevent complications.
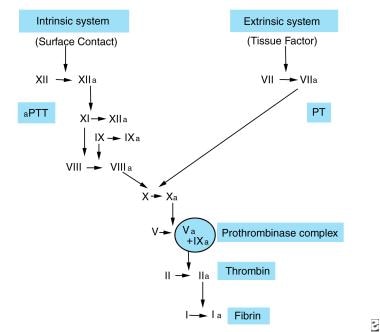
Factor VII, also known as proconvertin, is a protein involved in the coagulation cascade, which is a series of chemical reactions that leads to the formation of a blood clot. Factor VII is synthesized in the liver and is activated when it comes into contact with tissue factor, which is exposed when blood vessels are damaged. Activated Factor VII then activates Factor X, leading to the formation of thrombin and ultimately a fibrin clot.
Inherited deficiencies or dysfunctions of Factor VII can lead to an increased risk of bleeding, while elevated levels of Factor VII have been associated with an increased risk of thrombosis (blood clots).
Blood coagulation disorders, inherited, also known as coagulopathies, are genetic conditions that affect the body's ability to form blood clots in response to injury or damage to blood vessels. These disorders can lead to excessive bleeding or hemorrhage, and in some cases, abnormal clotting.
There are several types of inherited blood coagulation disorders, including:
1. Hemophilia A and B: These are X-linked recessive disorders that affect the production of factors VIII and IX, respectively, which are essential for normal blood clotting. People with hemophilia may experience prolonged bleeding after injury or surgery, and spontaneous bleeding into joints and muscles.
2. Von Willebrand disease: This is the most common inherited coagulation disorder, affecting both men and women. It results from a deficiency or abnormality of von Willebrand factor, a protein that helps platelets stick to damaged blood vessels and assists in the activation of factor VIII. People with von Willebrand disease may experience excessive bleeding after injury, surgery, or dental work.
3. Factor XI deficiency: This is an autosomal recessive disorder that affects the production of factor XI, a protein involved in the intrinsic pathway of blood coagulation. People with factor XI deficiency may have a mild to moderate bleeding tendency, particularly after surgery or trauma.
4. Rare coagulation factor deficiencies: There are several other rare inherited coagulation disorders that affect the production of other clotting factors, such as factors II, V, VII, X, and XIII. These conditions can lead to a range of bleeding symptoms, from mild to severe.
Inherited blood coagulation disorders are usually diagnosed through a combination of medical history, physical examination, and laboratory tests that measure the levels and function of clotting factors in the blood. Treatment may include replacement therapy with purified clotting factor concentrates, medications to control bleeding, and management of bleeding symptoms as they arise.
Factor VIIa is a protein involved in the coagulation cascade, which is a series of chemical reactions that leads to the formation of a blood clot. Factor VIIa is the activated form of factor VII, which is normally activated by tissue factor (TF) when there is damage to the blood vessels. Together, TF and Factor VIIa convert Factor X to its active form, Factor Xa, which then converts prothrombin to thrombin, leading to the formation of a fibrin clot.
In summary, Factor VIIa is an important protein in the coagulation cascade that helps to initiate the formation of a blood clot in response to injury.
Thromboplastin is a substance that activates the coagulation cascade, leading to the formation of a clot (thrombus). It's primarily found in damaged or injured tissues and blood vessels, as well as in platelets (thrombocytes). There are two types of thromboplastin:
1. Extrinsic thromboplastin (also known as tissue factor): This is a transmembrane glycoprotein that is primarily found in subendothelial cells and released upon injury to the blood vessels. It initiates the extrinsic pathway of coagulation by binding to and activating Factor VII, ultimately leading to the formation of thrombin and fibrin clots.
2. Intrinsic thromboplastin (also known as plasma thromboplastin or factor III): This term is used less frequently and refers to a labile phospholipid component present in platelet membranes, which plays a role in the intrinsic pathway of coagulation.
In clinical settings, the term "thromboplastin" often refers to reagents used in laboratory tests like the prothrombin time (PT) and activated partial thromboplastin time (aPTT). These reagents contain a source of tissue factor and calcium ions to initiate and monitor the coagulation process.
 Factor VII deficiency
Factor VII deficiency
Haemophilia C
Coagulation factor VII
Major trauma
Alaskan Klee Kai
Partial thromboplastin time
Deep vein thrombosis
Amputation
Recombinant factor VIIa
Emergency bleeding control
Katherine A. High
Haemophilia B
Nuclear mitochondrial DNA segment
Nosebleed
Hypoprothrombinemia
List of diseases (F)
Factor XIII deficiency
List of MeSH codes (C16)
List of MeSH codes (C15)
Phaedon Fessas
Factor V
L-type lectin domain
Bleeding
Imerslund-Gräsbeck syndrome
MCFD2
LMAN1
Vitamin K deficiency bleeding
POLR3C
POLR3F
POLR2K
 Factor VII deficiency: MedlinePlus Genetics
Factor VII deficiency: MedlinePlus Genetics
 Factor VII Deficiency: Practice Essentials, Background, Pathophysiology
Factor VII Deficiency: Practice Essentials, Background, Pathophysiology
Factor VII deficiency - Wikipedia
Pediatric Factor VII Deficiency: Practice Essentials, Pathophysiology, Epidemiology
Factor VII Deficiency | Profiles RNS
Pediatric Factor VII Deficiency: Background, Pathophysiology, Epidemiology
Pediatric Factor VII Deficiency Clinical Presentation: History, Physical, Causes
 Paw Print Genetics - Coagulation Factor VII Deficiency in the Alaskan Husky
Paw Print Genetics - Coagulation Factor VII Deficiency in the Alaskan Husky
 Factor VII deficiency | Hemophilia
Factor VII deficiency | Hemophilia Factor VII Deficiency - Irish Haemophilia Society
Factor VII Deficiency - Irish Haemophilia Society
 U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency
U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency
 Alaskan Klee Kai Facts - Wisdom Panel™ Dog Breeds
Alaskan Klee Kai Facts - Wisdom Panel™ Dog Breeds
 "Factor VII Deficiency and Second Trimester Abortion: A Case Report" by Katie Nguyen, Tamara Lynne Aqui et al.
"Factor VII Deficiency and Second Trimester Abortion: A Case Report" by Katie Nguyen, Tamara Lynne Aqui et al.
Hemab Therapeutics Presents New Preclinical Research Demonstrating Effects of Its Bispecific Antibody HMB-001 in Factor VII...
 Beagle | Breeds A to Z | The Kennel Club
Beagle | Breeds A to Z | The Kennel Club
 Dr. William Mitchell, MD, Pediatric Hematology-Oncology Specialist - Bronx, NY | Sharecare
Dr. William Mitchell, MD, Pediatric Hematology-Oncology Specialist - Bronx, NY | Sharecare
 A List of Available Canine and Feline Genetic Tests - TUFTSBG2005 - VIN
A List of Available Canine and Feline Genetic Tests - TUFTSBG2005 - VIN
JCI - Sex differences in thrombosis in mice are mediated by sex-specific growth hormone secretion patterns
 coagulation factor VIII - Side Effects, Uses, Dosage, Overdose, Pregnancy, Alcohol | RxWiki
coagulation factor VIII - Side Effects, Uses, Dosage, Overdose, Pregnancy, Alcohol | RxWiki
 SMART: EGF CA domain annotation
SMART: EGF CA domain annotation
SMART: EGF CA domain annotation
Questions & Answers
Factor VII assay: MedlinePlus Medical Encyclopedia
AKR1B10 | Cancer Genetics Web
 RX List database - use generic or medication brand name - GlobalRPH
RX List database - use generic or medication brand name - GlobalRPH
Amputation - Wikipedia
 coagulation factor VIIa (injection) | Cigna
coagulation factor VIIa (injection) | Cigna
 X-Linked PRA2 (Miniature Schnauzer Type)
- DNA Test - Orivet
X-Linked PRA2 (Miniature Schnauzer Type)
- DNA Test - Orivet
 ההסתדרות הרפואית בישראל
ההסתדרות הרפואית בישראל
Hemophilia7
- Coagulation factor VIIa is used to treat or prevent bleeding in people with hemophilia A or hemophilia B, or factor VII deficiency. (cigna.com)
- Recombinant factor VIIa (rFVIIa) has become available for treating people with hemophilia with inhibitors who experience bleeding or require surgery. (medscape.com)
- Originally, rFVIIa was developed for the treatment of bleeding complications in patients with hemophilia with alloantibodies (inhibitors) against exogenous factor VIII or IX. (medscape.com)
- We describe the uses of rFVIIa in conditions unrelated to hemophilia and the treatment of acquired inhibitors of factors VIII and IX. (medscape.com)
- Novo Nordisk's NovoSecure is a comprehensive patient support program for patients with hemophilia A, hemophilia A or B with inhibitors, factor VII deficiency, acquired hemophilia, Glanzmann's thrombasthenia, or factor XIII deficiency, regardless of product choice. (kelleycom.com)
- On September 5, 1995, a 13-year-old boy with mild hemophilia A (factor VIII deficiency) became acutely ill with nausea and vomiting after a 2-week period of fatigue, poor appetite, and low-grade fever. (cdc.gov)
- In response to this bulletin, two brothers with hemophilia A (aged 6 and 7 years) who had received this clotting factor concentrate were identified and tested for anti-HAV on November 17. (cdc.gov)
FVII Deficiency23
- Girolami A, Santarossa C, Cosi E, Ferrari S, Lombardi AM. Acquired Isolated FVII Deficiency: An Underestimated and Potentially Important Laboratory Finding. (medlineplus.gov)
- Greifswald Factor FVII Deficiency Study Group. (medlineplus.gov)
- citation needed] While in congenital disease symptoms may be present at birth or show up later, in patients with acquired FVII deficiency symptoms typically show up in later life. (wikipedia.org)
- citation needed] About 3-4% of patients with FVII deficiency may also experience thrombotic episodes. (wikipedia.org)
- Inherited or congenital FVII deficiency is passed on by autosomal recessive inheritance. (wikipedia.org)
- In persons with the congenital FVII deficiency the condition is lifelong. (wikipedia.org)
- In the acquired of FVII deficiency an insufficient amount of factor VII is produced by the liver due to liver disease, vitamin K deficiency, or certain medications (i.e. (wikipedia.org)
- Blood tests are needed to differentiate FVII deficiency from other bleeding disorders. (wikipedia.org)
- Inherited factor VII (FVII) deficiency is a rare autosomal recessive hemorrhagic disorder. (medscape.com)
- Most severe cases of factor VII (FVII) deficiency are diagnosed during childhood, often during the first 6 months of life. (medscape.com)
- Life-threatening hemorrhagic symptoms in severe FVII deficiency are prevented by frequent administration of fresh frozen plasma or recombinant FVIIa. (unife.it)
- This effect, which was dose-dependent, clearly demonstrated that impaired recognition by the U1-snRNA was the mechanism responsible for FVII deficiency. (unife.it)
- COPENHAGEN, DENMARK AND BOSTON, MASS., US - June 24, 2023 - Hemab Therapeutics , a clinical-stage biotechnology company developing the first prophylactic therapeutics for serious, underserved bleeding and thrombotic disorders, announced results today from preclinical research of HMB-001 in models of factor VII (FVII) deficiency at the International Society on Thrombosis and Haemostasis (ISTH) 2023 Congress in Montreal. (healthcap.eu)
- The new preclinical data presented today show HMB-001 successfully targeted and accumulated endogenous FVIIa to levels that would be expected to provide clinical benefit in FVII deficiency, supporting the potential for HMB-001 in an additional underserved bleeding disorder. (healthcap.eu)
- The preclinical research presented at ISTH, "HMB-001, a Bispecific anti-FVIIa/anti-TLT-1 Antibody Demonstrates Effect in Models of FVII Deficiency,"assessed key requirements for HMB-001 to function in FVII deficiency, specifically its ability to bind with FVII protein variants associated with moderate/severe deficiency and the potential of HMB-001 to accumulate FVIIa in an in-vivo non-human primate model of FVII deficiency. (healthcap.eu)
- A panel of 12 FVII variants from the European Association for Haemophilia and Allied Disorders (EAHAD) database was produced based on high prevalence and association with moderate/severe FVII deficiency phenotype as well as proximity to the HMB-001 binding site. (healthcap.eu)
- The ability of HMB-001 to accumulate endogenous FVIIa in FVII deficiency was assessed using small interfering RNA (siRNA) to knock down FVII/FVIIa to levels between 10 to 30 percent of normal in animal models (n=3). (healthcap.eu)
- These initial results suggest HMB-001 may have potential application as a treatment for FVII deficiency. (healthcap.eu)
- Factor VII (FVII) deficiency is characterized by normal activated partial thromboplastin time (aPTT) and prolonged prothrombin time (PT) values. (ima.org.il)
- To analyze correlations between PT, international normalized ratio (INR), and FVII:C in pediatric patients before otolaryngology surgery and to establish alternative methods for identifying FVII deficiency. (ima.org.il)
- We compared demographic and clinical parameters using Spearman correlation coefficient and receiver operating characteristic (ROC) curve analysis to determine the accuracy of PT and INR values to predict FVII deficiency. (ima.org.il)
- When PT is abnormal, determining FVII:C protein levels is needed for diagnosing FVII deficiency and considering surgical prophylactic treatment. (ima.org.il)
- Classification of Clinical Severity may use the severity classification for FVII deficiency proposed by Mariani et al 2005 . (eahad.org)
Autosomal recessive5
- Inherited deficiency of factor VII (FVII), the crucial enzyme triggering blood coagulation, is the most common of the rare coagulation disorders transmitted in an autosomal recessive manner. (medscape.com)
- An autosomal recessive characteristic or a coagulation disorder acquired in association with VITAMIN K DEFICIENCY. (umassmed.edu)
- Coagulation factor VII deficiency is inherited in an Autosomal Recessive manner in dogs meaning that they must receive two copies of the mutated gene (one from each parent) to develop the disease. (pawprintgenetics.com)
- Factor VII deficiency is an autosomal recessive disorder, which means that both parents must carry the defective gene in order to pass it on to their child. (haemophilia.ie)
- Factor VII deficiency is very rare, but like all autosomal recessive disorders, it is found more frequently in areas of the world where marriage between close relatives is common. (haemophilia.ie)
Mutations10
- The inherited form of factor VII deficiency, known as congenital factor VII deficiency, is caused by mutations in the F7 gene, which provides instructions for making a protein called coagulation factor VII. (medlineplus.gov)
- These mutations reduce the amount of coagulation factor VII in the bloodstream. (medlineplus.gov)
- Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. (medlineplus.gov)
- More than 100 mutations, mostly missense mutations, have been identified in the factor VII gene located on chromosome 13. (medscape.com)
- Although individuals with the lowest factor VII levels are most likely to be symptomatic, patients with identical mutations may have marked differences in clinical bleeding, suggesting that other factors may contribute to the clinical manifestations of factor VII deficiency. (medscape.com)
- Splicing mutations in clotting factors, a relatively frequent cause of severe bleeding, represent ideal models to test this strategy, because tiny increases in functional full-length protein levels in patients significantly ameliorate hemorrhagic phenotypes. (unife.it)
- These findings suggest compensatory U1-snRNAs as therapeutic tools in coagulation factor deficiencies caused by mutations at 5'ss, a frequent cause of severe defects. (unife.it)
- Mutations in the F10 -coding gene can cause factor X (FX) deficiency, leading to abnormal coagulation activity and severe tendency for hemorrhage. (karger.com)
- Therefore, identifying mutations in F10 is important for diagnosing congenital FX deficiency. (karger.com)
- Genetic analysis of these two mutations may help characterize the bleeding tendency and confirm congenital FX deficiency. (karger.com)
Disorders11
- HMB-001 was designed to be a first-in-class prophylactic treatment for Glanzmann Thrombasthenia (GT) with potential for other debilitating rare bleeding disorders, including factor VII deficiency. (healthcap.eu)
- The company's strategic guidance, Hemab 1-2-5 TM , targets the development of 5 clinical assets by 2025 to deliver long-awaited innovation for patients with high unmet need blood clotting disorders like Glanzmann Thrombasthenia, factor VII deficiency, Bernard Soulier Syndrome, Von Willebrand Disease and other serious disorders. (healthcap.eu)
- Recent research clearly indicates that the underlying causes of autism are neurobiological disorders and combinations of different factors, such as environmental and genetic factors, and abnormality in the communication between neurons, probably associated with an abnormal set of neuropeptides in the brain [ 3 - 9 ]. (hindawi.com)
- Iodine deficiency disorders (IDDs) refer to a wide range of health problems associated with iodine deficiency1 in a population. (who.int)
- In 1990, World Health Assembly Resolution WHA43.2 endorsed the goal of eliminating IDD as a public health problem.3 In 1993, WHO, UNICEF and the International Council for the Control of Iodine Deficiency Disorders (ICCIDD) recommended universal salt iodization as the main strategy to achieve elimination of IDD.4 In high-risk areas, iodized oil is recommended for the most vulnerable groups such as pregnant women and young children. (who.int)
- Consequently, in 2005 and 2007, the World Health Assembly resolutions WHA58.24 and WHA60.21 on sustaining the elimination of iodine deficiency disorders called on countries to establish multidisciplinary national coalitions to monitor the state of iodine nutrition every three years and to report progress to the World Health Assembly. (who.int)
- Elimination of iodine deficiency disorders will improve children's cognitive development, reduce stillbirths and reduce stunting. (who.int)
- The European Association for Haemophilia and Allied Disorders (EAHAD) Coagulation Factor Variant Databases: Important resources for haemostasis clinicians and researchers. (eahad.org)
- Загальні відомості про геморагічні вазопатії Bleeding may result from abnormalities in Platelets Coagulation factors Blood vessels Vascular bleeding disorders result from defects in blood vessels, typically causing cutaneous or mucosal. (msdmanuals.com)
- Because all coagulation factors are made in the liver (by hepatocytes and hepatic sinusoidal endothelial cells), both the prothrombin time (PT) and partial thromboplastin time (PTT) are prolonged in severe liver disorders. (msdmanuals.com)
- Гемофілія Hemophilias are common hereditary bleeding disorders caused by deficiencies of either clotting factor VIII or IX. (msdmanuals.com)
Hemorrhage4
- Management of acute hemorrhage primarily consists of factor VII replacement therapy to treat bleeding. (medscape.com)
- Management of this second-trimester abortion with a history of F7D was possible with effective communication and the organization of a multidisciplinary team to account for the risk of thrombosis versus hemorrhage and the availability of factor VII replacement therapy. (hcahealthcare.com)
- Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. (medscape.com)
- This has become the case for treatment of hemorrhage with recombinant factor VIIa (rFVIIa). (medscape.com)
Congenital factor6
- Mariani G, Herrmann FH, Bernardi F, Schved JF, Auerswald G, Ingerslev J. Clinical manifestations, management, and molecular genetics in congenital factor VII deficiency: the International Registry on Congenital Factor VII Deficiency (IRF7). (medlineplus.gov)
- Mandhyan R, Tiwari A, Cherian G. Congenital factor VII deficiency. (medscape.com)
- Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. (medscape.com)
- Surgery in patients with congenital factor VII deficiency: A single center experience. (medscape.com)
- Mariani G, Dolce A, Marchetti G, Bernardi F. Clinical picture and management of congenital factor VII deficiency. (medscape.com)
- Mariani G, Lapecorella M, Dolce A. Steps towards an effective treatment strategy in congenital factor VII deficiency. (medscape.com)
Genetic7
- Factor VII plasma levels are influenced by both environmental and genetic factors. (medscape.com)
- Genetic testing of the F7 gene in dogs will reliably determine whether a dog is a genetic Carrier of coagulation factor VII deficiency. (pawprintgenetics.com)
- The relationships between the Arterial and venous thrombosis, which clinical presentation and FVII levels and the clinically manifest as stroke, myocardial in- associated molecular genetic defects lack farction or pulmonary embolism are a ma- apparent consistency [ 7,10 ]. (who.int)
- Genetic factors levels vary significantly in the general pop- contribute significantly to the development ulation and are influenced by environmental of these diseases. (who.int)
- Dogs age at very different rates due to a number of genetic and environmental factors. (embarkvet.com)
- Body size is a strong genetic influence: for example, a seven year old Great Dane is at the start of his golden years, but a seven year old Pomeranian is just learning what "slow down" means. (embarkvet.com)
- We then factor in your dog's breed composition, information at certain genes that affect size, and their inbreeding coefficient to calculate genetic age. (embarkvet.com)
Iron-Deficiency3
- Iron deficiency can cause fatigue. (naturalfactors.com)
- Iron deficiency in Cambodia: the need for iron supplementation among preschool-aged children. (who.int)
- Only a small proportion of anemia in North East Thai school children is associated with iron deficiency. (who.int)
VIIa26
- Therapeutic options for major bleeds include recombinant activated factor VIIa (rFVIIa), plasma-derived factor VII, fresh frozen plasma, and prothrombin complex concentrates. (medscape.com)
- When the vascular lumen is damaged, tissue factor is exposed and then binds to the small amounts of circulating factors VIIa and VII. (medscape.com)
- This facilitates conversion of factor VII to factor VIIa. (medscape.com)
- Factor VIIa bound to tissue factor in the presence of calcium and phospholipids facilitates the conversion of factor IX to factor IXa and factor X to factor Xa. (medscape.com)
- Although this division is useful for understanding in vitro laboratory coagulation tests, no such division occurs in vivo because the tissue factor VIIa complex is a potent activator of factor IX and factor X. (medscape.com)
- Factor VII is then converted to factor VIIa. (medscape.com)
- The ability of factor VIIa to cleave other clotting factors depends on binding to its cofactor tissue factor (TF), which is expressed on the surface of endothelial cells and monocytes in response to injury or inflammation. (medscape.com)
- With formation of the TF/VIIa complex, factor VIIa rapidly activates clotting factors VII, IX, and X, initiating the coagulation cascade. (medscape.com)
- Five identified allelic polymorphisms also affect plasma levels of factor VII and factor VIIa, with variations of as much as 25-30% in levels of activity and antigen. (medscape.com)
- HMB-001 is bispecific antibody that binds and stabilizes endogenous factor VIIa (FVIIa) with one antibody arm and TLT-1 on activated platelets with the other arm. (healthcap.eu)
- What is the most important information I should know about coagulation factor VIIa? (cigna.com)
- If possible before you receive coagulation factor VIIa, tell your doctor about all your medical conditions and allergies. (cigna.com)
- What is coagulation factor VIIa? (cigna.com)
- Coagulation factor VIIa is a man-made protein similar to a natural protein in the body that helps the blood to clot. (cigna.com)
- Coagulation factor VIIa may also be used for purposes not listed in this medication guide. (cigna.com)
- What should I discuss with my healthcare provider before receiving coagulation factor VIIa? (cigna.com)
- You should not receive coagulation factor VIIa if you are allergic to it. (cigna.com)
- It is not known whether coagulation factor VIIa passes into breast milk or if it could harm a nursing baby. (cigna.com)
- How is coagulation factor VIIa given? (cigna.com)
- Coagulation factor VIIa is injected into a vein through an IV. (cigna.com)
- You may need frequent medical tests to help your doctor determine how long to treat you with coagulation factor VIIa. (cigna.com)
- Any medical care provider who treats you should know that you are using coagulation factor VIIa. (cigna.com)
- Because you will receive coagulation factor VIIa in a clinical setting, you are not likely to miss a dose. (cigna.com)
- What should I avoid after receiving coagulation factor VIIa? (cigna.com)
- What are the possible side effects of coagulation factor VIIa? (cigna.com)
- At the site of injury, tissue factor (TF) and factor VIIa activate factors X and IX. (medscape.com)
Gene9
- Factor VII is coded by the gene on band 13q34, closely located to the gene for factor X ( F10 ). (medscape.com)
- ABSTRACT Factor VII gene polymorphisms may contribute to elevations in factor VII coagulant (FVIIc) levels that have been associated with cardiovascular risk. (who.int)
- The Q353 allele of the factor VII gene polymorphism is associated with decreased factor VII and could be protective against cardiovascular disease. (who.int)
- 10976 in exon 8 in the catalytic region of ing factors that may increase cardiovascu- the FVII gene and an insertion of a decanu- lar disease. (who.int)
- Recently several reports have cleotide (designated as 0/10 bp) in the pro- focused on the association between the moter region of the gene at position -323 factor VII of the cascade coagulation and [ 13,14 ]. (who.int)
- We studied FVII gene poly- then activates factors IX and X leading to morphisms in healthy Tunisians with the the generation of thrombin [ 2 ]. (who.int)
- Atasay B, Arsan S, Gunlemez A, Kemahli S, Akar N. Factor V Leiden and prothrombin gene 20210A variant in neonatal thromboembolism and in healthy neonates and adults: a study in a single center. (medscape.com)
- Prevalence of factor V Leiden and prothrombin G20210A gene mutation. (medscape.com)
- Factor VII deficiency is caused by variants in the gene ( F7 ) that codes for coagulation factor VII. (eahad.org)
Proconvertin2
- Factor VII is a vitamin K-dependent serine protease glycoprotein (also known as stable factor or proconvertin) with a pivotal role in hemostasis and coagulation. (medscape.com)
- Factor VII deficiency is a bleeding disorder characterized by a lack in the production of Factor VII (FVII) (proconvertin), a protein that causes blood to clot in the coagulation cascade. (wikipedia.org)
Women with factor VII defici2
- Many women with factor VII deficiency have heavy or prolonged menstrual bleeding (menorrhagia). (medlineplus.gov)
- Excessive menstrual bleeding in women with factor VII deficiency may be controlled with hormonal contraceptives (birth control pills) or antifibrinolytic drugs. (haemophilia.ie)
Serum prothrombin conversion accelerator1
- In 1951, Alexander and colleagues identified factor VII as the key initiator of coagulation when they reported the first case of factor VII deficiency in a child and called it serum prothrombin conversion accelerator deficiency. (medscape.com)
Hereditary3
- Molecular characterization of hereditary factor VII deficiency in the beagle. (pawprintgenetics.com)
- Hereditary factor VII deficiency in the Alaskan Klee Kai dog. (pawprintgenetics.com)
- Withnall E, Giger U. Effects of recombinant human activated factor VII and canine fresh frozen plasma in Beagles with hereditary coagulation factor VII deficiency. (pawprintgenetics.com)
Thrombosis1
- Although factor VII deficiency is primarily associated with increased bleeding, some people with the condition have excessive blood clotting (thrombosis). (medlineplus.gov)
Prothrombin deficiency3
- Girolami A, Santarossa C, Cosi E, Ferrari S, Lombardi AM, Girolami B. Bleeding manifestations in heterozygotes with prothrombin deficiency or abnormalities vs. unaffected family members as observed during a long follow-up study. (medscape.com)
- Lancellotti S, Basso M, De Cristofaro R. Congenital prothrombin deficiency: an update. (medscape.com)
- Harel R, Shani D, Donohoe K. A case of congenital prothrombin deficiency and idiopathic thrombocytopenic purpura in a pregnant female. (medscape.com)
RFVIIa3
- This increase in the thrombin burst occurs after direct rFVIIa activation of factors IX and X on the surface of activated platelets (even in the absence of factor VIII or IX). (medscape.com)
- The rFVIIa seems to work in a TF-independent manner directly on factors IX and X on the phospholipid surface of activated platelets. (medscape.com)
- rFVIIa is able to activate factor X on phospholipid vesicles, activated platelets, or monocytes independent of TF, although the TF-independent generation of thrombin is much less efficient than the TF-dependent thrombin generation by rFVIIa. (medscape.com)
Disorder7
- Factor VII deficiency is a rare bleeding disorder that varies in severity among affected individuals. (medlineplus.gov)
- The noninherited form of the disorder, called acquired factor VII deficiency, is less common than the congenital form. (medlineplus.gov)
- Coagulation factor VII deficiency is an inherited bleeding disorder affecting dogs. (pawprintgenetics.com)
- Deficiency of this factor most commonly results in a mild bleeding disorder. (pawprintgenetics.com)
- Factor VII deficiency is an inherited bleeding disorder that is caused by a problem with factor VII. (haemophilia.ie)
- Iodine deficiency disorder is a public health problem in populations where the median urine iodine is less than 100 µg/l or where more than 5% of children aged 6 to 12 years have goitre. (who.int)
- 3 Resolution WHA43.2, The prevention and control of iodine deficiency disorder s . (who.int)
Abnormalities2
- Congenital deficiencies and abnormalities of prothrombin. (medscape.com)
- Immune system abnormalities may be caused partly by complement system factor I deficiency. (hindawi.com)
Symptoms4
- The symptoms of factor VII deficiency are different for everyone. (haemophilia.ie)
- As a general rule, the less factor VII a person has in his/her blood, the more frequent and/or severe the symptoms. (haemophilia.ie)
- People with very low levels of factor VII can have very serious symptoms. (haemophilia.ie)
- Patients who have normal initial test results, along with symptoms or signs of bleeding and a positive family history, should be tested for VWD by measuring plasma von Willebrand factor (VWF) antigen, ristocetin cofactor activity (an indirect test for large VWF multimers), VWF multimer pattern, and factor VIII levels. (msdmanuals.com)
Diseases4
- Factor I deficiency can be conferred by a C3 deficiency, since this also increases susceptibility to pyogenic infections by Neisseria meningitides, Haemophilus influenza, and Streptococcus pneumonia and increases the incidence of immune complex diseases due to impaired complement-mediated function [ 30 ]. (hindawi.com)
- The world health report 2002 2 describes in detail how, in most countries, a few major risk factors account for much of the morbidity and mortality, and for noncommunicable diseases, the most important risks included high blood pressure, high concentrations of cholesterol in the blood, low intake of fruit and vegetables, being overweight, physical inactivity and tobacco use. (who.int)
- In the poorest countries of the world, even though infectious diseases and undernutrition dominate their current disease burden, the major risk factors for chronic diseases are spreading. (who.int)
- However, in time, all the major risk factors for chronic noncommunicable diseases tend to cluster among the poorest communities and contribute substantially to inequities associated with social class. (who.int)
Genetics1
- Inherited factor VII deficiency: genetics and molecular pathology. (medscape.com)
Prevalence2
- The prevalence of factor VII deficiency (F7D) is 1 in 500,000. (hcahealthcare.com)
- ABSTRACT A cross-sectional study was made of the prevalence of HCV and associated risk factors in 382 multi- transfused patients and haemodialysis staff in Yadz province in 2006. (who.int)
Severity3
- The choice of treatment for patients with factor VII deficiency depends on the site and severity of bleeding and the baseline factor VII activity. (medscape.com)
- Dosage and duration of treatment with BeneFIX depend on the severity of the factor IX deficiency, the location and extent of bleeding, and the patient's clinical condition, age and recovery of factor IX. (globalrph.com)
- The extent of factor deficiency determines the probability and severity of bleeding. (msdmanuals.com)
Beagle1
- A novel missense mutation responsible for factor VII deficiency in research Beagle colonies. (pawprintgenetics.com)
Inhibitors1
- A mixing study was performed to eliminate the presence of coagulation factor inhibitors and lupus anticoagulant. (karger.com)
Concentrates1
- The results provide evidence that blood donor screening and use of virus-inactivated factor concentrates have lowered the risk of HCV infection among multi-transfused patients. (who.int)
Predictors of blee2
- Investigations to determine the contribution by factor VII polymorphisms, other hemostatic proteins, and environmental factors have not yielded specific predictors of bleeding risk. (medscape.com)
- Potential predictors of bleeding risk in inherited factorVII deficiency. (medscape.com)
Assay4
- A specific assay for factor VII, using known factor VII-deficient plasma, is required to confirm the diagnosis. (medscape.com)
- The factor VII assay is a blood test to measure the activity of factor VII. (medlineplus.gov)
- The potency (in IU) is determined using an in vitro one-stage clotting assay against the World Health Organization (WHO) International Standard for Factor IX concentrate. (globalrph.com)
- Monitor patients using a factor IX activity assay to ensure that the desired factor IX activity level has been achieved. (globalrph.com)
Clinical8
- [ 1 , 2 ] Clinical bleeding can widely vary and does not always correlate with the level of factor VII coagulant activity measured in plasma. (medscape.com)
- Correlations between the factor VII genotype, factor VII clotting activity and the clinical phenotype are not tight. (medscape.com)
- At present, classification based on clinical history (age and type of presentation) rather than on factor VII activity levels has proved to be more useful in predicting future risk of bleeding. (medscape.com)
- Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. (medscape.com)
- Dosing of BeneFIX may differ from that of plasma-derived factor IX products [see CLINICAL PHARMACOLOGY ]. (globalrph.com)
- Titrate the dose using the factor IX activity, pharmacokinetic parameters, such as half-life and recovery, as well as taking the clinical situation into consideration in order to adjust the dose as appropriate. (globalrph.com)
- There has been considerable enthusiasm to use MGMT as a predictive biomarker for GBM patients, with the long-term scope for its use as a biomarker to assign alkylating therapy to individual patients, and it is an important stratification factor in current clinical trials. (nature.com)
- Four of seven clinical specimens tested positive for norovirus. (cdc.gov)
19952
- During September-November 1995, three cases of hepatitis A in recipients of Alphanate (TM) * factor VIII concentrate (Alpha Therapeutic Corporation, Los Angeles, California) from lot number AP5014A were reported to CDC. (cdc.gov)
- During 1995, her only exposure to factor concentrate was use of 48 vials (approximately 24,000 units) of Alphanate (TM) from the implicated lot on September 19. (cdc.gov)
Infection2
19981
- 1998 Oct. 9(7):557-69. (medscape.com)
Molecular2
- Factor I has a molecular weight of about 88 kDa, consists of two disulfide-linked polypeptide chains (50 kDa and 38 kDa, respectively), and is synthesized as a single-chain precursor in the liver [ 24 , 25 ]. (hindawi.com)
- Factor I-mediated cleavage of the α chain of C3b liberates 3 fragments with molecular weights of 68 kDa, 43 kDa, and 2 kDa. (hindawi.com)
Liver4
- Factor VII is synthesized in the liver and secreted as a single-chain glycoprotein of 48 kd. (medscape.com)
- Factor VII is one of the vitamin K-dependent coagulation factors synthesized in the liver. (medscape.com)
- treatment of bleeding due to low levels of liver-dependent coagulation factors. (globalrph.com)
- Factor VII deficiency is associated with severe liver disease, vitamin K deficiency and broad spectrum antibiotic therapy, or may follow anticoagulant coumarin therapy. (drugfuture.com)
Thyroid1
- These health problems include goitre, stillbirth, stunted growth (cretinism), thyroid deficiency and mental defects (impaired neurocognitive development), and are preventable by ensuring adequate intake of iodine. (who.int)
Diagnosis1
- Specific factor VII assays are required for diagnosis. (medscape.com)
Neonatal1
- Pasmant E, Dumont B, Lacapere JJ, Dautzenberg MD, Bezeaud A. A severe neonatal presentation of factor II deficiency. (medscape.com)
Programme1
- See Citing Us (below) for information on our recent papers on the EAHAD F7 coagulation factor DB and the EAHAD-DB programme. (eahad.org)
Incidence3
- For example, the reported incidence of factor VII deficiency in Iran is 3 times higher than that in the United Kingdom or Italy. (medscape.com)
- Deficiency in factor I activity is associated with an increased incidence of infections in humans. (hindawi.com)
- The annual incidence of TB declined only gradually during the first 7 years of residence, from an initial 2,000 per 100,000 to 700 per 100,000. (cdc.gov)
Blood coagulation1
- The EAHAD blood coagulation factor VII variant database. (eahad.org)
VIII11
- How was your experience with coagulation factor VIII? (rxwiki.com)
- What tips would you provide a friend before taking coagulation factor VIII? (rxwiki.com)
- What are you taking coagulation factor VIII for? (rxwiki.com)
- How well did coagulation factor VIII work for you? (rxwiki.com)
- How likely would you be to recommend coagulation factor VIII to a friend? (rxwiki.com)
- Factor VIII should be used during pregnancy only if the possible benefit outweighs the possible risk to the unborn baby. (rxwiki.com)
- Factor VIII should be given to a pregnant woman only if clearly needed. (rxwiki.com)
- Take coagulation factor viii exactly as prescribed by your doctor. (rxwiki.com)
- Mice homozygous for a null allele exhibit decreased serum factor V and VIII and aspartate transaminase serum levels with accumulation of the proteins in the ER of hepatocytes. (jax.org)
- During the 6 weeks preceding illness, the patient had used 68 vials (approximately 34,000 units) from the implicated lot (i.e., lot number AP5014A) of Alphanate (TM) and nine vials from four lots of another brand of factor VIII concentrate. (cdc.gov)
- Von Willebrand disease is a common entity in which the associated deficiency of factor VIII is frequently insufficient to prolong the PTT. (msdmanuals.com)
Iodine2
Tissue5
- Tissue factor is an intrinsic membrane glycoprotein that is normally not exposed on the surface of intact blood vessels. (medscape.com)
- The epidermal growth factor domain has a calcium ion- binding site that to some degree mediates interaction with the tissue factor exposed at the site of vessel injury. (medscape.com)
- After a trauma factor VII initiates the process of coagulation in conjunction with tissue factor (TF/factor III) in the extrinsic pathway. (wikipedia.org)
- Factor VII/tissue factor complex activates factor IX and factor X. Factor IXa along with factor VIIIa results in formation of more factor Xa. (medscape.com)
- Upon contact with tissue ferences in FVII activity levels and in geno- factor exposed by vascular injury, FVII is type frequencies depend on the ethnic cleaved to its two-chain active form, which groups [ 15 ]. (who.int)
Vitamin K-dependent4
- The discovery of vitamin K-dependent factors evolved slowly, after the initial identification of the role of prothrombin in blood clotting 100 years ago. (medscape.com)
- Other vitamin K-dependent factors include prothrombin, factors IX and X, and proteins C and S. (medscape.com)
- Warfarin inhibits the activity of vitamin K epoxide reductase and prevents recycling of vitamin K back to the reduced form, thus interfering with the synthesis of factor VII and other vitamin K-dependent factors. (medscape.com)
- FACTOR VII is a Vitamin K dependent glycoprotein essential to the extrinsic pathway of coagulation. (umassmed.edu)
Mild2
- The heterozygous variants p.Ser362Asn or p.Tyr384Ter indicate mild FX deficiency, but the compound heterozygous mutation of the two causes severe congenital FX deficiency and bleeding. (karger.com)
- While extreme vitamin C deficiency is known to cause scurvy, it is less well known that mild to moderate deficiency can contribute to fatigue. (naturalfactors.com)