Fucosidosis
alpha-L-Fucosidase
Carbohydrate Metabolism, Inborn Errors
Disaccharidases
alpha-Mannosidosis
Fucose
alpha-N-Acetylgalactosaminidase
Fabry Disease
Angiokeratoma
alpha-Galactosidase
Trihexosylceramides
Enzyme Replacement Therapy
New Zealand
Spectrum of mutations in fucosidosis. (1/22)
Fucosidosis is a lysosomal storage disorder characterised by progressive psychomotor deterioration, angiokeratoma and growth retardation. It is due to deficient alpha-l-fucosidase activity leading to accumulation of fucose-containing glycolipids and glycoproteins in various tissues. Fucosidosis is extremely rare with less than 100 patients reported worldwide, although the disease occurs at a higher rate in Italy, in the Hispanic-American population of New Mexico and Colorado, and in Cuba. We present here a review study of the mutational spectrum of fucosidosis. Exon by exon mutation analysis of FUCA1, the structural gene of alpha-l-fucosidase, has identified the mutation(s) in nearly all fucosidosis patients investigated. The spectrum of the 22 mutations detected to date includes four missense mutations, 17 nonsense mutations consisting of seven stop codon mutations, six small deletions, two large deletions, one duplication, one small insertion and one splice site mutation. All these mutations lead to nearly absent enzymatic activity and severely reduced cross-reacting immunomaterial. The observed clinical variability is, therefore, not due to the nature of the fucosidosis mutation, but to secondary unknown factors. (+info)Glycoprotein lysosomal storage disorders: alpha- and beta-mannosidosis, fucosidosis and alpha-N-acetylgalactosaminidase deficiency. (2/22)
Glycoproteinoses belong to the lysosomal storage disorders group. The common feature of these diseases is the deficiency of a lysosomal protein that is part of glycan catabolism. Most of the lysosomal enzymes involved in the hydrolysis of glycoprotein carbohydrate chains are exo-glycosidases, which stepwise remove terminal monosaccharides. Thus, the deficiency of a single enzyme causes the blockage of the entire pathway and induces a storage of incompletely degraded substances inside the lysosome. Different mutations may be observed in a single disease and in all cases account for the nonexpression of lysosomal glycosidase activity. Different clinical phenotypes generally characterize a specific disorder, which rather must be described as a continuum in severity, suggesting that other biochemical or environmental factors influence the course of the disease. This review provides details on clinical features, genotype-phenotype correlations, enzymology and biochemical storage of four human glycoprotein lysosomal storage disorders, respectively alpha- and beta-mannosidosis, fucosidosis and alpha-N-acetylgalactosaminidase deficiency. Moreover, several animal disorders of glycoprotein metabolism have been found and constitute valuable models for the understanding of their human counterparts. (+info)MR brain imaging of fucosidosis type I. (3/22)
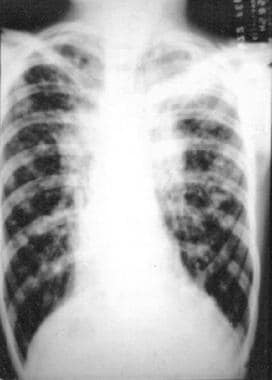
SUMMARY: Fucosidosis is a rare autosomal recessive lysosomal storage disease with the main clinical findings of progressive neuromotor deterioration, seizures, coarse facial features, dysostosis multiplex, angiokeratoma corporis diffusum, visceromegaly, recurrent respiratory infections, and growth retardation. Fucosidosis type I rapidly evolves toward a progressive neurologic deterioration and death. We report MR imaging findings of the brain of three patients with fucosidosis type I, including previously unreported findings, to expand the knowledge of the neuroradiologic spectrum of the disease. (+info)Four year follow-up of a case of fucosidosis treated with unrelated donor bone marrow transplantation. (4/22)
Fucosidosis is a rare autosomal recessive lysosomal disorder caused by alpha-fucosidase deficiency. We report a child with fucosidosis, second daughter of non-consanguineous parents, for whom biochemical diagnosis followed clinical evidence of the disease in her older sister. Based on previous experiences, the indication to transplant was considered. Since she lacked a matched sibling, an unrelated marrow donor was found. At pre-hematopoietic stem cell transplantation evaluation, first signs of neurological involvement were clinically detectable. MRI showed diffuse hypomyelination and auditory brainstem responses and somatic-sensorial evoked potentials were altered. Visual evoked potentials were normal, tortuosity in the retinal veins and peripapillary hemorrhages were detected. Bone marrow transplantation conditioning was with a regimen of busulphan, thiotepa and cyclophosphamide; in vivo Campath 1G, cyclosporin A and short course methotrexate were given to prevent graft-versus-host disease. The patient engrafted rapidly and her post-transplant course was complicated by moderate graft-versus-host disease, transient episodes of idiopathic thrombocytopenic purpura, repeated septic complications and recurrent episodes of Sweet's syndrome. Sequential short tandem repeat polymorphisms on peripheral blood and bone marrow cells documented the persistence of donor engraftment. Follow-up showed a progressive rise of enzymatic levels. Psychomotor development improved, as confirmed by evaluation of evoked potentials and by MRI scanning. (+info)Crystal structure of Thermotoga maritima alpha-L-fucosidase. Insights into the catalytic mechanism and the molecular basis for fucosidosis. (5/22)
Fucosylated glycoconjugates are involved in numerous biological events, and alpha-l-fucosidases, the enzymes responsible for their processing, are therefore of crucial importance. Deficiency in alpha-l-fucosidase activity is associated with fucosidosis, a lysosomal storage disorder characterized by rapid neurodegeneration, resulting in severe mental and motor deterioration. To gain insight into alpha-l-fucosidase function at the molecular level, we have determined the crystal structure of Thermotoga maritima alpha-l-fucosidase. This enzyme assembles as a hexamer and displays a two-domain fold, composed of a catalytic (beta/alpha)(8)-like domain and a C-terminal beta-sandwich domain. The structures of an enzyme-product complex and of a covalent glycosyl-enzyme intermediate, coupled with kinetic and mutagenesis studies, allowed us to identify the catalytic nucleophile, Asp(244), and the Bronsted acid/base, Glu(266). Because T. maritima alpha-l-fucosidase occupies a unique evolutionary position, being far more closely related to the mammalian enzymes than to any other prokaryotic homolog, a structural model of the human enzyme was built to document the structural consequences of the genetic mutations associated with fucosidosis. (+info)Fucosidosis with hypothyroidism: a case report. (6/22)
Fucosidosis is a rare, autosomal recessive lysosomal storage disorder caused by a severe deficiency of alpha-L-fucosidase. Here we present a 27-month-old male who was referred to us for evaluation of developmental delay, which was first detected at age six months. His past medical history was also remarkable for recurrent pulmonary infections and myoclonic seiures. His family history revealed that he was the first living child from a consanguineous marriage. He had a younger sister who died at five months of age from pneumonia who had facial resemblance to the proband, developmental delay and a congenital heart defect. Physical examination revealed length: 81 cm (25-50p), weight: 10.2 kg (25-50p), and head circumference: 49 cm (50-75p). He had a coarse face, hepatomegaly and generalized spasticity. His initial laboratory examination revealed negative urine screening column chromatography for mucopolysaccharidosis. His X-ray findings were consistent with mild form of dysostosis multiplex. Based on clinical and laboratory features, fucosidosis was suspected. Fucosidase enzyme activity was zero. In addition to fucosidosis, thyroid function tests indicated primary hypothyroidism. This is, to the best of our knowledge, the fourth case of fucosidosis diagnosed in Turkey. (+info)Characterization and 400-MHz 1H-NMR analysis of urinary fucosyl glycoasparagines in fucosidosis. (7/22)
Fucosyl glycoasparagines accumulating in the urine of a patient with fucosidosis were isolated using reverse-phase HPLC. Structural analysis of 25 glycoasparagines was carried out by combination of methylation and 400-MHz 1H-NMR spectroscopy analyses. The compounds represent different steps in the incomplete catabolism of N-glycosidically linked glycans, as the result of an alpha-L-fucosidase deficiency. All of the glycoasparagines possess a fucose residue alpha-1,6-linked to the GlcNAc 1 residue attached to asparagine. Fucose residues on the peripheral branches were linked either alpha-1,3 to GlcNAc residues (X determinant) or alpha-1,2 to galactose residues (H determinant). The present study allows precise assignments of the NMR parameters for most of the fucosyl linkages occurring in N-glycosidically linked glycans of the N-acetyllactosamine type. (+info)Distribution of saposin proteins (sphingolipid activator proteins) in lysosomal storage and other diseases. (8/22)
Saposins (A, B, C, and D) are small glycoproteins required for the hydrolysis of sphingolipids by specific lysosomal hydrolases. Concentrations of these saposins in brain, liver, and spleen from normal humans as well as patients with lysosomal storage disease were determined. A quantitative HPLC method was used for saposin A, C, and D and a stimulation assay was used for saposin B. In normal tissues, saposin D was the most abundant of the four saposins. Massive accumulations of saposins, especially saposin A (about 80-fold increase over normal), were found in brain of patients with Tay-Sachs disease or infantile Sandhoff disease. In spleen of adult patients with Gaucher disease, saposin A and D accumulations (60- and 17-fold, respectively, over normal) were higher than that of saposin C (about 16-fold over normal). Similar massive accumulations of saposins A and D were found in liver of patients with fucosidosis (about 70- and 20-fold, respectively, over normal). Saposin D was the primary saposin stored in the liver of a patient with Niemann-Pick disease (about 30-fold over normal). Moderate increases of saposins B and D were found in a patient with GM1 gangliosidosis. Normal or near normal levels of all saposins were found in patients with Krabbe disease, metachromatic leukodystrophy, Fabry disease, adrenoleukodystrophy, I-cell disease, mucopolysaccharidosis types 2 and 3B, or Jansky-Bielschowsky disease. The implications of the storage of saposins in these diseases are discussed. (+info)Fucosidosis is a rare inherited metabolic disorder caused by the deficiency of the enzyme alpha-L-fucosidase. This enzyme is responsible for breaking down complex sugars called glycoproteins and glycolipids in the body. Without sufficient levels of this enzyme, these substances accumulate in various tissues and organs, leading to progressive cellular damage and impaired function.
The condition is characterized by a wide range of symptoms, including coarse facial features, developmental delays, intellectual disability, seizures, vision and hearing loss, cardiac problems, and skeletal abnormalities. There are two main types of fucosidosis, type 1 and type 2, which differ in the age of onset and severity of symptoms.
Fucosidosis is an autosomal recessive disorder, meaning that an individual must inherit two copies of the defective gene, one from each parent, to develop the condition. It is typically diagnosed through enzyme assays and genetic testing. Currently, there is no cure for fucosidosis, and treatment is focused on managing symptoms and improving quality of life.
Alpha-L-Fucosidase is an enzyme that catalyzes the hydrolysis of the terminal alpha-L-fucose residues from glycoproteins, glycolipids, and other substrates. This enzyme plays a crucial role in the degradation and recycling of complex carbohydrates found on the surface of cells and in various biological fluids. Deficiencies in alpha-L-fucosidase activity can lead to genetic disorders such as fucosidosis, which is characterized by the accumulation of fucose-containing glycoproteins and glycolipids in various tissues and organs, resulting in progressive neurological deterioration and other systemic manifestations.
Inborn errors of carbohydrate metabolism refer to genetic disorders that affect the body's ability to break down and process carbohydrates, which are sugars and starches that provide energy for the body. These disorders are caused by defects in enzymes or transport proteins that play a critical role in the metabolic pathways involved in carbohydrate metabolism.
There are several types of inborn errors of carbohydrate metabolism, including:
1. Galactosemia: This disorder affects the body's ability to metabolize the sugar galactose, which is found in milk and other dairy products. It is caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase.
2. Glycogen storage diseases: These disorders affect the body's ability to store and break down glycogen, which is a complex carbohydrate that serves as a source of energy for the body. There are several types of glycogen storage diseases, each caused by a deficiency in a different enzyme involved in glycogen metabolism.
3. Hereditary fructose intolerance: This disorder affects the body's ability to metabolize the sugar fructose, which is found in fruits and sweeteners. It is caused by a deficiency of the enzyme aldolase B.
4. Pentose phosphate pathway disorders: These disorders affect the body's ability to metabolize certain sugars and generate energy through the pentose phosphate pathway. They are caused by defects in enzymes involved in this pathway.
Symptoms of inborn errors of carbohydrate metabolism can vary widely depending on the specific disorder and its severity. Treatment typically involves dietary restrictions, supplementation with necessary enzymes or cofactors, and management of complications. In some cases, enzyme replacement therapy or even organ transplantation may be considered.
Disaccharidases are a group of enzymes found in the brush border of the small intestine. They play an essential role in digesting complex carbohydrates into simpler sugars, which can then be absorbed into the bloodstream. The three main disaccharidases are:
1. Maltase-glucoamylase: This enzyme breaks down maltose (a disaccharide formed from two glucose molecules) and maltotriose (a trisaccharide formed from three glucose molecules) into individual glucose units.
2. Sucrase: This enzyme is responsible for breaking down sucrose (table sugar, a disaccharide composed of one glucose and one fructose molecule) into its component monosaccharides, glucose and fructose.
3. Lactase: This enzyme breaks down lactose (a disaccharide formed from one glucose and one galactose molecule) into its component monosaccharides, glucose and galactose.
Deficiencies in these disaccharidases can lead to various digestive disorders, such as lactose intolerance (due to lactase deficiency), sucrase-isomaltase deficiency, or congenital sucrase-isomaltase deficiency (CSID). These conditions can cause symptoms like bloating, diarrhea, and abdominal cramps after consuming foods containing the specific disaccharide.
Alpha-mannosidosis is a rare inherited metabolic disorder caused by the deficiency of the enzyme alpha-mannosidase. This enzyme is responsible for breaking down complex sugar molecules called mannose-rich oligosaccharides, which are found on the surface of many different types of cells in the body.
When the alpha-mannosidase enzyme is deficient or not working properly, these sugar molecules accumulate inside the lysosomes (the recycling centers of the cell) and cause damage to various tissues and organs, leading to a range of symptoms.
The severity of the disease can vary widely, depending on the amount of functional alpha-mannosidase enzyme activity present in an individual's cells. Three main types of alpha-mannosidosis have been described: mild, moderate, and severe. The severe form is usually diagnosed in infancy or early childhood and is characterized by developmental delay, intellectual disability, coarse facial features, skeletal abnormalities, hearing loss, and recurrent respiratory infections.
The moderate form of the disease may not be diagnosed until later in childhood or even adulthood, and it is generally milder than the severe form. Symptoms can include mild to moderate intellectual disability, skeletal abnormalities, hearing loss, and speech difficulties. The mild form of alpha-mannosidosis may not cause any noticeable symptoms until much later in life, and some individuals with this form of the disease may never experience any significant health problems.
Currently, there is no cure for alpha-mannosidosis, and treatment is focused on managing the symptoms of the disease. Enzyme replacement therapy (ERT) has shown promise in treating some forms of the disorder, but it is not yet widely available. Bone marrow transplantation has also been used to treat alpha-mannosidosis, with varying degrees of success.
Dysostosis is a term used to describe a group of genetic disorders that are characterized by abnormal development and formation of one or more bones in the body. The condition is typically present at birth (congenital) and can affect any bone, but it most commonly involves the bones of the skull, face, hands, and feet.
The term "dysostosis" comes from the Greek words "dys," meaning difficult or abnormal, and "osteon," meaning bone. Dysostoses are usually caused by mutations in specific genes that regulate bone development. These genetic changes can be inherited from one or both parents or can occur spontaneously during fetal development.
There are many different types of dysostoses, each with its own set of symptoms and characteristics. Some common examples include:
1. Cleidocranial Dysplasia: This is a rare genetic disorder that affects the development of the skull and collarbones (cleido). People with cleidocranial dysplasia may have a larger than normal head, wide-set eyes, a prominent forehead, and underdeveloped or missing collarbones.
2. Acrocephalopolysyndactyly Type II: Also known as ACPS II or Greig cephalopolysyndactyly syndrome, this disorder is characterized by a pointed skull (acrocephaly), extra fingers and toes (polydactyly), and wide-set eyes.
3. Osteogenesis Imperfecta: This is a group of genetic disorders that affect the body's production of collagen, a protein that helps to strengthen bones. People with osteogenesis imperfecta have fragile bones that break easily, often as a result of minor trauma.
4. Diastrophic Dysplasia: This is a rare genetic disorder that affects the development of the bones and cartilage in the body. People with diastrophic dysplasia may have short limbs, a deformed spine, and a characteristic "hitchhiker's thumb" appearance.
5. Thanatophoric Dysplasia: This is a severe genetic disorder that affects the development of the bones in the body. People with thanatophoric dysplasia have very short limbs, a small chest, and a deformed skull. The condition is often fatal in infancy or early childhood.
These are just a few examples of the many different types of skeletal dysplasias that exist. While some forms of these disorders can be managed with medical treatment and therapy, others may require surgery or other interventions to help improve quality of life. In some cases, genetic counseling and testing may be recommended for individuals who are considering starting a family and have a history of skeletal dysplasia in their family.
Fucose is a type of sugar molecule that is often found in complex carbohydrates known as glycans, which are attached to many proteins and lipids in the body. It is a hexose sugar, meaning it contains six carbon atoms, and is a type of L-sugar, which means that it rotates plane-polarized light in a counterclockwise direction.
Fucose is often found at the ends of glycan chains and plays important roles in various biological processes, including cell recognition, signaling, and interaction. It is also a component of some blood group antigens and is involved in the development and function of the immune system. Abnormalities in fucosylation (the addition of fucose to glycans) have been implicated in various diseases, including cancer, inflammation, and neurological disorders.
Alpha-N-Acetylgalactosaminidase (also known as alpha-GalNAcase) is an enzyme that belongs to the class of glycoside hydrolases. Its systematic name is N-acetyl-alpha-galactosaminide galactosaminohydrolase. This enzyme is responsible for catalyzing the hydrolysis of the terminal, non-reducing N-acetyl-D-galactosamine residues in gangliosides and glycoproteins.
Gangliosides are sialic acid-containing glycosphingolipids found in animal tissues, especially in the nervous system. Glycoproteins are proteins that contain oligosaccharide chains (glycans) covalently attached to their polypeptide backbone.
Deficiency or dysfunction of alpha-N-Acetylgalactosaminidase can lead to various genetic disorders, such as Schindler and Kanzaki diseases, which are characterized by the accumulation of gangliosides and glycoproteins in lysosomes, leading to progressive neurological deterioration.
Fabry disease is a rare X-linked inherited lysosomal storage disorder caused by mutations in the GLA gene, which encodes the enzyme alpha-galactosidase A. This enzyme deficiency leads to the accumulation of glycosphingolipids, particularly globotriaosylceramide (Gb3 or GL-3), in various tissues and organs throughout the body. The accumulation of these lipids results in progressive damage to multiple organ systems, including the heart, kidneys, nerves, and skin.
The symptoms of Fabry disease can vary widely among affected individuals, but common manifestations include:
1. Pain: Acroparesthesias (burning or tingling sensations) in the hands and feet, episodic pain crises, chronic pain, and neuropathy.
2. Skin: Angiokeratomas (small, red, rough bumps on the skin), hypohidrosis (decreased sweating), and anhydrosis (absent sweating).
3. Gastrointestinal: Abdominal pain, diarrhea, constipation, nausea, and vomiting.
4. Cardiovascular: Left ventricular hypertrophy (enlargement of the heart muscle), cardiomyopathy, ischemic heart disease, arrhythmias, and valvular abnormalities.
5. Renal: Proteinuria (protein in the urine), hematuria (blood in the urine), chronic kidney disease, and end-stage renal disease.
6. Nervous system: Hearing loss, tinnitus, vertigo, stroke, and cognitive decline.
7. Ocular: Corneal opacities, cataracts, and retinal vessel abnormalities.
8. Pulmonary: Chronic cough, bronchial hyperresponsiveness, and restrictive lung disease.
9. Reproductive system: Erectile dysfunction in males and menstrual irregularities in females.
Fabry disease affects both males and females, but the severity of symptoms is generally more pronounced in males due to the X-linked inheritance pattern. Early diagnosis and treatment with enzyme replacement therapy (ERT) or chaperone therapy can help manage the progression of the disease and improve quality of life.
Angiokeratoma is a cutaneous condition characterized by the presence of small, dilated blood vessels (capillaries) in the upper dermis, which are covered by thickened epidermis. These lesions appear as dark red to black papules or plaques on the skin surface. They can occur spontaneously or as a result of an underlying medical condition such as Fabry disease. Angiokeratomas are typically asymptomatic but may occasionally cause mild discomfort or itching. They most commonly affect the lower extremities, particularly the buttocks and genital region, but can also appear on other parts of the body.
Alpha-galactosidase is an enzyme that breaks down complex carbohydrates, specifically those containing alpha-galactose molecules. This enzyme is found in humans, animals, and microorganisms. In humans, a deficiency of this enzyme can lead to a genetic disorder known as Fabry disease, which is characterized by the accumulation of these complex carbohydrates in various tissues and organs, leading to progressive damage. Alpha-galactosidase is also used as a medication for the treatment of Fabry disease, where it is administered intravenously to help break down the accumulated carbohydrates and alleviate symptoms.
Trihexosylceramides are a type of glycosphingolipids, which are complex lipids found in animal tissues. They consist of a ceramide molecule (a sphingosine and fatty acid) with three hexose sugars attached to it in a specific sequence, typically glucose-galactose-galactose.
Trihexosylceramides are further classified into two types based on the type of ceramide they contain: lactosylceramide (Gal-Glc-Cer) and isoglobotrihexosylceramide (GalNAcβ1-4Galβ1-4Glc-Cer).
These lipids are important components of the cell membrane and play a role in various biological processes, including cell recognition, signal transduction, and cell adhesion. Abnormal accumulation of trihexosylceramides has been implicated in certain diseases, such as Gaucher disease and Tay-Sachs disease, which are caused by deficiencies in enzymes involved in their breakdown.
Enzyme Replacement Therapy (ERT) is a medical treatment approach in which functional copies of a missing or deficient enzyme are introduced into the body to compensate for the lack of enzymatic activity caused by a genetic disorder. This therapy is primarily used to manage lysosomal storage diseases, such as Gaucher disease, Fabry disease, Pompe disease, and Mucopolysaccharidoses (MPS), among others.
In ERT, the required enzyme is produced recombinantly in a laboratory using biotechnological methods. The purified enzyme is then administered to the patient intravenously at regular intervals. Once inside the body, the exogenous enzyme is taken up by cells, particularly those affected by the disorder, and helps restore normal cellular functions by participating in essential metabolic pathways.
ERT aims to alleviate disease symptoms, slow down disease progression, improve quality of life, and increase survival rates for patients with lysosomal storage disorders. However, it does not cure the underlying genetic defect responsible for the enzyme deficiency.
I'm sorry for any confusion, but "New Zealand" is not a medical term or concept. It is a country located in the southwestern Pacific Ocean, known for its stunning landscapes, unique wildlife, and as the filming location for the "Lord of the Rings" films. If you have any questions related to medicine or health, I'd be happy to try and help answer those for you!
 Fucosidosis - Wikipedia
Fucosidosis - Wikipedia Fucosidosis: MedlinePlus Genetics
Fucosidosis: MedlinePlus Genetics What is the definition of Fucosidosis? | Dictionary.net
What is the definition of Fucosidosis? | Dictionary.net A capillary electrophoresis procedure for the screening of oligosaccharidoses and related diseases
A capillary electrophoresis procedure for the screening of oligosaccharidoses and related diseases A Complete List of Available Canine and Feline Genetic Tests - TUFTSBG2003 - VIN
A Complete List of Available Canine and Feline Genetic Tests - TUFTSBG2003 - VIN English Springer Spaniel Breed Profile - Your Dog
English Springer Spaniel Breed Profile - Your Dog Beskrivelse av diagnosen - Frambu
Beskrivelse av diagnosen - Frambu Pathology
Pathology Lipid Storage Disorders: Background, Pathophysiology, Mode of Inheritance
Lipid Storage Disorders: Background, Pathophysiology, Mode of Inheritance Fabry disease | DermNet
Fabry disease | DermNet Ethical Guidelines for Breeders
Ethical Guidelines for Breeders International Classification of Diseases - Endocrine, Nutritional and Metabolic Diseases, and Immunity Disorders
International Classification of Diseases - Endocrine, Nutritional and Metabolic Diseases, and Immunity Disorders Дефицит фосфофруктокиназы (PFKD) - AnimaLabs©
Дефицит фосфофруктокиназы (PFKD) - AnimaLabs© Sepsecs Mouse Gene Details | Sep (O-phosphoserine) tRNA:Sec (selenocysteine) tRNA synthase | International Mouse Phenotyping...
Sepsecs Mouse Gene Details | Sep (O-phosphoserine) tRNA:Sec (selenocysteine) tRNA synthase | International Mouse Phenotyping... VSAAC Chair Biographies - AVBC
VSAAC Chair Biographies - AVBC Coxa valga (Concept Id: C0239137)
- MedGen - NCBI
Coxa valga (Concept Id: C0239137)
- MedGen - NCBI Gentest nach Rasse
Gentest nach Rasse Metabolic Testing | CENTOGENE CentoMetabolic MOx: centogene.com
Metabolic Testing | CENTOGENE CentoMetabolic MOx: centogene.com