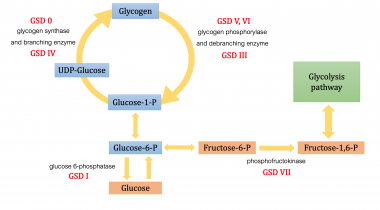
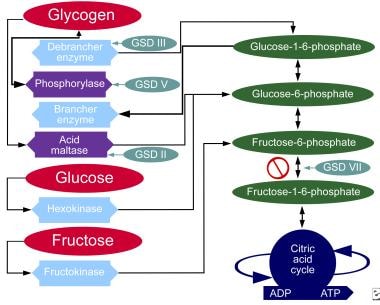
Glycogen Storage Disease Type I
Glycogen Storage Disease Type III
Glycogen Storage Disease
Glycogen Storage Disease Type IV
Glycogen Storage Disease Type II
Glycogen Storage Disease Type VII
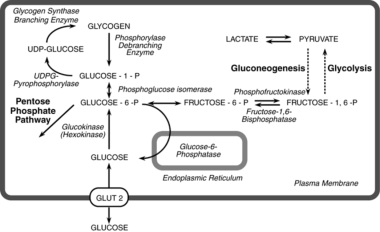
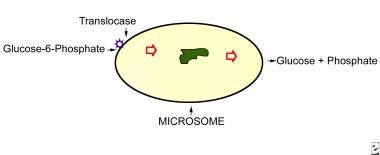
Glucose-6-Phosphatase
Gaucher Disease
Glycogen Storage Disease Type VI
Glycogen
alpha-Glucosidases
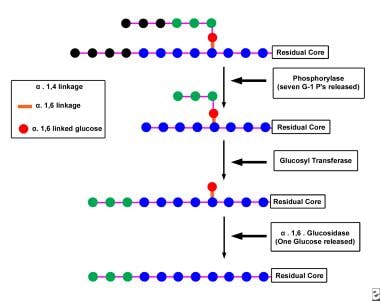
Glycogen Debranching Enzyme System
Glycogen Storage Disease Type V
Glycogen Storage Disease Type VIII
Glucan 1,4-alpha-Glucosidase
Inactivation of the glucose 6-phosphate transporter causes glycogen storage disease type 1b. (1/169)
Glycogen storage disease type 1b (GSD-1b) is proposed to be caused by a deficiency in microsomal glucose 6-phosphate (G6P) transport, causing a loss of glucose-6-phosphatase activity and glucose homeostasis. However, for decades, this disorder has defied molecular characterization. In this study, we characterize the structural organization of the G6P transporter gene and identify mutations in the gene that segregate with the GSD-1b disorder. We report the functional characterization of the recombinant G6P transporter and demonstrate that mutations uncovered in GSD-1b patients disrupt G6P transport. Our results, for the first time, define a molecular basis for functional deficiency in GSD-1b and raise the possibility that the defective G6P transporter contributes to neutropenia and neutrophil/monocyte dysfunctions characteristic of GSD-1b patients. (+info)Identification of protein components of the microsomal glucose 6-phosphate transporter by photoaffinity labelling. (2/169)
The glucose-6-phosphatase system catalyses the terminal step of hepatic glucose production from both gluconeogenesis and glycogenolysis and is thus a key regulatory factor of blood glucose homoeostasis. To identify the glucose 6-phosphate transporter T1, we have performed photoaffinity labelling of human and rat liver microsomes by using the specific photoreactive glucose-6-phosphate translocase inhibitors S 0957 and S 1743. Membrane proteins of molecular mass 70, 55, 33 and 31 kDa were labelled in human microsomes by [3H]S 0957, whereas in rat liver microsomes bands at 95, 70, 57, 54, 50, 41, 33 and 31 kDa were detectable. The photoprobe [3H]S 1743 led to the predominant labelling of a 57 kDa and a 50 kDa protein in the rat. Stripping of microsomes with 0.3% CHAPS retains the specific binding of T1 inhibitors; photoaffinity labelling of such CHAPS-treated microsomes resulted in the labelling of membrane proteins of molecular mass 55, 33 and 31 kDa in human liver and 50, 33 and 31 kDa in rat liver. Photoaffinity labelling of human liver tissue samples from a healthy individual and from liver samples of patients with a diagnosed glycogen-storage disease type 1b (GSD type 1b; von Gierke's disease) revealed the absence of the 55 kDa protein from one of the patients with GSD type 1. These findings support the identity of the glucose 6-phosphate transporter T1, with endoplasmic reticulum protein of molecular mass 50 kDa in rat liver and 55 kDa in human liver. (+info)Complete genomic structure and mutational spectrum of PHKA2 in patients with x-linked liver glycogenosis type I and II. (3/169)
X-linked liver glycogenosis (XLG) is probably the most frequent glycogen-storage disease. XLG can be divided into two subtypes: XLG I, with a deficiency in phosphorylase kinase (PHK) activity in peripheral blood cells and liver; and XLG II, with normal in vitro PHK activity in peripheral blood cells and with variable activity in liver. Both types of XLG are caused by mutations in the same gene, PHKA2, that encodes the regulatory alpha subunit of PHK. To facilitate mutation analysis in PHKA2, we determined its genomic structure. The gene consists of 33 exons, spanning >/=65 kb. By SSCP analysis of the different PHKA2 exons, we identified five new XLG I mutations, one new XLG II mutation, and one mutation present in both a patient with XLG I and a patient with XLG II, bringing the total to 19 XLG I and 12 XLG II mutations. Most XLG I mutations probably lead to truncation or disruption of the PHKA2 protein. In contrast, all XLG II mutations are missense mutations or small in-frame deletions and insertions. These results suggest that the biochemical differences between XLG I and XLG II might be due to the different nature of the disease-causing mutations in PHKA2. XLG I mutations may lead to absence of the alpha subunit, which causes an unstable PHK holoenzyme and deficient enzyme activity, whereas XLG II mutations may lead to in vivo deregulation of PHK, which might be difficult to demonstrate in vitro. (+info)The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. (4/169)
The purpose of this work was to test the hypothesis that mutations in the putative glucose 6-phosphate translocase gene would account for most of the cases of GSD I that are not explained by mutations in the phosphohydrolase gene, ie that are not type Ia. Twenty-three additional families diagnosed as having GSD I non-a (GSDIb, Ic or Id) have now been analysed. The 9exons of the gene were amplified by PCR and mutations searched both by SSCP and heteroduplex analysis. Except for one family in which only one mutation was found, all patients had two allelic mutations in the gene encoding the putative glucose 6-phosphate translocase. Sixteen of the mutations are new and they are all predicted to lead to non-functional proteins. All investigated patients had some degree of neutropenia or neutrophil dysfunction and the clinical phenotype of the four new patients who had been diagnosed as GSD Ic and the one diagnosed as GSD Id was no different from the GSD Ib patients. Since these patients, and the four type Ic patients from two families previously studied, shared several mutations with GSD Ib patients, we conclude that their basic defect is in the putative glucose 6-phosphate translocase and that they should be reclassified as GSD Ib. Isolated defects in microsomal Pi transporter or in microsomal glucose transporter must be very rare or have phenotypes that are not recognised as GSD I, so that in practice there are only two subtypes of GSD I (GSD Ia and GSD Ib). (+info)Mutations in the glucose-6-phosphate transporter (G6PT) gene in patients with glycogen storage diseases type 1b and 1c. (5/169)
Glycogen storage diseases type 1 (GSD 1) are a group of autosomal recessive disorders characterized by impairment of terminal steps of glycogenolysis and gluconeogenesis. Mutations of the glucose-6-phosphatase gene are responsible for the most frequent form of GSD 1, the subtype 1a, while mutations of the glucose-6-phosphate transporter gene (G6PT) have recently been shown to cause the non 1a forms of GSD, namely the 1b and 1c subtypes. Here, we report on the analysis by single-stranded conformation polymorphism (SSCP) and/or DNA sequencing of the exons of the G6PT in 14 patients diagnosed either as affected by the GSD 1b or 1c subtypes. Mutations in the G6PT gene were found in all patients. Four of the detected mutations were novel mutations, while the others were previously described. Our results confirm that the GSD 1b and 1c forms are due to mutations in the same gene, i.e. the G6PT gene. We also show that the same kind of mutation can be associated or not with evident clinical complications such as neutrophil impairment. Since no correlation between the type and position of the mutation and the severity of the disease was found, other unknown factors may cause the expression of symptoms, such as neutropenia, which dramatically influence the severity of the disease. (+info)Liver transplantation for glycogen storage disease types I, III, and IV. (6/169)
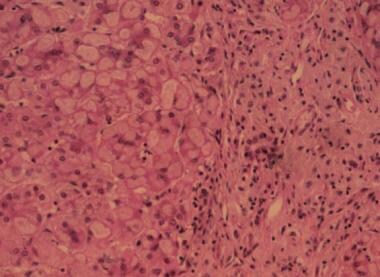
Glycogen storage disease (GSD) types I, III, and IV can be associated with severe liver disease. The possible development of hepatocellular carcinoma and/or hepatic failure make these GSDs potential candidates for liver transplantation. Early diagnosis and initiation of effective dietary therapy have dramatically improved the outcome of GSD type I by reducing the incidence of liver adenoma and renal insufficiency. Nine type I and 3 type III patients have received liver transplants because of poor metabolic control, multiple liver adenomas, or progressive liver failure. Metabolic abnormalities were corrected in all GSD type I and type III patients, while catch-up growth was reported only in two patients. Whether liver transplantation results in reversal and/or prevention of renal disease remains unclear. Neutropenia persisted in both GSDIb patients post liver transplantation necessitating continuous granulocyte colony stimulating factor treatment. Thirteen GSD type IV patients were liver transplanted because of progressive liver cirrhosis and failure. All but one patient have not had neuromuscular or cardiac complications during follow-up periods for as long as 13 years. Four have died within a week and 5 years after transplantation. Caution should be taken in selecting GSD type IV candidates for liver transplantation because of the variable phenotype, which may include life-limiting extrahepatic manifestations. It remains to be evaluated, whether a genotype-phenotype correlation exists for GSD type IV, which may aid in the decision making. CONCLUSION: Liver transplantation should be considered for patients with glycogen storage disease who have developed liver malignancy or hepatic failure, and for type IV patients with the classical and progressive hepatic form. (+info)Correction of glycogen storage disease type 1a in a mouse model by gene therapy. (7/169)
Glycogen storage disease type 1a (GSD-1a), characterized by hypoglycemia, liver and kidney enlargement, growth retardation, hyperlipidemia, and hyperuricemia, is caused by a deficiency in glucose-6-phosphatase (G6Pase), a key enzyme in glucose homeostasis. To evaluate the feasibility of gene replacement therapy for GSD-1a, we have infused adenoviral vector containing the murine G6Pase gene (Ad-mG6Pase) into G6Pase-deficient (G6Pase(-/-)) mice that manifest symptoms characteristic of human GSD-1a. Whereas <15% of G6Pase(-/-) mice under glucose therapy survived weaning, a 100% survival rate was achieved when G6Pase(-/-) mice were infused with Ad-mG6Pase, 90% of which lived to 3 months of age. Hepatic G6Pase activity in Ad-mG6Pase-infused mice was restored to 19% of that in G6Pase(+/+) mice at 7-14 days post-infusion; the activity persisted for at least 70 days. Ad-mG6Pase infusion also greatly improved growth of G6Pase(-/-) mice and normalized plasma glucose, cholesterol, triglyceride, and uric acid profiles. Furthermore, liver and kidney enlargement was less pronounced with near-normal levels of glycogen depositions in both organs. Our data demonstrate that a single administration of a recombinant adenoviral vector can alleviate the pathological manifestations of GSD-1a in mice, suggesting that this disorder in humans can potentially be corrected by gene therapy. (+info)New lessons in the regulation of glucose metabolism taught by the glucose 6-phosphatase system. (8/169)
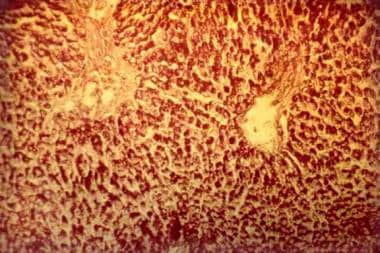
The operation of glucose 6-phosphatase (EC 3.1.3.9) (Glc6Pase) stems from the interaction of at least two highly hydrophobic proteins embedded in the ER membrane, a heavily glycosylated catalytic subunit of m 36 kDa (P36) and a 46-kDa putative glucose 6-phosphate (Glc6P) translocase (P46). Topology studies of P36 and P46 predict, respectively, nine and ten transmembrane domains with the N-terminal end of P36 oriented towards the lumen of the ER and both termini of P46 oriented towards the cytoplasm. P36 gene expression is increased by glucose, fructose 2,6-bisphosphate (Fru-2,6-P2) and free fatty acids, as well as by glucocorticoids and cyclic AMP; the latter are counteracted by insulin. P46 gene expression is affected by glucose, insulin and cyclic AMP in a manner similar to P36. Accordingly, several response elements for glucocorticoids, cyclic AMP and insulin regulated by hepatocyte nuclear factors were found in the Glc6Pase promoter. Mutations in P36 and P46 lead to glycogen storage disease (GSD) type-1a and type-1 non a (formerly 1b and 1c), respectively. Adenovirus-mediated overexpression of P36 in hepatocytes and in vivo impairs glycogen metabolism and glycolysis and increases glucose production; P36 overexpression in INS-1 cells results in decreased glycolysis and glucose-induced insulin secretion. The nature of the interaction between P36 and P46 in controling Glc6Pase activity remains to be defined. The latter might also have functions other than Glc6P transport that are related to Glc6P metabolism. (+info)Glycogen Storage Disease Type I (GSD I) is a rare inherited metabolic disorder caused by deficiency of the enzyme glucose-6-phosphatase, which is necessary for the liver to release glucose into the bloodstream. This leads to an accumulation of glycogen in the liver and abnormally low levels of glucose in the blood (hypoglycemia).
There are two main subtypes of GSD I: Type Ia and Type Ib. In Type Ia, there is a deficiency of both glucose-6-phosphatase enzyme activity in the liver, kidney, and intestine, leading to hepatomegaly (enlarged liver), hypoglycemia, lactic acidosis, hyperlipidemia, and growth retardation. Type Ib is characterized by a deficiency of glucose-6-phosphatase enzyme activity only in the neutrophils, leading to recurrent bacterial infections.
GSD I requires lifelong management with frequent feedings, high-carbohydrate diet, and avoidance of fasting to prevent hypoglycemia. In some cases, treatment with continuous cornstarch infusions or liver transplantation may be necessary.
Glycogen Storage Disease Type III, also known as Cori or Forbes disease, is a rare inherited metabolic disorder caused by deficiency of the debranching enzyme amylo-1,6-glucosidase, which is responsible for breaking down glycogen in the liver and muscles. This results in an abnormal accumulation of glycogen in these organs leading to its associated symptoms.
There are two main types: Type IIIa affects both the liver and muscles, while Type IIIb affects only the liver. Symptoms can include hepatomegaly (enlarged liver), hypoglycemia (low blood sugar), hyperlipidemia (high levels of fats in the blood), and growth retardation. In Type IIIa, muscle weakness and cardiac problems may also occur.
The diagnosis is usually made through biochemical tests and genetic analysis. Treatment often involves dietary management with frequent meals to prevent hypoglycemia, and in some cases, enzyme replacement therapy. However, there is no cure for this condition and life expectancy can be reduced depending on the severity of the symptoms.
Glycogen storage disease (GSD) is a group of rare inherited metabolic disorders that affect the body's ability to break down and store glycogen, a complex carbohydrate that serves as the primary form of energy storage in the body. These diseases are caused by deficiencies or dysfunction in enzymes involved in the synthesis, degradation, or transport of glycogen within cells.
There are several types of GSDs, each with distinct clinical presentations and affected organs. The most common type is von Gierke disease (GSD I), which primarily affects the liver and kidneys. Other types include Pompe disease (GSD II), McArdle disease (GSD V), Cori disease (GSD III), Andersen disease (GSD IV), and others.
Symptoms of GSDs can vary widely depending on the specific type, but may include:
* Hypoglycemia (low blood sugar)
* Growth retardation
* Hepatomegaly (enlarged liver)
* Muscle weakness and cramping
* Cardiomyopathy (heart muscle disease)
* Respiratory distress
* Developmental delays
Treatment for GSDs typically involves dietary management, such as frequent feedings or a high-protein, low-carbohydrate diet. In some cases, enzyme replacement therapy may be used to manage symptoms. The prognosis for individuals with GSDs depends on the specific type and severity of the disorder.
Glycogen Storage Disease Type IV (GSD IV), also known as Andersen's disease, is a rare inherited metabolic disorder that affects the body's ability to break down glycogen, a complex carbohydrate that serves as a source of energy for the body.
In GSD IV, there is a deficiency in the enzyme called glycogen branching enzyme (GBE), which is responsible for adding branches to the glycogen molecule during its synthesis. This results in an abnormal form of glycogen that accumulates in various organs and tissues, particularly in the liver, heart, and muscles.
The accumulation of this abnormal glycogen can lead to progressive damage and failure of these organs, resulting in a variety of symptoms such as muscle weakness, hypotonia, hepatomegaly (enlarged liver), cardiomyopathy (heart muscle disease), and developmental delay. The severity of the disease can vary widely, with some individuals experiencing milder symptoms while others may have a more severe and rapidly progressing form of the disorder.
Currently, there is no cure for GSD IV, and treatment is focused on managing the symptoms and slowing down the progression of the disease. This may include providing nutritional support, addressing specific organ dysfunction, and preventing complications.
Glycogen Storage Disease Type II, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar that serves as energy storage, within lysosomes. When GAA is deficient, glycogen accumulates in various tissues, particularly in muscle cells, leading to their dysfunction and damage.
The severity of Pompe Disease can vary significantly, depending on the amount of functional enzyme activity remaining. The classic infantile-onset form presents within the first few months of life with severe muscle weakness, hypotonia, feeding difficulties, and respiratory insufficiency. This form is often fatal by 1-2 years of age if left untreated.
A later-onset form, which can present in childhood, adolescence, or adulthood, has a more variable clinical course. Affected individuals may experience progressive muscle weakness, respiratory insufficiency, and cardiomyopathy, although the severity and rate of progression are generally less pronounced than in the infantile-onset form.
Enzyme replacement therapy with recombinant human GAA is available for the treatment of Pompe Disease and has been shown to improve survival and motor function in affected individuals.
Glycogen Storage Disease Type VII, also known as Tarui's disease, is a rare inherited metabolic disorder caused by a deficiency of the enzyme phosphofructokinase (PFK), which is required for glycogenolysis – the breakdown of glycogen to glucose-1-phosphate and ultimately into glucose. This enzyme deficiency results in the accumulation of glycogen, particularly in muscle and red blood cells, leading to symptoms such as exercise-induced muscle cramps, myoglobinuria (the presence of myoglobin in the urine), and hemolytic anemia. The disease can also cause muscle weakness, fatigue, and dark-colored urine after strenuous exercise. It is inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
Glucose-6-phosphatase is an enzyme that plays a crucial role in the regulation of glucose metabolism. It is primarily located in the endoplasmic reticulum of cells in liver, kidney, and intestinal mucosa. The main function of this enzyme is to remove the phosphate group from glucose-6-phosphate (G6P), converting it into free glucose, which can then be released into the bloodstream and used as a source of energy by cells throughout the body.
The reaction catalyzed by glucose-6-phosphatase is as follows:
Glucose-6-phosphate + H2O → Glucose + Pi (inorganic phosphate)
This enzyme is essential for maintaining normal blood glucose levels, particularly during periods of fasting or starvation. In these situations, the body needs to break down stored glycogen in the liver and convert it into glucose to supply energy to the brain and other vital organs. Glucose-6-phosphatase is a key enzyme in this process, allowing for the release of free glucose into the bloodstream.
Deficiencies or mutations in the gene encoding glucose-6-phosphatase can lead to several metabolic disorders, such as glycogen storage disease type I (von Gierke's disease) and other related conditions. These disorders are characterized by an accumulation of glycogen and/or fat in various organs, leading to impaired glucose metabolism, growth retardation, and increased risk of infection and liver dysfunction.
Gaucher disease is an inherited metabolic disorder caused by the deficiency of the enzyme glucocerebrosidase. This enzyme is responsible for breaking down a complex fatty substance called glucocerebroside, found in the cells of various tissues throughout the body. When the enzyme is not present in sufficient quantities or is entirely absent, glucocerebroside accumulates inside the lysosomes (cellular organelles responsible for waste material breakdown) of certain cell types, particularly within white blood cells called macrophages. This buildup of lipids leads to the formation of characteristic lipid-laden cells known as Gaucher cells.
There are three main types of Gaucher disease, classified based on the absence or presence and severity of neurological symptoms:
1. Type 1 (non-neuronopathic) - This is the most common form of Gaucher disease, accounting for approximately 95% of cases. It primarily affects the spleen, liver, and bone marrow but does not typically involve the central nervous system. Symptoms may include an enlarged spleen and/or liver, low red blood cell counts (anemia), low platelet counts (thrombocytopenia), bone pain and fractures, and fatigue.
2. Type 2 (acute neuronopathic) - This rare and severe form of Gaucher disease affects both visceral organs and the central nervous system. Symptoms usually appear within the first six months of life and progress rapidly, often leading to death before two years of age due to neurological complications.
3. Type 3 (subacute neuronopathic) - This form of Gaucher disease affects both visceral organs and the central nervous system but has a slower progression compared to type 2. Symptoms may include those seen in type 1, as well as neurological issues such as seizures, eye movement abnormalities, and cognitive decline.
Gaucher disease is inherited in an autosomal recessive manner, meaning that an individual must inherit two defective copies of the gene (one from each parent) to develop the condition. Treatment options for Gaucher disease include enzyme replacement therapy (ERT), substrate reduction therapy (SRT), and chaperone therapy, depending on the type and severity of the disease.
Glycogen Storage Disease Type VI, also known as Hers disease, is a rare inherited metabolic disorder caused by deficiency of the liver enzyme called glycogen phosphorylase. This enzyme is responsible for breaking down glycogen, which is a stored form of glucose, into glucose-1-phosphate during the process of glycogenolysis.
In GSD Type VI, the lack of this enzyme leads to an abnormal accumulation of glycogen in the liver, causing hepatomegaly (enlarged liver) and elevated liver enzymes. The symptoms of this condition are usually milder compared to other types of GSD, and may include fatigue, weakness, and hypoglycemia (low blood sugar), especially after prolonged fasting or physical exertion.
The diagnosis of GSD Type VI is typically made through biochemical tests that measure the activity of the glycogen phosphorylase enzyme in liver tissue, as well as genetic testing to identify mutations in the gene responsible for the enzyme's production. Treatment may involve dietary management, such as frequent feeding and avoidance of prolonged fasting, to prevent hypoglycemia. In some cases, medication may be necessary to manage symptoms and prevent complications.
Glycogen is a complex carbohydrate that serves as the primary form of energy storage in animals, fungi, and bacteria. It is a polysaccharide consisting of long, branched chains of glucose molecules linked together by glycosidic bonds. Glycogen is stored primarily in the liver and muscles, where it can be quickly broken down to release glucose into the bloodstream during periods of fasting or increased metabolic demand.
In the liver, glycogen plays a crucial role in maintaining blood glucose levels by releasing glucose when needed, such as between meals or during exercise. In muscles, glycogen serves as an immediate energy source for muscle contractions during intense physical activity. The ability to store and mobilize glycogen is essential for the proper functioning of various physiological processes, including athletic performance, glucose homeostasis, and overall metabolic health.
Alpha-glucosidases are a group of enzymes that break down complex carbohydrates into simpler sugars, such as glucose, by hydrolyzing the alpha-1,4 and alpha-1,6 glycosidic bonds in oligosaccharides, disaccharides, and polysaccharides. These enzymes are located on the brush border of the small intestine and play a crucial role in carbohydrate digestion and absorption.
Inhibitors of alpha-glucosidases, such as acarbose and miglitol, are used in the treatment of type 2 diabetes to slow down the digestion and absorption of carbohydrates, which helps to reduce postprandial glucose levels and improve glycemic control.
The Glycogen Debranching Enzyme System, also known as glycogen debranching enzyme or Amy-1, is a crucial enzyme complex in human biochemistry. It plays an essential role in the metabolism of glycogen, which is a large, branched polymer of glucose that serves as the primary form of energy storage in animals and fungi.
The Glycogen Debranching Enzyme System consists of two enzymatic activities: a transferase and an exo-glucosidase. The transferase activity transfers a segment of a branched glucose chain to another part of the same or another glycogen molecule, while the exo-glucosidase activity cleaves the remaining single glucose units from the outer branches of the glycogen molecule.
This enzyme system is responsible for removing the branched structures of glycogen, allowing the linear chains to be further degraded by other enzymes into glucose molecules that can be used for energy production or stored for later use. Defects in this enzyme complex can lead to several genetic disorders, such as Glycogen Storage Disease Type III (Cori's disease) and Type IV (Andersen's disease), which are characterized by the accumulation of abnormal glycogen molecules in various tissues.
Glycogen Storage Disease Type V, also known as McArdle's disease, is a genetic disorder that affects the body's ability to break down glycogen, a complex carbohydrate stored in muscles, into glucose, which provides energy for muscle contraction.
This condition results from a deficiency of the enzyme myophosphorylase, which is responsible for breaking down glycogen into glucose-1-phosphate within the muscle fibers. Without sufficient myophosphorylase activity, muscles become easily fatigued and may cramp or become rigid during exercise due to a lack of available energy.
Symptoms typically appear in childhood or adolescence and can include muscle weakness, stiffness, cramps, and myoglobinuria (the presence of myoglobin, a protein found in muscle cells, in the urine) following exercise. Diagnosis is usually confirmed through genetic testing and enzyme assays. Treatment typically involves avoiding strenuous exercise and ensuring adequate hydration and rest before and after physical activity. In some cases, dietary modifications such as high-protein or high-carbohydrate intake may be recommended to help manage symptoms.
Glycogen Storage Disease Type VIII, also known as Phosphorylase Kinase Deficiency, is a rare genetic metabolic disorder that affects the production and breakdown of glycogen in the body. Glycogen is a complex carbohydrate that serves as the primary form of energy storage in the body.
In this condition, there is a deficiency or dysfunction of the enzyme phosphorylase kinase (PhK), which plays a crucial role in activating glycogen phosphorylase, an enzyme responsible for breaking down glycogen into glucose-1-phosphate during periods of increased energy demand.
The deficiency or dysfunction of PhK leads to the abnormal accumulation of glycogen in various tissues, particularly in the liver and muscles. This accumulation can result in hepatomegaly (enlarged liver), hypoglycemia (low blood sugar levels), growth retardation, and muscle weakness.
Glycogen Storage Disease Type VIII is inherited in an autosomal recessive manner, meaning that an individual must inherit two defective copies of the gene, one from each parent, to develop the condition. There are four subtypes of GSD Type VIII, classified based on the specific genetic mutation and the severity of symptoms.
Treatment for Glycogen Storage Disease Type VIII typically involves managing the symptoms and complications associated with the disorder, such as providing a high-carbohydrate diet to prevent hypoglycemia and addressing any liver or muscle dysfunction. Regular monitoring by a healthcare team experienced in metabolic disorders is essential for optimizing treatment and ensuring appropriate management of this complex condition.
Glucan 1,4-alpha-glucosidase, also known as amyloglucosidase or glucoamylase, is an enzyme that catalyzes the hydrolysis of 1,4-glycosidic bonds in starch and other oligo- and polysaccharides, breaking them down into individual glucose molecules. This enzyme specifically acts on the alpha (1->4) linkages found in amylose and amylopectin, two major components of starch. It is widely used in various industrial applications, including the production of high fructose corn syrup, alcoholic beverages, and as a digestive aid in some medical supplements.
 Glycogen storage disease type I
Glycogen storage disease type I
Glycogen storage disease type III
Glycogen storage disease type VI
Glycogen storage disease type V
Glycogen storage disease type 0
Glycogen storage disease type IV
Glycogen storage disease type II
Glycogen storage disease type IX
Glycogen storage disease
Glycogen branching enzyme
Glycogen debranching enzyme
Glycogen synthase
Phosphofructokinase deficiency
Glycogen-branching enzyme deficiency
Lafora disease
Liver disease
Glucose 6-phosphatase
Shin Joong Oh
Glucose-6-phosphate exchanger SLC37A4
Barbara Illingworth Brown
Pseudoathletic appearance
Medical genetics of Jews
G6PC
Corn starch
Peter J. Taub
Acid alpha-glucosidase
PHKG2
Glucose cycle
Glycogen phosphorylase
Danon disease
Glycogen storage disease type I - Wikipedia
 Glycogen storage disease type IV: MedlinePlus Genetics
Glycogen storage disease type IV: MedlinePlus Genetics
 Glycogen Storage Disease Type IV
Glycogen Storage Disease Type IV
 Type IV Glycogen Storage Disease: Practice Essentials, Pathophysiology, Prognosis
Type IV Glycogen Storage Disease: Practice Essentials, Pathophysiology, Prognosis
Genetics of Glycogen-Storage Disease Type II (Pompe Disease) Medication: Enzyme replacement, Pharmacologic Chaperones
 Biochemical and molecular investigation of two Korean patients with glycogen storage disease type III
Biochemical and molecular investigation of two Korean patients with glycogen storage disease type III
Association of the congenital neuromuscular form of glycogen storage disease type IV with a large deletion and recurrent...
Type V Glycogen Storage Disease: Practice Essentials, Pathophysiology, Epidemiology
 Glycogen storage disease type II (Pompe disease)
Glycogen storage disease type II (Pompe disease)
 Glycogen storage disease type VI
Glycogen storage disease type VI
 Glycogen storage disease type III diagnosis and management guidelines. | Read by QxMD
Glycogen storage disease type III diagnosis and management guidelines. | Read by QxMD
 Glycogen Storage Disease Type 1b - CureGSD1b
Glycogen Storage Disease Type 1b - CureGSD1b
Glycogen-Storage Disease Type 0 Differential Diagnoses
Glycogen Storage Disease Type V | Profiles RNS
 Glycogen Storage Disease Type IIIa, (GSD IIIa)
Glycogen Storage Disease Type IIIa, (GSD IIIa)
Type V Glycogen Storage Disease: Background, Pathophysiology, Epidemiology
 Glycogen storage disease type I: Video & Anatomy | Osmosis
Glycogen storage disease type I: Video & Anatomy | Osmosis
Molecular Genetics of Type 1 Glycogen Storage Diseases. | Read by QxMD
 Understanding glycogen storage disease type 1b and its impacts. - Sci Ani
Understanding glycogen storage disease type 1b and its impacts. - Sci Ani
 Glycogen Storage Disease, Type 1A (G6PC), 9 Variants | ARUP Laboratories Test Directory
Glycogen Storage Disease, Type 1A (G6PC), 9 Variants | ARUP Laboratories Test Directory
 Glycogen debranching enzyme - wikidoc
Glycogen debranching enzyme - wikidoc
Pulmonary arterial hypertension and type-I glycogen-storage disease: the serotonin hypothesis - The Lincoln Repository
 Pharmacological and nutritional treatment for McArdle disease (Glycogen Storage Disease type V) | Evidence-Based Medicine...
Pharmacological and nutritional treatment for McArdle disease (Glycogen Storage Disease type V) | Evidence-Based Medicine...
 A glycogen storage disease type 1a patient with type 2 diabetes | BMC Medical Genomics | Peer Review
A glycogen storage disease type 1a patient with type 2 diabetes | BMC Medical Genomics | Peer Review
 PAA849Hu02 | Polyclonal Antibody to Glycogen Phosphorylase, Liver (PYGL) | Homo sapiens (Human) USCN(Wuhan USCN Business Co.,...
PAA849Hu02 | Polyclonal Antibody to Glycogen Phosphorylase, Liver (PYGL) | Homo sapiens (Human) USCN(Wuhan USCN Business Co.,...
 Glycogen Storage Disease Type VII (GSD VII) | Syndromes: Rapid Recognition and Perioperative Implications |...
Glycogen Storage Disease Type VII (GSD VII) | Syndromes: Rapid Recognition and Perioperative Implications |...
 Genes to Cells: Vol 18, No 12
Genes to Cells: Vol 18, No 12
Cirrhosis: Practice Essentials, Overview, Etiology
Cirrhosis: Practice Essentials, Overview, Etiology
 Metabolic Crises | SpringerLink
Metabolic Crises | SpringerLink
Mutations9
- GBE1 gene mutations that cause GSD IV lead to a shortage (deficiency) of the glycogen branching enzyme. (medlineplus.gov)
- Glycogen storage disease type IV (GSD IV), or Andersen disease, is an autosomal recessive disorder caused by mutations in the gene-encoding glycogen-branching enzyme necessary for normal glycogen metabolism. (medscape.com)
- This case report also highlights the need for a more comprehensive search for large deletion mutations associated with glycogen storage disease type IV, especially if routine GBE1 gene sequencing results are equivocal. (nih.gov)
- GSD type V is an autosomal recessive disease resulting from mutations in the PYGM gene that encodes for the muscle isoform of glycogen phosphorylase (myophosphorylase). (medscape.com)
- Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0. (medscape.com)
- Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. (medscape.com)
- [1] When glycogen breakdown is compromised by mutations in the glycogen debranching enzyme, metabolic diseases such as Glycogen storage disease type III can result. (wikidoc.org)
- Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. (springer.com)
- Mutations in this gene cause glycogen storage disease type 9D, also known as X-linked muscle glycogenosis. (nih.gov)
Phosphorylase8
- Glycogen storage disease type VI is a type of glycogen storage disease caused by a deficiency in liver glycogen phosphorylase . (chemeurope.com)
- Together with phosphorylase , glycogen debranching enzymes function in glycogen breakdown and glucose mobilization. (wikidoc.org)
- When phosphorylase has digested a glycogen branch down to four glucose residues, it will not remove further residues. (wikidoc.org)
- Glycogen debranching enzymes assist phosphorylase, the primary enzyme involved in glycogen breakdown , mobilize glycogen stores. (wikidoc.org)
- Phosphorylase can only cleave α-1,4- glycosidic bond between adjacent glucose molecules in glycogen but branches exist as α-1,6 linkages. (wikidoc.org)
- because 1 in 10 residues is branched, cleavage by phosphorylase alone would not be sufficient in mobilizing glycogen stores. (wikidoc.org)
- Thus the debranching enzymes, transferase and α-1,6- glucosidase converts the branched glycogen structure into a linear one, paving the way for further cleavage by phosphorylase. (wikidoc.org)
- Magnetic Luminex Assay Kit for Glycogen Phosphorylase, Liver (PYGL) ,etc. (uscnk.com)
Caused by a deficiency4
- Glycogen storage disease type III (GSD-III) is an inborn error of glycogen metabolism caused by a deficiency of the glycogen debranching enzyme, amylo-1,6-glucosidase,4-α-glucanotransferase (AGL). (degruyter.com)
- Anderson disease, also known as glycogen storage disease type IV (MIM 232500), is a rare autosomal recessive disorder caused by a deficiency of glycogen branching enzyme. (nih.gov)
- Glycogen storage disease type 1 (GSD-1), also known as von Gierke disease, is caused by a deficiency in the activity of the enzyme glucose-6-phosphatase (G6Pase). (qxmd.com)
- A case of pulmonary arterial hypertension in a patient with type-Ia glycogen-storage disease, a rare autosomal recessive disorder caused by a deficiency of glucose-6-phosphatase is reported in this study. (lincoln.ac.uk)
Defective glycogen synthase1
- Interestingly, GSD type 0 also is described and is a disorder causing glycogen deficiency due to defective glycogen synthase. (medscape.com)
Accumulation4
- thus, an enzyme deficiency results in glycogen accumulation in specific tissues. (medscape.com)
- Carbohydrate metabolic pathways are blocked, leading to excess glycogen accumulation in affected tissues and/or disturbances in energy production. (medscape.com)
- Because free glucose is the product of the hepatic glucose-6 phosphatase reaction, either type leads to accumulation of liver glycogen, accompanied by fasting hypoglycemia. (lu.se)
- At the opportunity, the pathologist visualized glycogen accumulation in vesicles inside the cardiac fibers1. (bvsalud.org)
Form of glycogen2
- Any glucose that is not used immediately for energy is held in reserve in the liver, muscles, and kidneys in the form of glycogen and is released when needed by the body. (msdmanuals.com)
- About 1 in 25,000 infants has some form of glycogen storage disease. (msdmanuals.com)
Gene5
- People with one copy of the faulty gene are carriers of the disease and have no symptoms. (wikipedia.org)
- As with other autosomal recessive diseases, each child born to two carriers of the disease has a 25% chance of inheriting both copies of the faulty gene and manifesting the disease. (wikipedia.org)
- The GBE1 gene provides instructions for making the glycogen branching enzyme. (medlineplus.gov)
- A novel mutation in the glycogen synthase 2 gene in a child with glycogen storage disease type 0. (medscape.com)
- Stuehler B, Reichert J, Stremmel W, Schaefer M. Analysis of the human homologue of the canine copper toxicosis gene MURR1 in Wilson disease patients. (medscape.com)
Deficiency of glycogen1
- Infantile hypoglycaemia due to inherited deficiency of glycogen synthetase in liver. (medscape.com)
Metabolism of glycogen2
- Glycogen storage disease (GSD) type IIIa is a disorder that affects the metabolism of glycogen. (wisdompanel.com)
- that occur when there is a defect in the enzymes that are involved in the metabolism of glycogen, often resulting in growth abnormalities, weakness, a large liver, low blood sugar, and confusion. (msdmanuals.com)
Break down glycogen2
- The inability of muscle cells to break down glycogen for energy leads to muscle weakness and wasting. (medlineplus.gov)
- Glycogen storage diseases are caused by the lack of an enzyme needed to change glucose into glycogen and break down glycogen into glucose. (msdmanuals.com)
Glucose from glycogen2
- With intense exercise, glucose from glycogen stores in muscle becomes the predominant resource. (medscape.com)
- Patients with glycogen storage disease type I are unable to release glucose from glycogen. (lu.se)
Congenital3
- The congenital muscular type of GSD IV is usually not evident before birth but develops in early infancy. (medlineplus.gov)
- Infants with the congenital muscular type of GSD IV typically survive only a few months. (medlineplus.gov)
- 121 Mendelian pathogenic or likely pathogenic variants associated with 31 inherited diseases were detected, among these hearing loss, congenital hypothyroidism, methylmalonic acidemia, methylmalonic acidemia with homocystinuria, phenylketonuria(PKU) and benign hyperphenylalaninemia accounted for half of the carrier variants. (researchsquare.com)
Neuromuscular6
- The fatal perinatal neuromuscular type is the most severe form of GSD IV, with signs developing before birth. (medlineplus.gov)
- Infants with the fatal perinatal neuromuscular type of GSD IV have very low muscle tone (severe hypotonia) and muscle wasting (atrophy). (medlineplus.gov)
- The childhood neuromuscular type of GSD IV develops in late childhood and is characterized by myopathy and dilated cardiomyopathy. (medlineplus.gov)
- Individuals with the fatal perinatal neuromuscular type tend to produce less than 5 percent of usable enzyme, while those with the childhood neuromuscular type may have around 20 percent of enzyme function. (medlineplus.gov)
- The childhood neuromuscular subtype is rare and the course is variable, ranging from onset in the second decade with a mild disease course to a more severe, progressive course resulting in death in the third decade. (nih.gov)
- This management guideline specifically addresses evaluation and diagnosis across multiple organ systems (cardiovascular, gastrointestinal/nutrition, hepatic, musculoskeletal, and neuromuscular) involved in glycogen storage disease type III. (qxmd.com)
Hepatic10
- The progressive hepatic type is the most common form of GSD IV. (medlineplus.gov)
- Children with the progressive hepatic type of GSD IV often die of liver failure in early childhood. (medlineplus.gov)
- The non-progressive hepatic type of GSD IV has many of the same features as the progressive hepatic type, but the liver disease is not as severe. (medlineplus.gov)
- In the non-progressive hepatic type, hepatomegaly and liver disease are usually evident in early childhood, but affected individuals typically do not develop cirrhosis. (medlineplus.gov)
- Patient complaints probably relate to end-organ injuries of Andersen disease, such as hepatic failure, cardiomyopathy, or muscular atrophy. (medscape.com)
- Aynsley-Green A, Williamson DH, Gitzelmann R. Asymptomatic hepatic glycogen-synthetase deficiency. (medscape.com)
- Hepatic glycogen synthetase deficiency. (medscape.com)
- Effect of growth hormone treatment on hypoglycemia in a patient with both hepatic glycogen synthase and isolated growth hormone deficiencies. (medscape.com)
- Glucose homeostasis in adulthood and in pregnancy in a patient with hepatic glycogen synthetase deficiency. (medscape.com)
- Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. (medscape.com)
Synthase deficiency4
- Spiegel R, Mahamid J, Orho-Melander M, Miron D, Horovitz Y. The variable clinical phenotype of liver glycogen synthase deficiency. (medscape.com)
- Liver glycogen synthase deficiency: a rarely diagnosed entity. (medscape.com)
- Laberge AM, Mitchell GA, van de Werve G. Long-term follow-up of a new case of liver glycogen synthase deficiency. (medscape.com)
- Rutledge SL, Atchison J, Bosshard NU, Steinmann B. Case report: liver glycogen synthase deficiency--a cause of ketotic hypoglycemia. (medscape.com)
Abnormal glycogen3
- Abnormal glycogen molecules called polyglucosan bodies accumulate in cells, leading to damage and cell death. (medlineplus.gov)
- Other GSDs do not have this abnormal glycogen structure. (medscape.com)
- For types I, III, and VI, symptoms are low levels of sugar in the blood ( hypoglycemia ) and protrusion of the abdomen (because excess or abnormal glycogen may enlarge the liver). (msdmanuals.com)
GSDs3
- Although at least 14 unique GSDs are discussed in the literature, the four that cause clinically significant muscle weakness are Pompe disease ( GSD type II , acid maltase deficiency), Cori disease ( GSD type III , debranching enzyme deficiency), McArdle disease ( GSD type V , myophosphorylase deficiency), and Tarui disease ( GSD type VII , phosphofructokinase deficiency). (medscape.com)
- In general, no specific treatment exists to cure glycogen storage diseases (GSDs). (medscape.com)
- Glycogen storage diseases (GSDs) are a group of inborn errors of metabolism, typically caused by enzyme defects, resulting in a buildup of glycogen in the liver, muscles, and other organs. (arupconsult.com)
Clinical15
- The clinical manifestations of glycogen storage disease type IV (GSD IV) discussed in this entry span a continuum of different subtypes with variable ages of onset, severity, and clinical features. (nih.gov)
- Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. (medscape.com)
- Biochemical and molecular investigation of two Korean patients with glycogen storage disease type III" Clinical Chemistry and Laboratory Medicine , vol. 46, no. 9, 2008, pp. 1245-1249. (degruyter.com)
- Glycogen storage disease type IV has a broad clinical spectrum ranging from a perinatal lethal form to a nonprogressive later-onset disease in adults. (nih.gov)
- Heterozygotes usually do not manifest clinical features of the disease. (medscape.com)
- Glycogen storage disease type III is a rare disease of variable clinical severity affecting primarily the liver, heart, and skeletal muscle. (qxmd.com)
- Glycogen storage disease type III manifests a wide clinical spectrum. (qxmd.com)
- Please note: It is possible that disease signs similar to the ones caused by the GSD mutation could develop due to a different genetic or clinical cause. (wisdompanel.com)
- The disease presents with both clinical and biochemical heterogeneity consistent with the existence of two major subgroups, GSD-1a and GSD-1b, which have been confirmed at the molecular genetic level. (qxmd.com)
- Clinical features similar to GSD type V. Temporary weakness and painful muscle cramps occur after exercise. (mhmedical.com)
- Three clinical forms have been described: classic, infantile onset, and late onset type. (mhmedical.com)
- Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. (medscape.com)
- Soni D, Shukla G, Singh S, Goyal V, Behari M. Cardiovascular and sudomotor autonomic dysfunction in Wilson's disease--limited correlation with clinical severity. (medscape.com)
- This classification is fundamental to the type of marketing authorization (MA), and therefore to the controls to be performed, from preclinical stages through clinical trials to pharmacovigilance, to meet the safety requirements for patients. (frontiersin.org)
- Hepatomegaly is the clinical hallmark of disease. (lu.se)
Degradation3
- It is caused by deficient activity of glycogen debranching enzyme, which is a key enzyme in glycogen degradation. (qxmd.com)
- glucose cannot be used as a source of energy and glycogen accumulates because of impaired degradation and/or excess synthesis. (mhmedical.com)
- Protein targeting to glycogen (PTG) is a scaffolding protein that targets protein phosphatase 1α (PP1α) to glycogen, and links it to enzymes involved in glycogen synthesis and degradation. (jci.org)
McArdle4
- These inherited enzyme defects usually present in childhood, although some, such as McArdle disease and Pompe disease, have separate adult-onset forms. (medscape.com)
- Myophosphorylase, the deficient enzyme in McArdle disease, is found in muscle tissue. (medscape.com)
- One hallmark of McArdle disease is weakness with exertion. (medscape.com)
- Pharmacological and Nutritional Treatment for McArdle Disease (Glycogen Storage Disease Type V)." Evidence-Based Medicine Guidelines , Duodecim Medical Publications Limited, 2019. (unboundmedicine.com)
Pompe Disease12
- Pompe disease). (medscape.com)
- Enzyme replacement therapies are available for all age groups (ie, infantile [early onset] or late onset [juvenile/adult]) affected by Pompe disease. (medscape.com)
- Replaces rhGAA, which is deficient or lacking in persons with Pompe disease. (medscape.com)
- Myozyme has been shown to improve ventilator-free survival in patients with infantile-onset Pompe disease compared with untreated historical controls. (medscape.com)
- It has not been adequately studied for treatment of other forms of Pompe disease. (medscape.com)
- Lumizyme is indicated for infantile-onset Pompe disease and also for late (non-infantile) Pompe disease. (medscape.com)
- Indicated for treatment of patients aged 1 year and older with late-onset Pompe disease. (medscape.com)
- Indicated in combination with miglustat (Opfolda) for adults with late-onset Pompe disease (lysosomal acid alpha-glucosidase [GAA] deficiency) who weigh ≥40 kg and are not improving on their current enzyme replacement therapy (ERT). (medscape.com)
- Pompe disease: early diagnosis and early treatment make a difference. (medscape.com)
- A French multicenter Phase 4 open label extension study of long -term safety and efficacy in patients with Pompe disease who previously participated in avalglucosidase development studies in France. (institut-myologie.org)
- to investigate nursing team knowledge and practices regarding care for children with Pompe disease in intensive care. (bvsalud.org)
- Pompe Disease (PD) was discovered in 1932 by pathologist Joannes Cassianus Pompe, during the autopsy of a seven-month-old child who died from idiopathic myocardial hypertrophy. (bvsalud.org)
Glycogenosis1
- Glycogenosis type VII. (mhmedical.com)
Disorder8
- Glycogen storage disease type IV (GSD IV) is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body's cells. (medlineplus.gov)
- People with this type of the disorder can also have hypotonia and muscle weakness (myopathy). (medlineplus.gov)
- Generally, the severity of the disorder is linked to the amount of functional glycogen branching enzyme that is produced. (medlineplus.gov)
- Glycogen Storage Disease Type 1b (GSD1b) is a rare genetic disorder that has a huge impact on patients' lives and the lives of their families. (sciani.com)
- RBCK1-related disease: A rare multisystem disorder with polyglucosan storage, auto-inflammation, recurrent infections, skeletal, and cardiac myopathy-Four additional patients and a review of the current literature. (nih.gov)
- Glycogen Storage Disease Type Ia (GSD Ia) is a severe metabolic disorder causing critically low blood sugar levels and liver enlargement. (wisdompanel.com)
- von Willebrand's Disease (vWD) type 1 is a clotting disorder that usually causes mild bleeding tendencies in affected dogs though some may have more severe signs. (wisdompanel.com)
- NORD is not a medical provider or health care facility and thus can neither diagnose any disease or disorder nor endorse or recommend any specific medical treatments. (rarediseases.org)
Tyrosinemia1
- Reversibility of cirrhotic regenerative liver nodules upon NTBC treatment in a child with tyrosinemia type I. Acta Paediatr. (springer.com)
Autosomal1
- This disease is autosomal recessive meaning that two copies of the mutation are needed for disease signs to occur. (wisdompanel.com)
Genetics2
- Molecular Genetics of Type 1 Glycogen Storage Diseases. (qxmd.com)
- Kieffer DA, Medici V. Wilson disease: at the crossroads between genetics and epigenetics-A review of the evidence. (medscape.com)
Lipid2
- These data suggest that PTG plays a critical role in glycogen synthesis and is necessary to maintain the appropriate metabolic balance for the partitioning of fuel substrates between glycogen and lipid. (jci.org)
- The metabolic myopathies (MM) are a group of muscle disorders resulting from failed energy production related to defects in glycogen, lipid or mitochondrial metabolism. (pediatriconcall.com)
Diagnosis12
- The diagnosis is established in a proband by the demonstration of glycogen branching enzyme (GBE) deficiency in liver, muscle, or skin fibroblasts or the identification of biallelic pathogenic variants in GBE1 on molecular genetic testing. (nih.gov)
- If the GBE1 pathogenic variants have been identified in an affected family member, test at-risk relatives to allow for early diagnosis and management of disease manifestations. (nih.gov)
- Lin CY, Hwang B, Hsiao KJ, Jin YR. Pompe's disease in Chinese and prenatal diagnosis by determination of alpha-glucosidase activity. (medscape.com)
- Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. (medscape.com)
- Glycogen storage disease type III diagnosis and management guidelines. (qxmd.com)
- This guideline for the management of glycogen storage disease type III was developed as an educational resource for health care providers to facilitate prompt and accurate diagnosis and appropriate management of patients. (qxmd.com)
- A guideline that will facilitate the accurate diagnosis and appropriate management of individuals with glycogen storage disease type III was developed. (qxmd.com)
- This guideline will help health care providers recognize patients with all forms of glycogen storage disease type III, expedite diagnosis, and minimize stress and negative sequelae from delayed diagnosis and inappropriate management. (qxmd.com)
- The diagnosis and management of the acutely ill child with suspected metabolic disease can present a formidable challenge to even the most astute clinician. (springer.com)
- Diagnosis and treatment of Wilson disease: an update. (medscape.com)
- Schilsky ML. Wilson disease: diagnosis, treatment, and follow-up. (medscape.com)
- Most articles standardize to single equal sign top level parent headings: Disease Entity, Diagnosis, Management, Additional Resources, References that are more responsive to mobile friendly style changes. (aao.org)
Buildup1
- Because of the glycogen buildup, GSD I patients typically present with enlarged livers from non-alcoholic fatty liver disease. (wikipedia.org)
Enzymes4
- Together with phosphorylases , debranching enzymes mobilize glucose reserves from glycogen deposits in the muscles and liver. (wikidoc.org)
- Proteins that catalyze both functions are referred to as glycogen debranching enzymes (GDEs). (wikidoc.org)
- When glucosyltransferase and glucosidase are catalyzed by distinct enzymes, "glycogen debranching enzyme" usually refers to the glucosidase enzyme . (wikidoc.org)
- Normally your enzymes break carbohydrates down into glucose (a type of sugar). (medlineplus.gov)
Biochemical1
- Metabolic disease may present in a fulminate fashion to the pediatric intensivist with profound biochemical disturbances, encephalopathy and even cardiac failure. (springer.com)
Tissues1
- One of the four glycogen storage diseases characterized by phosphofructokinase deficiency in the muscles and associated with abnormal deposition of glycogen in muscle tissues, exercise intolerance, and anemia. (mhmedical.com)
Hypoglycemia5
- Because glycogenolysis is the principal metabolic mechanism by which the liver supplies glucose to the body during fasting, both deficiencies cause severe hypoglycemia and, over time, excess glycogen storage in the liver and (in some cases) in the kidneys. (wikipedia.org)
- However, after birth, the inability to maintain blood glucose from stored glycogen in the liver causes measurable hypoglycemia in no more than 1-2 hours after feedings. (wikipedia.org)
- Fasting blood glucose testing is indicated because hypoglycemia sometimes can be found in some types of GSD. (medscape.com)
- Meticulous adherence to a dietary regimen to maintain a euglycemic state and prevent the formation of excessive glycogen may reduce the liver size, prevent hypoglycemia, reduce symptoms, and allow growth and development. (medscape.com)
- Individuals with glycogen storage disease type III present with hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation. (qxmd.com)
Cleave1
- It binds to mannose-6-phosphate receptors and then is transported into lysosomes, then undergoes proteolytic cleavage that results in increased enzymatic activity and ability to cleave glycogen. (medscape.com)
GSD1B2
- Sophie's Hope Foundation and CureGSD1b, in partnership with Sanguine Biosciences, is reaching out to raise awareness about an at-home research opportunity for adult patients and children diagnosed with Glycogen storage disease type 1B (GSD1B). (curegsd1b.org)
- Over the past 18 months we have worked closely with doctors, researchers, drug developers, policy makers and other rare disease organizations to get a better understanding of how we move the needle forward for GSD1b care and new treatments. (curegsd1b.org)
Gierke2
- The disease was named after German doctor Edgar von Gierke, who first described it in 1929. (wikipedia.org)
- The von Gierke disease (GSD type Ia, glucose-6-phosphatase deficiency) causes clinically significant end-organ disease with substantial morbidity. (medscape.com)
Liver disease6
- Children with this type develop a form of liver disease called cirrhosis that often is irreversible. (medlineplus.gov)
- however, they are likely to survive without progression of the liver disease and may not show cardiac, skeletal muscle, or neurologic involvement. (nih.gov)
- Those with type IIIa have symptoms related to liver disease and progressive muscle (cardiac and skeletal) involvement that varies in age of onset, rate of disease progression, and severity. (qxmd.com)
- Those with type IIIb primarily have symptoms related to liver disease. (qxmd.com)
- Other individuals have a multitude of the most severe symptoms of end-stage liver disease and a limited chance for survival. (medscape.com)
- Copper: its role in the pathogenesis of liver disease. (medscape.com)
MeSH1
- Glycogen Storage Disease Type V" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (sdsu.edu)
Blood-glucose levels1
- Glycogen breakdown is highly regulated in the body, especially in the liver , by various hormones including insulin and glucagon , to maintain a homeostatic balance of blood-glucose levels. (wikidoc.org)
Rare diseases2
- Rare-X is a collaborative platform for global data sharing and analysis to accelerate treatments for rare diseases. (curegsd1b.org)
- Rare Disease PHGKB is an online, continuously updated, searchable database of published scientific literature, CDC and NIH resources, and other information that address the public health impact and translation of genomic and other precision health discoveries into improved health outcomes related to rare diseases. (cdc.gov)
Maple Syrup U1
- Maple syrup urine disease: it has come a long way. (springer.com)
Acid maltase de1
- Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. (medscape.com)
Proteins1
- Carbohydrates Carbohydrates, proteins, and fats are the main types of macronutrients in food (nutrients that are required daily in large quantities). (msdmanuals.com)
Protein2
- Ricin is a potent (type 2) ribosome-inactivating protein toxin produced in the seeds of the castor bean plant Ricinus communis . (mdpi.com)
- Genes Genes are segments of deoxyribonucleic acid (DNA) that contain the code for a specific protein that functions in one or more types of cells in the body or the code for functional ribonucleic. (msdmanuals.com)
Deficient1
- Gordon N. Classic diseases revisited: carbohydrate-deficient glycoprotein syndromes. (springer.com)
Cirrhosis1
- Specific medical therapies may be applied to many liver diseases in an effort to diminish symptoms and to prevent or forestall the development of cirrhosis. (medscape.com)
Fatigue1
- Fatigue develops when the glycogen supply is exhausted. (medscape.com)
Centers for Diseas2
Skeletal1
- These mice have reduced glycogen stores in adipose tissue, liver, heart, and skeletal muscle, corresponding with decreased glycogen synthase activity and glycogen synthesis rate. (jci.org)
IIIa1
- Glycogen storage disease type IIIa in curly-coated retrievers. (wisdompanel.com)
Severe2
- To test this hypothesis, plasma serotonin concentrations were prospectively measured in 13 patients with type-Ia glycogen-storage disease, one patient with severe pulmonary hypertension and type-Ia glycogen-storage disease, 16 patients displaying severe pulmonary arterial hypertension, and 26 normal healthy controls. (lincoln.ac.uk)
- It is concluded that type-Ia glycogen-storage disease may be another condition in which abnormal handling of serotonin is one event in a multistep process leading to severe pulmonary arterial hypertension. (lincoln.ac.uk)
Abnormalities1
- Chitkara DK, Nurko S, Shoffner JM, Buie T, Flores A. Abnormalities in gastrointestinal motility are associated with diseases of oxidative phosphorylation in children. (springer.com)