Glycogen Storage Disease Type IV
Glycogen Storage Disease Type I
1,4-alpha-Glucan Branching Enzyme
Glycogen Storage Disease Type III
Glycogen Storage Disease
Glycogen Storage Disease Type II
Glycogen Storage Disease Type VII
Glucose-6-Phosphatase
Glycogen Storage Disease Type VI
Glycogen
alpha-Glucosidases
Glycogen Debranching Enzyme System
Glycogen Storage Disease Type V
Glycogen Storage Disease Type VIII
Glucan 1,4-alpha-Glucosidase
Antiporters
Glucose-6-Phosphate
Amylopectinosis in fetal and neonatal Quarter Horses. (1/22)
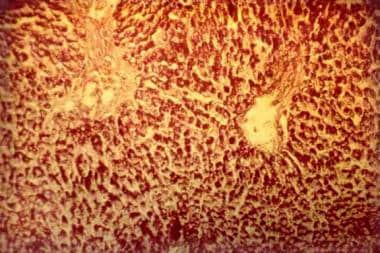
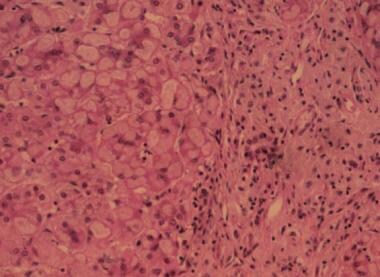
Three Quarter Horses, a stillborn filly (horse No. 1), a female fetus aborted at approximately 6 months of gestation (horse No. 2), and a 1-month-old colt that had been weak at birth (horse No. 3), had myopathy characterized histologically by large spherical or ovoid inclusions in skeletal and cardiac myofibers. Smaller inclusions were also found in brain and spinal cord and in some cells of all other tissues examined. These inclusions were basophilic, red-purple after staining with periodic acid-Schiff (both before and after digestion with diastase), and moderately dark blue after staining with toluidine blue. The inclusions did not react when stained with Congo red. Staining with iodine ranged from pale blue to black. Their ultrastructural appearance varied from amorphous to somewhat filamentous. On the basis of staining characteristics and diastase resistance, we concluded that these inclusions contained amylopectin. A distinctly different kind of inclusion material was also present in skeletal muscle and tongue of horse Nos. 1 and 3. These inclusions were crystalline with a sharply defined ultrastructural periodicity. The crystals were eosinophilic and very dark blue when stained with toluidine blue but did not stain with iodine. Crystals sometimes occurred freely within the myofibers but more often were encased by deposits of amylopectin. This combination of histologic and ultrastructural features characterizes a previously unreported storage disease in fetal and neonatal Quarter Horses, with findings similar to those of glycogen storage disease type IV. We speculate that a severe inherited loss of glycogen brancher enzyme activity may be responsible for these findings. The relation of amylopectinosis to the death of the foals is unknown. (+info)Liver transplantation for glycogen storage disease types I, III, and IV. (2/22)
Glycogen storage disease (GSD) types I, III, and IV can be associated with severe liver disease. The possible development of hepatocellular carcinoma and/or hepatic failure make these GSDs potential candidates for liver transplantation. Early diagnosis and initiation of effective dietary therapy have dramatically improved the outcome of GSD type I by reducing the incidence of liver adenoma and renal insufficiency. Nine type I and 3 type III patients have received liver transplants because of poor metabolic control, multiple liver adenomas, or progressive liver failure. Metabolic abnormalities were corrected in all GSD type I and type III patients, while catch-up growth was reported only in two patients. Whether liver transplantation results in reversal and/or prevention of renal disease remains unclear. Neutropenia persisted in both GSDIb patients post liver transplantation necessitating continuous granulocyte colony stimulating factor treatment. Thirteen GSD type IV patients were liver transplanted because of progressive liver cirrhosis and failure. All but one patient have not had neuromuscular or cardiac complications during follow-up periods for as long as 13 years. Four have died within a week and 5 years after transplantation. Caution should be taken in selecting GSD type IV candidates for liver transplantation because of the variable phenotype, which may include life-limiting extrahepatic manifestations. It remains to be evaluated, whether a genotype-phenotype correlation exists for GSD type IV, which may aid in the decision making. CONCLUSION: Liver transplantation should be considered for patients with glycogen storage disease who have developed liver malignancy or hepatic failure, and for type IV patients with the classical and progressive hepatic form. (+info)Hepatocellular carcinoma in glycogen storage disease type IV. (3/22)
A 13 year old patient with juvenile type IV glycogen storage disease died of the complications of hepatocellular carcinoma. To our knowledge this is the first reported case of hepatocellular carcinoma in association with type IV glycogen storage disease. (+info)Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. (4/22)
Deposition of aggregated protein into neurofilament-rich cytoplasmic inclusion bodies is a common cytopathological feature of neurodegenerative disease. How-or indeed whether-protein aggregation and inclusion body formation cause neurotoxicity are presently unknown. Here, we show that the capacity of superoxide dismutase (SOD) to aggregate into biochemically distinct, high molecular weight, insoluble protein complexes (IPCs) is a gain of function associated with mutations linked to autosomal dominant familial amyotrophic lateral sclerosis. SOD IPCs are detectable in spinal cord extracts from transgenic mice expressing mutant SOD several months before inclusion bodies and motor neuron pathology are apparent. Sequestration of mutant SOD into cytoplasmic inclusion bodies resembling aggresomes requires retrograde transport on microtubules. These data indicate that aggregation and inclusion body formation are mechanistically and temporally distinct processes. (+info)Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. (5/22)
Inwardly rectifying K(+) (Kir) channels are important regulators of resting membrane potential and cell excitability. The activity of Kir channels is critically dependent on the integrity of channel interactions with phosphatidylinositol 4,5-bisphosphate (PIP(2)). Here we identify and characterize channel-PIP(2) interactions that are conserved among Kir family members. We find basic residues that interact with PIP(2), two of which have been associated with Andersen's and Bartter's syndromes. We show that several naturally occurring mutants decrease channel-PIP(2) interactions, leading to disease. (+info)Andersen mutations of KCNJ2 suppress the native inward rectifier current IK1 in a dominant-negative fashion. (6/22)
OBJECTIVE: The Andersen's syndrome is a hereditary disease, which is characterized by cardiac arrhythmias, periodic paralysis and dysmorphic features. Recently, mutations of the KCNJ2 gene, which encodes the inward rectifying potassium channel subunit Kir2.1, have been identified in affected individuals. However, the functional effects of these mutations have not yet been fully elucidated. METHODS AND RESULTS: To clarify this situation we generated known Andersen disease mutants of KCNJ2 which did not yield any measurable K(+) currents in CHO cells indicating that the Andersen mutants failed to form functional homomultimeric complexes. EGFP-tagged KCNJ2 wild-type and mutant channels distributed in a similar homogeneous pattern in the cell membrane suggesting that protein trafficking was not altered by the Andersen mutations but rather implicating that the mutations rendered the KCNJ2 channel non-functional. In heterologous coexpression experiments the Andersen mutants exerted a dominant-negative effect on wild-type KCNJ2. However, the extent of suppression varied between the different KCNJ2 mutants. Given our results in CHO cells, we expressed the disease mutant KCNJ2-S136F in neonate rat cardiomyocytes using adenoviral gene transfer to test the effect of Andersen mutants on native I(K1). I(K1) density was indeed significantly reduced in KCNJ2-S136F-infected cells (n=9) compared to control cells (n=9) over a voltage range from -70 to -150 mV (P<0.05). CONCLUSION: These results support that Kir2.x channels are a critical component of native I(K1) in neonate rat cardiomyocytes and that a dominant-negative suppression of I(K1) in native cells is the pathophysiological correlate of the Andersen's syndrome. (+info)A complex rearrangement in GBE1 causes both perinatal hypoglycemic collapse and late-juvenile-onset neuromuscular degeneration in glycogen storage disease type IV of Norwegian forest cats. (7/22)
Deficiency of glycogen branching enzyme (GBE) activity causes glycogen storage disease type IV (GSD IV), an autosomal recessive error of metabolism. Abnormal glycogen accumulates in myocytes, hepatocytes, and neurons, causing variably progressive, benign to lethal organ dysfunctions. A naturally occurring orthologue of human GSD IV was described previously in Norwegian forest cats (NFC). Here, we report that while most affected kittens die at or soon after birth, presumably due to hypoglycemia, survivors of the perinatal period appear clinically normal until onset of progressive neuromuscular degeneration at 5 months of age. Molecular investigation of affected cats revealed abnormally spliced GBE1 mRNA products and lack of GBE cross-reactive material in liver and muscle. Affected cats are homozygous for a complex rearrangement of genomic DNA in GBE1, constituted by a 334 bp insertion at the site of a 6.2 kb deletion that extends from intron 11 to intron 12 (g. IVS11+1552_IVS12-1339 del6.2kb ins334 bp), removing exon 12. An allele-specific, PCR-based test demonstrates that the rearrangement segregates with the disease in the GSD IV kindred and is not found in unrelated normal cats. Screening of 402 privately owned NFC revealed 58 carriers and 4 affected cats. The molecular characterization of feline GSD IV will enhance further studies of GSD IV pathophysiology and development of novel therapies in this unique animal model. (+info)Neuromuscular forms of glycogen branching enzyme deficiency. (8/22)
Deficiency of glycogen branching enzyme is causative of Glycogen Storage Disease type IV (GSD-IV), a rare autosomal recessive disorder of the glycogen synthesis, characterized by the accumulation of amylopectin-like polysaccharide, also known as polyglucosan, in almost all tissues. Its clinical presentation is variable and involves the liver or the neuromuscular system and different mutations in the GBE1 gene, located on chromosome 3, have been identified in both phenotypes. This review will addresses the neuromuscular clinical variants, focusing on the molecular genetics aspects of this disorder. (+info)Glycogen Storage Disease Type IV (GSD IV), also known as Andersen's disease, is a rare inherited metabolic disorder that affects the body's ability to break down glycogen, a complex carbohydrate that serves as a source of energy for the body.
In GSD IV, there is a deficiency in the enzyme called glycogen branching enzyme (GBE), which is responsible for adding branches to the glycogen molecule during its synthesis. This results in an abnormal form of glycogen that accumulates in various organs and tissues, particularly in the liver, heart, and muscles.
The accumulation of this abnormal glycogen can lead to progressive damage and failure of these organs, resulting in a variety of symptoms such as muscle weakness, hypotonia, hepatomegaly (enlarged liver), cardiomyopathy (heart muscle disease), and developmental delay. The severity of the disease can vary widely, with some individuals experiencing milder symptoms while others may have a more severe and rapidly progressing form of the disorder.
Currently, there is no cure for GSD IV, and treatment is focused on managing the symptoms and slowing down the progression of the disease. This may include providing nutritional support, addressing specific organ dysfunction, and preventing complications.
Glycogen Storage Disease Type I (GSD I) is a rare inherited metabolic disorder caused by deficiency of the enzyme glucose-6-phosphatase, which is necessary for the liver to release glucose into the bloodstream. This leads to an accumulation of glycogen in the liver and abnormally low levels of glucose in the blood (hypoglycemia).
There are two main subtypes of GSD I: Type Ia and Type Ib. In Type Ia, there is a deficiency of both glucose-6-phosphatase enzyme activity in the liver, kidney, and intestine, leading to hepatomegaly (enlarged liver), hypoglycemia, lactic acidosis, hyperlipidemia, and growth retardation. Type Ib is characterized by a deficiency of glucose-6-phosphatase enzyme activity only in the neutrophils, leading to recurrent bacterial infections.
GSD I requires lifelong management with frequent feedings, high-carbohydrate diet, and avoidance of fasting to prevent hypoglycemia. In some cases, treatment with continuous cornstarch infusions or liver transplantation may be necessary.
1,4-Alpha-Glucan Branching Enzyme (GBE) is an enzyme that plays a crucial role in the synthesis of glycogen, a complex carbohydrate that serves as the primary form of energy storage in animals and fungi. GBE catalyzes the transfer of a segment of a linear glucose chain (alpha-1,4 linkage) to an alpha-1,6 position on another chain, creating branches in the glucan molecule. This branching process enhances the solubility and compactness of glycogen, allowing it to be stored more efficiently within cells.
Defects in GBE are associated with a group of genetic disorders known as glycogen storage diseases type IV (GSD IV), also called Andersen's disease. This autosomal recessive disorder is characterized by the accumulation of abnormally structured glycogen in various tissues, particularly in the liver and muscles, leading to progressive liver failure, muscle weakness, cardiac complications, and sometimes neurological symptoms.
Glycogen Storage Disease Type III, also known as Cori or Forbes disease, is a rare inherited metabolic disorder caused by deficiency of the debranching enzyme amylo-1,6-glucosidase, which is responsible for breaking down glycogen in the liver and muscles. This results in an abnormal accumulation of glycogen in these organs leading to its associated symptoms.
There are two main types: Type IIIa affects both the liver and muscles, while Type IIIb affects only the liver. Symptoms can include hepatomegaly (enlarged liver), hypoglycemia (low blood sugar), hyperlipidemia (high levels of fats in the blood), and growth retardation. In Type IIIa, muscle weakness and cardiac problems may also occur.
The diagnosis is usually made through biochemical tests and genetic analysis. Treatment often involves dietary management with frequent meals to prevent hypoglycemia, and in some cases, enzyme replacement therapy. However, there is no cure for this condition and life expectancy can be reduced depending on the severity of the symptoms.
Glycogen storage disease (GSD) is a group of rare inherited metabolic disorders that affect the body's ability to break down and store glycogen, a complex carbohydrate that serves as the primary form of energy storage in the body. These diseases are caused by deficiencies or dysfunction in enzymes involved in the synthesis, degradation, or transport of glycogen within cells.
There are several types of GSDs, each with distinct clinical presentations and affected organs. The most common type is von Gierke disease (GSD I), which primarily affects the liver and kidneys. Other types include Pompe disease (GSD II), McArdle disease (GSD V), Cori disease (GSD III), Andersen disease (GSD IV), and others.
Symptoms of GSDs can vary widely depending on the specific type, but may include:
* Hypoglycemia (low blood sugar)
* Growth retardation
* Hepatomegaly (enlarged liver)
* Muscle weakness and cramping
* Cardiomyopathy (heart muscle disease)
* Respiratory distress
* Developmental delays
Treatment for GSDs typically involves dietary management, such as frequent feedings or a high-protein, low-carbohydrate diet. In some cases, enzyme replacement therapy may be used to manage symptoms. The prognosis for individuals with GSDs depends on the specific type and severity of the disorder.
Glycogen Storage Disease Type II, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar that serves as energy storage, within lysosomes. When GAA is deficient, glycogen accumulates in various tissues, particularly in muscle cells, leading to their dysfunction and damage.
The severity of Pompe Disease can vary significantly, depending on the amount of functional enzyme activity remaining. The classic infantile-onset form presents within the first few months of life with severe muscle weakness, hypotonia, feeding difficulties, and respiratory insufficiency. This form is often fatal by 1-2 years of age if left untreated.
A later-onset form, which can present in childhood, adolescence, or adulthood, has a more variable clinical course. Affected individuals may experience progressive muscle weakness, respiratory insufficiency, and cardiomyopathy, although the severity and rate of progression are generally less pronounced than in the infantile-onset form.
Enzyme replacement therapy with recombinant human GAA is available for the treatment of Pompe Disease and has been shown to improve survival and motor function in affected individuals.
Glycogen Storage Disease Type VII, also known as Tarui's disease, is a rare inherited metabolic disorder caused by a deficiency of the enzyme phosphofructokinase (PFK), which is required for glycogenolysis – the breakdown of glycogen to glucose-1-phosphate and ultimately into glucose. This enzyme deficiency results in the accumulation of glycogen, particularly in muscle and red blood cells, leading to symptoms such as exercise-induced muscle cramps, myoglobinuria (the presence of myoglobin in the urine), and hemolytic anemia. The disease can also cause muscle weakness, fatigue, and dark-colored urine after strenuous exercise. It is inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
Glucose-6-phosphatase is an enzyme that plays a crucial role in the regulation of glucose metabolism. It is primarily located in the endoplasmic reticulum of cells in liver, kidney, and intestinal mucosa. The main function of this enzyme is to remove the phosphate group from glucose-6-phosphate (G6P), converting it into free glucose, which can then be released into the bloodstream and used as a source of energy by cells throughout the body.
The reaction catalyzed by glucose-6-phosphatase is as follows:
Glucose-6-phosphate + H2O → Glucose + Pi (inorganic phosphate)
This enzyme is essential for maintaining normal blood glucose levels, particularly during periods of fasting or starvation. In these situations, the body needs to break down stored glycogen in the liver and convert it into glucose to supply energy to the brain and other vital organs. Glucose-6-phosphatase is a key enzyme in this process, allowing for the release of free glucose into the bloodstream.
Deficiencies or mutations in the gene encoding glucose-6-phosphatase can lead to several metabolic disorders, such as glycogen storage disease type I (von Gierke's disease) and other related conditions. These disorders are characterized by an accumulation of glycogen and/or fat in various organs, leading to impaired glucose metabolism, growth retardation, and increased risk of infection and liver dysfunction.
Glycogen Storage Disease Type VI, also known as Hers disease, is a rare inherited metabolic disorder caused by deficiency of the liver enzyme called glycogen phosphorylase. This enzyme is responsible for breaking down glycogen, which is a stored form of glucose, into glucose-1-phosphate during the process of glycogenolysis.
In GSD Type VI, the lack of this enzyme leads to an abnormal accumulation of glycogen in the liver, causing hepatomegaly (enlarged liver) and elevated liver enzymes. The symptoms of this condition are usually milder compared to other types of GSD, and may include fatigue, weakness, and hypoglycemia (low blood sugar), especially after prolonged fasting or physical exertion.
The diagnosis of GSD Type VI is typically made through biochemical tests that measure the activity of the glycogen phosphorylase enzyme in liver tissue, as well as genetic testing to identify mutations in the gene responsible for the enzyme's production. Treatment may involve dietary management, such as frequent feeding and avoidance of prolonged fasting, to prevent hypoglycemia. In some cases, medication may be necessary to manage symptoms and prevent complications.
Glycogen is a complex carbohydrate that serves as the primary form of energy storage in animals, fungi, and bacteria. It is a polysaccharide consisting of long, branched chains of glucose molecules linked together by glycosidic bonds. Glycogen is stored primarily in the liver and muscles, where it can be quickly broken down to release glucose into the bloodstream during periods of fasting or increased metabolic demand.
In the liver, glycogen plays a crucial role in maintaining blood glucose levels by releasing glucose when needed, such as between meals or during exercise. In muscles, glycogen serves as an immediate energy source for muscle contractions during intense physical activity. The ability to store and mobilize glycogen is essential for the proper functioning of various physiological processes, including athletic performance, glucose homeostasis, and overall metabolic health.
Alpha-glucosidases are a group of enzymes that break down complex carbohydrates into simpler sugars, such as glucose, by hydrolyzing the alpha-1,4 and alpha-1,6 glycosidic bonds in oligosaccharides, disaccharides, and polysaccharides. These enzymes are located on the brush border of the small intestine and play a crucial role in carbohydrate digestion and absorption.
Inhibitors of alpha-glucosidases, such as acarbose and miglitol, are used in the treatment of type 2 diabetes to slow down the digestion and absorption of carbohydrates, which helps to reduce postprandial glucose levels and improve glycemic control.
The Glycogen Debranching Enzyme System, also known as glycogen debranching enzyme or Amy-1, is a crucial enzyme complex in human biochemistry. It plays an essential role in the metabolism of glycogen, which is a large, branched polymer of glucose that serves as the primary form of energy storage in animals and fungi.
The Glycogen Debranching Enzyme System consists of two enzymatic activities: a transferase and an exo-glucosidase. The transferase activity transfers a segment of a branched glucose chain to another part of the same or another glycogen molecule, while the exo-glucosidase activity cleaves the remaining single glucose units from the outer branches of the glycogen molecule.
This enzyme system is responsible for removing the branched structures of glycogen, allowing the linear chains to be further degraded by other enzymes into glucose molecules that can be used for energy production or stored for later use. Defects in this enzyme complex can lead to several genetic disorders, such as Glycogen Storage Disease Type III (Cori's disease) and Type IV (Andersen's disease), which are characterized by the accumulation of abnormal glycogen molecules in various tissues.
Glycogen Storage Disease Type V, also known as McArdle's disease, is a genetic disorder that affects the body's ability to break down glycogen, a complex carbohydrate stored in muscles, into glucose, which provides energy for muscle contraction.
This condition results from a deficiency of the enzyme myophosphorylase, which is responsible for breaking down glycogen into glucose-1-phosphate within the muscle fibers. Without sufficient myophosphorylase activity, muscles become easily fatigued and may cramp or become rigid during exercise due to a lack of available energy.
Symptoms typically appear in childhood or adolescence and can include muscle weakness, stiffness, cramps, and myoglobinuria (the presence of myoglobin, a protein found in muscle cells, in the urine) following exercise. Diagnosis is usually confirmed through genetic testing and enzyme assays. Treatment typically involves avoiding strenuous exercise and ensuring adequate hydration and rest before and after physical activity. In some cases, dietary modifications such as high-protein or high-carbohydrate intake may be recommended to help manage symptoms.
Glycogen Storage Disease Type VIII, also known as Phosphorylase Kinase Deficiency, is a rare genetic metabolic disorder that affects the production and breakdown of glycogen in the body. Glycogen is a complex carbohydrate that serves as the primary form of energy storage in the body.
In this condition, there is a deficiency or dysfunction of the enzyme phosphorylase kinase (PhK), which plays a crucial role in activating glycogen phosphorylase, an enzyme responsible for breaking down glycogen into glucose-1-phosphate during periods of increased energy demand.
The deficiency or dysfunction of PhK leads to the abnormal accumulation of glycogen in various tissues, particularly in the liver and muscles. This accumulation can result in hepatomegaly (enlarged liver), hypoglycemia (low blood sugar levels), growth retardation, and muscle weakness.
Glycogen Storage Disease Type VIII is inherited in an autosomal recessive manner, meaning that an individual must inherit two defective copies of the gene, one from each parent, to develop the condition. There are four subtypes of GSD Type VIII, classified based on the specific genetic mutation and the severity of symptoms.
Treatment for Glycogen Storage Disease Type VIII typically involves managing the symptoms and complications associated with the disorder, such as providing a high-carbohydrate diet to prevent hypoglycemia and addressing any liver or muscle dysfunction. Regular monitoring by a healthcare team experienced in metabolic disorders is essential for optimizing treatment and ensuring appropriate management of this complex condition.
Glucan 1,4-alpha-glucosidase, also known as amyloglucosidase or glucoamylase, is an enzyme that catalyzes the hydrolysis of 1,4-glycosidic bonds in starch and other oligo- and polysaccharides, breaking them down into individual glucose molecules. This enzyme specifically acts on the alpha (1->4) linkages found in amylose and amylopectin, two major components of starch. It is widely used in various industrial applications, including the production of high fructose corn syrup, alcoholic beverages, and as a digestive aid in some medical supplements.
Antiporters, also known as exchange transporters, are a type of membrane transport protein that facilitate the exchange of two or more ions or molecules across a biological membrane in opposite directions. They allow for the movement of one type of ion or molecule into a cell while simultaneously moving another type out of the cell. This process is driven by the concentration gradient of one or both of the substances being transported. Antiporters play important roles in various physiological processes, including maintaining electrochemical balance and regulating pH levels within cells.
Glucose-6-phosphate (G6P) is a vital intermediate compound in the metabolism of glucose, which is a simple sugar that serves as a primary source of energy for living organisms. G6P plays a critical role in both glycolysis and gluconeogenesis pathways, contributing to the regulation of blood glucose levels and energy production within cells.
In biochemistry, glucose-6-phosphate is defined as:
A hexose sugar phosphate ester formed by the phosphorylation of glucose at the 6th carbon atom by ATP in a reaction catalyzed by the enzyme hexokinase or glucokinase. This reaction is the first step in both glycolysis and glucose storage (glycogen synthesis) processes, ensuring that glucose can be effectively utilized for energy production or stored for later use.
G6P serves as a crucial metabolic branch point, leading to various pathways such as:
1. Glycolysis: In the presence of sufficient ATP and NAD+ levels, G6P is further metabolized through glycolysis to generate pyruvate, which enters the citric acid cycle for additional energy production in the form of ATP, NADH, and FADH2.
2. Gluconeogenesis: During periods of low blood glucose levels, G6P can be synthesized back into glucose through the gluconeogenesis pathway, primarily occurring in the liver and kidneys. This process helps maintain stable blood glucose concentrations and provides energy to cells when dietary intake is insufficient.
3. Pentose phosphate pathway (PPP): A portion of G6P can be shunted into the PPP, an alternative metabolic route that generates NADPH, ribose-5-phosphate for nucleotide synthesis, and erythrose-4-phosphate for aromatic amino acid production. The PPP is essential in maintaining redox balance within cells and supporting biosynthetic processes.
Overall, glucose-6-phosphate plays a critical role as a central metabolic intermediate, connecting various pathways to regulate energy homeostasis, redox balance, and biosynthesis in response to cellular demands and environmental cues.
A liver cell adenoma is a benign tumor that develops in the liver and is composed of cells similar to those normally found in the liver (hepatocytes). These tumors are usually solitary, but multiple adenomas can occur, especially in women who have taken oral contraceptives for many years. Liver cell adenomas are typically asymptomatic and are often discovered incidentally during imaging studies performed for other reasons. In rare cases, they may cause symptoms such as abdominal pain or discomfort, or complications such as bleeding or rupture. Treatment options include monitoring with periodic imaging studies or surgical removal of the tumor.
 Glycogen storage disease type IV
Glycogen storage disease type IV Glycogen storage disease type IV: MedlinePlus Genetics
Glycogen storage disease type IV: MedlinePlus Genetics Glycogen Storage Disease Type IV
Glycogen Storage Disease Type IV Type IV Glycogen Storage Disease: Practice Essentials, Pathophysiology, Prognosis
Type IV Glycogen Storage Disease: Practice Essentials, Pathophysiology, Prognosis Glycogen storage disease type III: AGL gene (exons 3, 4, 21, 24, 28, 31, 33 and 35) - index case - GenoMed
Glycogen storage disease type III: AGL gene (exons 3, 4, 21, 24, 28, 31, 33 and 35) - index case - GenoMed FIFes A&R regler Appendix I - Felis Danica
FIFes A&R regler Appendix I - Felis Danica Wolman Disease - Supra-Regional Assay Service
Wolman Disease - Supra-Regional Assay Service Reference Materials for Hereditary Genetic Disorders and HLA Testing | CDC
Reference Materials for Hereditary Genetic Disorders and HLA Testing | CDC Liver transplantation in glycogen storage disease: a single-center experience | Orphanet Journal of Rare Diseases | Full Text
Liver transplantation in glycogen storage disease: a single-center experience | Orphanet Journal of Rare Diseases | Full Text Portal hypertension (Concept Id: C0020541)
- MedGen - NCBI
Portal hypertension (Concept Id: C0020541)
- MedGen - NCBI Feline PCR Genetic Tests - Veterinary Immunohistochemistry (IHC) Database
Feline PCR Genetic Tests - Veterinary Immunohistochemistry (IHC) Database List of Rare Diseases | A-Z Database | NORD
List of Rare Diseases | A-Z Database | NORD HEALTH TESTING
HEALTH TESTING LABOKLIN (UK)| Genetic Diseases | Cats| Osteochondrodysplasia (Scottish Fold Osteodystrophy)
LABOKLIN (UK)| Genetic Diseases | Cats| Osteochondrodysplasia (Scottish Fold Osteodystrophy) Glycogen storage disease type II (Pompe disease)
Glycogen storage disease type II (Pompe disease) Sheffield Diagnostic Genetics Service - Constitutional
Sheffield Diagnostic Genetics Service - Constitutional Items where Year is 1991 - D-Scholarship@Pitt
Items where Year is 1991 - D-Scholarship@Pitt