Glycogen Storage Disease Type III
Glycogen Debranching Enzyme System
Glycogen Storage Disease Type I
Glycogen Storage Disease
Glycogen Storage Disease Type IV
Glycogen Storage Disease Type II
Glycogen Storage Disease Type VII
Glucose-6-Phosphatase
Glycogen Storage Disease Type VI
Glycogen
alpha-Glucosidases
Glycogen Storage Disease Type V
Glycogen Storage Disease Type VIII
Glucan 1,4-alpha-Glucosidase
Antiporters
Glucose-6-Phosphate
1,4-alpha-Glucan Branching Enzyme
Glycogen Storage Disease Type IIb
Fructose-1,6-Diphosphatase Deficiency
Enzyme Replacement Therapy
Different clinical aspects of debrancher deficiency myopathy. (1/32)
OBJECTIVE: To characterise the main clinical phenotypes of debrancher deficiency myopathy and to increase awareness for this probably underdiagnosed disorder. METHODS: The diagnosis of debrancher deficiency was established by laboratory tests, EMG, and muscle and liver biopsy. RESULTS: Four patients with debrancher deficiency myopathy were identified in the Tyrol, a federal state of Austria with half a million inhabitants. Clinical appearance was highly variable. The following phenotypes were differentiated: (1) adult onset distal myopathy; (2) subacute myopathy of the respiratory muscles; (3) severe generalised myopathy; and (4) minimal variant myopathy. Exercise intolerance was uncommon. The clinical course was complicated by advanced liver dysfunction in two patients and by severe cardiomyopathy in one. All had raised creatine kinase concentrations (263 to 810 U/l), myogenic and neurogenic features on EMG, and markedly decreased debrancher enzyme activities in muscle or liver biopsy specimens. The findings were substantiated by a review of 79 previously published cases with neuromuscular debrancher deficiency. CONCLUSIONS: This study illustrates the heterogeneity of neuromuscular manifestations in debrancher deficiency. Based on the clinical appearance, age at onset, and course of disease four phenotypes may be defined which differ in prognosis, frequency of complications, and response to therapy. (+info)Liver transplantation for glycogen storage disease types I, III, and IV. (2/32)
Glycogen storage disease (GSD) types I, III, and IV can be associated with severe liver disease. The possible development of hepatocellular carcinoma and/or hepatic failure make these GSDs potential candidates for liver transplantation. Early diagnosis and initiation of effective dietary therapy have dramatically improved the outcome of GSD type I by reducing the incidence of liver adenoma and renal insufficiency. Nine type I and 3 type III patients have received liver transplants because of poor metabolic control, multiple liver adenomas, or progressive liver failure. Metabolic abnormalities were corrected in all GSD type I and type III patients, while catch-up growth was reported only in two patients. Whether liver transplantation results in reversal and/or prevention of renal disease remains unclear. Neutropenia persisted in both GSDIb patients post liver transplantation necessitating continuous granulocyte colony stimulating factor treatment. Thirteen GSD type IV patients were liver transplanted because of progressive liver cirrhosis and failure. All but one patient have not had neuromuscular or cardiac complications during follow-up periods for as long as 13 years. Four have died within a week and 5 years after transplantation. Caution should be taken in selecting GSD type IV candidates for liver transplantation because of the variable phenotype, which may include life-limiting extrahepatic manifestations. It remains to be evaluated, whether a genotype-phenotype correlation exists for GSD type IV, which may aid in the decision making. CONCLUSION: Liver transplantation should be considered for patients with glycogen storage disease who have developed liver malignancy or hepatic failure, and for type IV patients with the classical and progressive hepatic form. (+info)Molecular genetic basis and prevalence of glycogen storage disease type IIIA in the Faroe Islands. (3/32)
Glycogen storage disease type IIIA (GSD IIIA) is caused by mutations of the amyloglucosidase gene (AGL). For most populations, none of the AGL mutations described to date is particularly frequent. In this paper, we report that six children with GSD IIIA from the Faroe Islands were found to be homozygous for the novel nonsense mutation c.1222C>T (R408X) of the AGL gene. This mutation is easily detected by restriction enzyme digest with NsiI after mismatch PCR. Investigating five intragenic polymorphisms, we could show that this mutation was always associated with the same haplotype. The c.1222C>T mutation could be detected on two chromosomes of another 50 unselected GSD IIIA patients of other European or North American origin which means that this mutation plays a minor role worldwide. From the fact that we are currently aware of a total of 14 GSD IIIA cases in the Faroese population of 45 000, the observed prevalence is 1 : 3100. While the novel AGL mutation c.1222C>T was not detectable among 198 German newborns, nine out of 272 children from the Faroese neonatal screening program were found to be heterozygous for this mutation. Thus, the calculated prevalence is 1 : 3600 (95% CI 1:700-1:6400). We conclude that due to a founder effect, the Faroe Islands have the highest prevalence of GSD IIIA world-wide. The detection of the molecular defect has facilitated the diagnosis and has offered the opportunity for prenatal diagnosis in this patient group. (+info)Liver transplantation-associated hypercalcemia followed by acute renal dysfunction. (4/32)
A 34-year-old woman with liver insufficiency due to glycogen storage disease III underwent a living spousal liver transplantation. Soon after the successful operation, moderate hypercalcemia along with hyperbilirubinemia emerged without clarified reasons. The hypercalcemia persisted for over a month despite calcitonin treatment and the serum calcium level surged to 13.2 mg/dl with albumin correction. Renal dysfunction was indicated by an acute increase in serum creatinine (approximately 0.8 to approximately 2.8 mg/ml), which was assumed to be hypercalcemia-induced and was effectively treated with bisphosphonate, pamidronate (30 mg, i.v.). Recent topics related to transplantation-associated hypercalcemia are discussed. (+info)Glycogen storage disease type III in Inuit children. (5/32)
Glycogen storage disease type III (GSD III) was diagnosed in 4 Inuit children (3 confirmed, 1 suspected case) at our institution over the last decade. This rare autosomal recessive disease, which results from a deficiency of the debranching enzyme required for complete degradation of the glycogen molecule, has not been previously described in this population. The possible clinical presentations are heterogeneous, as is the spectrum of severity of this disease. The long-term sequelae can be severe, including recurrent hypoglycemia, hepatic cirrhosis and progressive muscle weakness. These 4 cases would suggest an increased prevalence of GSD III in the Inuit population. Therefore, it is important for health care providers caring for this population to consider and recognize this rare but serious disease. (+info)Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? (6/32)
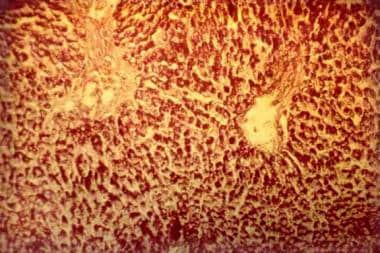
BACKGROUND/AIMS: Glycogen storage disease III (GSD III) is caused by a deficiency of glycogen-debranching enzyme which causes an incomplete glycogenolysis resulting in glycogen accumulation with abnormal structure (short outer chains resembling limit dextrin) in liver and muscle. Hepatic involvement is considered mild, self-limiting and improves with age. With increased survival, a few cases of liver cirrhosis and hepatocellular carcinoma (HCC) have been reported. METHODS: A systematic review of 45 cases of GSD III at our center (20 months to 67 years of age) was reviewed for HCC, 2 patients were identified. A literature review of HCC in GSD III was performed and findings compared to our patients. CONCLUSIONS: GSD III patients are at risk for developing HCC. Cirrhosis was present in all cases and appears to be responsible for HCC transformation There are no reliable biomarkers to monitor for HCC in GSD III. Systematic evaluation of liver disease needs be continued in all patients, despite lack of symptoms. Development of guidelines to allow for systematic review and microarray studies are needed to better delineate the etiology of the hepatocellular carcinoma in patients with GSD III. (+info)Long-term results of living donor liver transplantation for glycogen storage disorders in children. (7/32)
Liver transplantation (LT) may be indicated in glycogen storage disorders (GSD) when medical treatment fails to control the metabolic problems or when hepatic adenomas develop. We present our institutional experience with living donor LT (LDLT) for children with GSD. A total of 244 patients underwent primary LDLT at our institution from June 1994 to December 2005. A total of 12 (5%) children (8 female and 4 male) were afflicted with GSD and were not responsive to medical treatment. Nine patients had GSD type I and 3 had GSD type III. The median age at the time of transplantation was 7.27 yr (range, 2.4-15.7). All patients presented with metabolic abnormalities, including hypoglycemia, and lactic acidosis. In addition, 4 patients presented with growth retardation. A total of 11 patients received left lobe grafts and 1 received a right lobe graft. The mean graft-to-recipient weight ratio was 1.25 (range, 0.89-1.61). Two patients had hepatic vein stenoses that were treated by balloon dilatation; 1 patient had bile leak, which settled spontaneously. The overall surgical morbidity rate was 25%. Three patients had hepatic adenomas in the explanted liver. There was a single mortality at 2 months posttransplantation due to acute pancreatitis and sepsis. The mean follow up was 47.45 months. The metabolic abnormalities were corrected and renal function remained normal. In patients with growth retardation, catch-up growth was achieved posttransplantation. In conclusion, LDLT is a viable option to restore normal metabolic balance in patients with GSD when medical treatment fails. Long-term follow-up after LT for GSD shows excellent graft and patient survival. (+info)A Japanese patient with cardiomyopathy caused by a novel mutation R285X in the AGL gene. (8/32)
Left ventricular hypertrophy (LVH) is primarily or secondarily caused by a cardiovascular or systemic disease. The pattern of LVH is distinctive in hypertrophic or metabolic cardiomyopathy and differs from that seen in LVH caused by hypertension or aortic stenosis. A 42-year-old Japanese man had LVH similar to that with hypertrophic cardiomyopathy. The patient was diagnosed with glycogen storage disease type IIIa (GSD-IIIa). Echocardiography showed that he had severe LVH, and concomitant hepatomegaly and hypoglycemia, which led to measurement of glycogen debranching enzyme (GDE) activity; it was undetectable. Sequence analysis of the AGL gene encoding GDE showed a novel nonsense mutation: a C-to-T transition at codon 285 in exon 8, resulting in substitution of the arginine codon by the stop codon (R285X). The patient was homozygous for the mutation. Cardiomyopathy in this patient was caused by a nonsense mutation in the AGL gene. Five other Japanese GSD-IIIa patients over 30 years of age have all presented with cardiomyopathy, as well as hepatomegaly and hypoglycemia. Patients with LVH associated with hepatomegaly and hypoglycemia should undergo biochemical and genetic analyses for GSD-IIIa. (+info)Glycogen Storage Disease Type III, also known as Cori or Forbes disease, is a rare inherited metabolic disorder caused by deficiency of the debranching enzyme amylo-1,6-glucosidase, which is responsible for breaking down glycogen in the liver and muscles. This results in an abnormal accumulation of glycogen in these organs leading to its associated symptoms.
There are two main types: Type IIIa affects both the liver and muscles, while Type IIIb affects only the liver. Symptoms can include hepatomegaly (enlarged liver), hypoglycemia (low blood sugar), hyperlipidemia (high levels of fats in the blood), and growth retardation. In Type IIIa, muscle weakness and cardiac problems may also occur.
The diagnosis is usually made through biochemical tests and genetic analysis. Treatment often involves dietary management with frequent meals to prevent hypoglycemia, and in some cases, enzyme replacement therapy. However, there is no cure for this condition and life expectancy can be reduced depending on the severity of the symptoms.
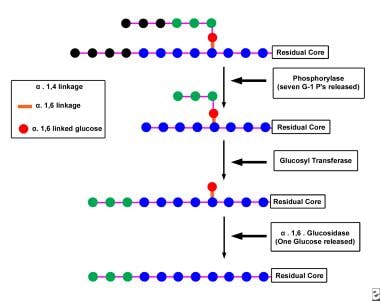
The Glycogen Debranching Enzyme System, also known as glycogen debranching enzyme or Amy-1, is a crucial enzyme complex in human biochemistry. It plays an essential role in the metabolism of glycogen, which is a large, branched polymer of glucose that serves as the primary form of energy storage in animals and fungi.
The Glycogen Debranching Enzyme System consists of two enzymatic activities: a transferase and an exo-glucosidase. The transferase activity transfers a segment of a branched glucose chain to another part of the same or another glycogen molecule, while the exo-glucosidase activity cleaves the remaining single glucose units from the outer branches of the glycogen molecule.
This enzyme system is responsible for removing the branched structures of glycogen, allowing the linear chains to be further degraded by other enzymes into glucose molecules that can be used for energy production or stored for later use. Defects in this enzyme complex can lead to several genetic disorders, such as Glycogen Storage Disease Type III (Cori's disease) and Type IV (Andersen's disease), which are characterized by the accumulation of abnormal glycogen molecules in various tissues.
Glycogen Storage Disease Type I (GSD I) is a rare inherited metabolic disorder caused by deficiency of the enzyme glucose-6-phosphatase, which is necessary for the liver to release glucose into the bloodstream. This leads to an accumulation of glycogen in the liver and abnormally low levels of glucose in the blood (hypoglycemia).
There are two main subtypes of GSD I: Type Ia and Type Ib. In Type Ia, there is a deficiency of both glucose-6-phosphatase enzyme activity in the liver, kidney, and intestine, leading to hepatomegaly (enlarged liver), hypoglycemia, lactic acidosis, hyperlipidemia, and growth retardation. Type Ib is characterized by a deficiency of glucose-6-phosphatase enzyme activity only in the neutrophils, leading to recurrent bacterial infections.
GSD I requires lifelong management with frequent feedings, high-carbohydrate diet, and avoidance of fasting to prevent hypoglycemia. In some cases, treatment with continuous cornstarch infusions or liver transplantation may be necessary.
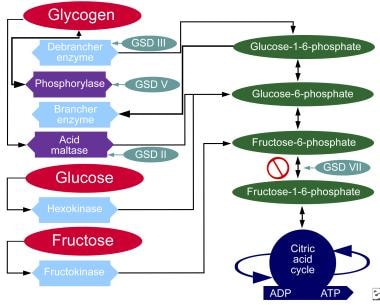
Glycogen storage disease (GSD) is a group of rare inherited metabolic disorders that affect the body's ability to break down and store glycogen, a complex carbohydrate that serves as the primary form of energy storage in the body. These diseases are caused by deficiencies or dysfunction in enzymes involved in the synthesis, degradation, or transport of glycogen within cells.
There are several types of GSDs, each with distinct clinical presentations and affected organs. The most common type is von Gierke disease (GSD I), which primarily affects the liver and kidneys. Other types include Pompe disease (GSD II), McArdle disease (GSD V), Cori disease (GSD III), Andersen disease (GSD IV), and others.
Symptoms of GSDs can vary widely depending on the specific type, but may include:
* Hypoglycemia (low blood sugar)
* Growth retardation
* Hepatomegaly (enlarged liver)
* Muscle weakness and cramping
* Cardiomyopathy (heart muscle disease)
* Respiratory distress
* Developmental delays
Treatment for GSDs typically involves dietary management, such as frequent feedings or a high-protein, low-carbohydrate diet. In some cases, enzyme replacement therapy may be used to manage symptoms. The prognosis for individuals with GSDs depends on the specific type and severity of the disorder.
Glycogen Storage Disease Type IV (GSD IV), also known as Andersen's disease, is a rare inherited metabolic disorder that affects the body's ability to break down glycogen, a complex carbohydrate that serves as a source of energy for the body.
In GSD IV, there is a deficiency in the enzyme called glycogen branching enzyme (GBE), which is responsible for adding branches to the glycogen molecule during its synthesis. This results in an abnormal form of glycogen that accumulates in various organs and tissues, particularly in the liver, heart, and muscles.
The accumulation of this abnormal glycogen can lead to progressive damage and failure of these organs, resulting in a variety of symptoms such as muscle weakness, hypotonia, hepatomegaly (enlarged liver), cardiomyopathy (heart muscle disease), and developmental delay. The severity of the disease can vary widely, with some individuals experiencing milder symptoms while others may have a more severe and rapidly progressing form of the disorder.
Currently, there is no cure for GSD IV, and treatment is focused on managing the symptoms and slowing down the progression of the disease. This may include providing nutritional support, addressing specific organ dysfunction, and preventing complications.
Glycogen Storage Disease Type II, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar that serves as energy storage, within lysosomes. When GAA is deficient, glycogen accumulates in various tissues, particularly in muscle cells, leading to their dysfunction and damage.
The severity of Pompe Disease can vary significantly, depending on the amount of functional enzyme activity remaining. The classic infantile-onset form presents within the first few months of life with severe muscle weakness, hypotonia, feeding difficulties, and respiratory insufficiency. This form is often fatal by 1-2 years of age if left untreated.
A later-onset form, which can present in childhood, adolescence, or adulthood, has a more variable clinical course. Affected individuals may experience progressive muscle weakness, respiratory insufficiency, and cardiomyopathy, although the severity and rate of progression are generally less pronounced than in the infantile-onset form.
Enzyme replacement therapy with recombinant human GAA is available for the treatment of Pompe Disease and has been shown to improve survival and motor function in affected individuals.
Glycogen Storage Disease Type VII, also known as Tarui's disease, is a rare inherited metabolic disorder caused by a deficiency of the enzyme phosphofructokinase (PFK), which is required for glycogenolysis – the breakdown of glycogen to glucose-1-phosphate and ultimately into glucose. This enzyme deficiency results in the accumulation of glycogen, particularly in muscle and red blood cells, leading to symptoms such as exercise-induced muscle cramps, myoglobinuria (the presence of myoglobin in the urine), and hemolytic anemia. The disease can also cause muscle weakness, fatigue, and dark-colored urine after strenuous exercise. It is inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
Glucose-6-phosphatase is an enzyme that plays a crucial role in the regulation of glucose metabolism. It is primarily located in the endoplasmic reticulum of cells in liver, kidney, and intestinal mucosa. The main function of this enzyme is to remove the phosphate group from glucose-6-phosphate (G6P), converting it into free glucose, which can then be released into the bloodstream and used as a source of energy by cells throughout the body.
The reaction catalyzed by glucose-6-phosphatase is as follows:
Glucose-6-phosphate + H2O → Glucose + Pi (inorganic phosphate)
This enzyme is essential for maintaining normal blood glucose levels, particularly during periods of fasting or starvation. In these situations, the body needs to break down stored glycogen in the liver and convert it into glucose to supply energy to the brain and other vital organs. Glucose-6-phosphatase is a key enzyme in this process, allowing for the release of free glucose into the bloodstream.
Deficiencies or mutations in the gene encoding glucose-6-phosphatase can lead to several metabolic disorders, such as glycogen storage disease type I (von Gierke's disease) and other related conditions. These disorders are characterized by an accumulation of glycogen and/or fat in various organs, leading to impaired glucose metabolism, growth retardation, and increased risk of infection and liver dysfunction.
Glycogen Storage Disease Type VI, also known as Hers disease, is a rare inherited metabolic disorder caused by deficiency of the liver enzyme called glycogen phosphorylase. This enzyme is responsible for breaking down glycogen, which is a stored form of glucose, into glucose-1-phosphate during the process of glycogenolysis.
In GSD Type VI, the lack of this enzyme leads to an abnormal accumulation of glycogen in the liver, causing hepatomegaly (enlarged liver) and elevated liver enzymes. The symptoms of this condition are usually milder compared to other types of GSD, and may include fatigue, weakness, and hypoglycemia (low blood sugar), especially after prolonged fasting or physical exertion.
The diagnosis of GSD Type VI is typically made through biochemical tests that measure the activity of the glycogen phosphorylase enzyme in liver tissue, as well as genetic testing to identify mutations in the gene responsible for the enzyme's production. Treatment may involve dietary management, such as frequent feeding and avoidance of prolonged fasting, to prevent hypoglycemia. In some cases, medication may be necessary to manage symptoms and prevent complications.
Glycogen is a complex carbohydrate that serves as the primary form of energy storage in animals, fungi, and bacteria. It is a polysaccharide consisting of long, branched chains of glucose molecules linked together by glycosidic bonds. Glycogen is stored primarily in the liver and muscles, where it can be quickly broken down to release glucose into the bloodstream during periods of fasting or increased metabolic demand.
In the liver, glycogen plays a crucial role in maintaining blood glucose levels by releasing glucose when needed, such as between meals or during exercise. In muscles, glycogen serves as an immediate energy source for muscle contractions during intense physical activity. The ability to store and mobilize glycogen is essential for the proper functioning of various physiological processes, including athletic performance, glucose homeostasis, and overall metabolic health.
Alpha-glucosidases are a group of enzymes that break down complex carbohydrates into simpler sugars, such as glucose, by hydrolyzing the alpha-1,4 and alpha-1,6 glycosidic bonds in oligosaccharides, disaccharides, and polysaccharides. These enzymes are located on the brush border of the small intestine and play a crucial role in carbohydrate digestion and absorption.
Inhibitors of alpha-glucosidases, such as acarbose and miglitol, are used in the treatment of type 2 diabetes to slow down the digestion and absorption of carbohydrates, which helps to reduce postprandial glucose levels and improve glycemic control.
Glycogen Storage Disease Type V, also known as McArdle's disease, is a genetic disorder that affects the body's ability to break down glycogen, a complex carbohydrate stored in muscles, into glucose, which provides energy for muscle contraction.
This condition results from a deficiency of the enzyme myophosphorylase, which is responsible for breaking down glycogen into glucose-1-phosphate within the muscle fibers. Without sufficient myophosphorylase activity, muscles become easily fatigued and may cramp or become rigid during exercise due to a lack of available energy.
Symptoms typically appear in childhood or adolescence and can include muscle weakness, stiffness, cramps, and myoglobinuria (the presence of myoglobin, a protein found in muscle cells, in the urine) following exercise. Diagnosis is usually confirmed through genetic testing and enzyme assays. Treatment typically involves avoiding strenuous exercise and ensuring adequate hydration and rest before and after physical activity. In some cases, dietary modifications such as high-protein or high-carbohydrate intake may be recommended to help manage symptoms.
Glycogen Storage Disease Type VIII, also known as Phosphorylase Kinase Deficiency, is a rare genetic metabolic disorder that affects the production and breakdown of glycogen in the body. Glycogen is a complex carbohydrate that serves as the primary form of energy storage in the body.
In this condition, there is a deficiency or dysfunction of the enzyme phosphorylase kinase (PhK), which plays a crucial role in activating glycogen phosphorylase, an enzyme responsible for breaking down glycogen into glucose-1-phosphate during periods of increased energy demand.
The deficiency or dysfunction of PhK leads to the abnormal accumulation of glycogen in various tissues, particularly in the liver and muscles. This accumulation can result in hepatomegaly (enlarged liver), hypoglycemia (low blood sugar levels), growth retardation, and muscle weakness.
Glycogen Storage Disease Type VIII is inherited in an autosomal recessive manner, meaning that an individual must inherit two defective copies of the gene, one from each parent, to develop the condition. There are four subtypes of GSD Type VIII, classified based on the specific genetic mutation and the severity of symptoms.
Treatment for Glycogen Storage Disease Type VIII typically involves managing the symptoms and complications associated with the disorder, such as providing a high-carbohydrate diet to prevent hypoglycemia and addressing any liver or muscle dysfunction. Regular monitoring by a healthcare team experienced in metabolic disorders is essential for optimizing treatment and ensuring appropriate management of this complex condition.
Glucan 1,4-alpha-glucosidase, also known as amyloglucosidase or glucoamylase, is an enzyme that catalyzes the hydrolysis of 1,4-glycosidic bonds in starch and other oligo- and polysaccharides, breaking them down into individual glucose molecules. This enzyme specifically acts on the alpha (1->4) linkages found in amylose and amylopectin, two major components of starch. It is widely used in various industrial applications, including the production of high fructose corn syrup, alcoholic beverages, and as a digestive aid in some medical supplements.
Antiporters, also known as exchange transporters, are a type of membrane transport protein that facilitate the exchange of two or more ions or molecules across a biological membrane in opposite directions. They allow for the movement of one type of ion or molecule into a cell while simultaneously moving another type out of the cell. This process is driven by the concentration gradient of one or both of the substances being transported. Antiporters play important roles in various physiological processes, including maintaining electrochemical balance and regulating pH levels within cells.
Glucose-6-phosphate (G6P) is a vital intermediate compound in the metabolism of glucose, which is a simple sugar that serves as a primary source of energy for living organisms. G6P plays a critical role in both glycolysis and gluconeogenesis pathways, contributing to the regulation of blood glucose levels and energy production within cells.
In biochemistry, glucose-6-phosphate is defined as:
A hexose sugar phosphate ester formed by the phosphorylation of glucose at the 6th carbon atom by ATP in a reaction catalyzed by the enzyme hexokinase or glucokinase. This reaction is the first step in both glycolysis and glucose storage (glycogen synthesis) processes, ensuring that glucose can be effectively utilized for energy production or stored for later use.
G6P serves as a crucial metabolic branch point, leading to various pathways such as:
1. Glycolysis: In the presence of sufficient ATP and NAD+ levels, G6P is further metabolized through glycolysis to generate pyruvate, which enters the citric acid cycle for additional energy production in the form of ATP, NADH, and FADH2.
2. Gluconeogenesis: During periods of low blood glucose levels, G6P can be synthesized back into glucose through the gluconeogenesis pathway, primarily occurring in the liver and kidneys. This process helps maintain stable blood glucose concentrations and provides energy to cells when dietary intake is insufficient.
3. Pentose phosphate pathway (PPP): A portion of G6P can be shunted into the PPP, an alternative metabolic route that generates NADPH, ribose-5-phosphate for nucleotide synthesis, and erythrose-4-phosphate for aromatic amino acid production. The PPP is essential in maintaining redox balance within cells and supporting biosynthetic processes.
Overall, glucose-6-phosphate plays a critical role as a central metabolic intermediate, connecting various pathways to regulate energy homeostasis, redox balance, and biosynthesis in response to cellular demands and environmental cues.
A liver cell adenoma is a benign tumor that develops in the liver and is composed of cells similar to those normally found in the liver (hepatocytes). These tumors are usually solitary, but multiple adenomas can occur, especially in women who have taken oral contraceptives for many years. Liver cell adenomas are typically asymptomatic and are often discovered incidentally during imaging studies performed for other reasons. In rare cases, they may cause symptoms such as abdominal pain or discomfort, or complications such as bleeding or rupture. Treatment options include monitoring with periodic imaging studies or surgical removal of the tumor.
1,4-Alpha-Glucan Branching Enzyme (GBE) is an enzyme that plays a crucial role in the synthesis of glycogen, a complex carbohydrate that serves as the primary form of energy storage in animals and fungi. GBE catalyzes the transfer of a segment of a linear glucose chain (alpha-1,4 linkage) to an alpha-1,6 position on another chain, creating branches in the glucan molecule. This branching process enhances the solubility and compactness of glycogen, allowing it to be stored more efficiently within cells.
Defects in GBE are associated with a group of genetic disorders known as glycogen storage diseases type IV (GSD IV), also called Andersen's disease. This autosomal recessive disorder is characterized by the accumulation of abnormally structured glycogen in various tissues, particularly in the liver and muscles, leading to progressive liver failure, muscle weakness, cardiac complications, and sometimes neurological symptoms.
Glycogen Storage Disease Type IIb, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex carbohydrate, into glucose within lysosomes. When GAA activity is lacking, glycogen accumulates in various tissues, including muscle and nerve cells, leading to cellular dysfunction and damage.
Type IIb Pompe Disease is characterized by progressive muscle weakness and hypertrophy (enlargement) of the heart muscle (cardiomyopathy). This form of the disease typically presents in infancy or early childhood and can progress rapidly, often resulting in severe cardiac complications and respiratory failure if left untreated.
Early diagnosis and treatment with enzyme replacement therapy (ERT) can significantly improve outcomes for individuals with Type IIb Pompe Disease. ERT involves administering recombinant human GAA to replace the deficient enzyme, helping to reduce glycogen accumulation in tissues and alleviate symptoms.
Fructose-1,6-diphosphatase deficiency is a rare inherited metabolic disorder that affects the body's ability to metabolize carbohydrates, particularly fructose and glucose. This enzyme deficiency results in an accumulation of certain metabolic intermediates, which can cause a variety of symptoms, including hypoglycemia (low blood sugar), lactic acidosis, hyperventilation, and seizures. The condition is typically diagnosed in infancy or early childhood and is treated with a diet low in fructose and other sugars that can't be metabolized properly due to the enzyme deficiency. If left untreated, the disorder can lead to serious complications, such as brain damage and death.
Enzyme Replacement Therapy (ERT) is a medical treatment approach in which functional copies of a missing or deficient enzyme are introduced into the body to compensate for the lack of enzymatic activity caused by a genetic disorder. This therapy is primarily used to manage lysosomal storage diseases, such as Gaucher disease, Fabry disease, Pompe disease, and Mucopolysaccharidoses (MPS), among others.
In ERT, the required enzyme is produced recombinantly in a laboratory using biotechnological methods. The purified enzyme is then administered to the patient intravenously at regular intervals. Once inside the body, the exogenous enzyme is taken up by cells, particularly those affected by the disorder, and helps restore normal cellular functions by participating in essential metabolic pathways.
ERT aims to alleviate disease symptoms, slow down disease progression, improve quality of life, and increase survival rates for patients with lysosomal storage disorders. However, it does not cure the underlying genetic defect responsible for the enzyme deficiency.
Monosaccharide transport proteins are a type of membrane transport protein that facilitate the passive or active transport of monosaccharides, such as glucose, fructose, and galactose, across cell membranes. These proteins play a crucial role in the absorption, distribution, and metabolism of carbohydrates in the body.
There are two main types of monosaccharide transport proteins: facilitated diffusion transporters and active transporters. Facilitated diffusion transporters, also known as glucose transporters (GLUTs), passively transport monosaccharides down their concentration gradient without the need for energy. In contrast, active transporters, such as the sodium-glucose cotransporter (SGLT), use energy in the form of ATP to actively transport monosaccharides against their concentration gradient.
Monosaccharide transport proteins are found in various tissues throughout the body, including the intestines, kidneys, liver, and brain. They play a critical role in maintaining glucose homeostasis by regulating the uptake and release of glucose into and out of cells. Dysfunction of these transporters has been implicated in several diseases, such as diabetes, cancer, and neurological disorders.