Homocystinuria
Cystathionine beta-Synthase
Homocystine
Cystathionine
Pyridoxine
Betaine
Metabolism, Inborn Errors
Amino Acid Metabolism, Inborn Errors
Anemia, Megaloblastic
Marfan Syndrome
Hydro-Lyases
Renal Aminoacidurias
Livedo Reticularis
5-Methyltetrahydrofolate-Homocysteine S-Methyltransferase
Vitamin B 12
Neonatal Screening
Methylmalonic Acid
The molecular basis of cystathionine beta-synthase deficiency in Dutch patients with homocystinuria: effect of CBS genotype on biochemical and clinical phenotype and on response to treatment. (1/142)
Homocystinuria due to cystathionine beta-synthase (CBS) deficiency, inherited as an autosomal recessive trait, is the most prevalent inborn error of methionine metabolism. Its diverse clinical expression may include ectopia lentis, skeletal abnormalities, mental retardation, and premature arteriosclerosis and thrombosis. This variability is likely caused by considerable genetic heterogeneity. We investigated the molecular basis of CBS deficiency in 29 Dutch patients from 21 unrelated pedigrees and studied the possibility of a genotype-phenotype relationship with regard to biochemical and clinical expression and response to homocysteine-lowering treatment. Clinical symptoms and biochemical parameters were recorded at diagnosis and during long-term follow-up. Of 10 different mutations detected in the CBS gene, 833T-->C (I278T) was predominant, present in 23 (55%) of 42 independent alleles. At diagnosis, homozygotes for this mutation (n=12) tended to have higher homocysteine levels than those seen in patients with other genotypes (n=17), but similar clinical manifestations. During follow-up, I278T homozygotes responded more efficiently to homocysteine-lowering treatment. After 378 patient-years of treatment, only 2 vascular events were recorded; without treatment, at least 30 would have been expected (P<.01). This intervention in Dutch patients significantly reduces the risk of cardiovascular disease and other sequelae of classical homocystinuria syndrome. (+info)Deletion of the regulatory domain in the pyridoxal phosphate-dependent heme protein cystathionine beta-synthase alleviates the defect observed in a catalytic site mutant. (2/142)
The most common cause of severely elevated homocysteine or homocystinuria is inherited disorders in cystathionine beta-synthase. The latter enzyme is a unique hemeprotein that catalyzes pyridoxal phosphate (PLP)-dependent condensation of serine and homocysteine to give cystathionine, thus committing homocysteine to catabolism. A point mutation, V168M, has been described in a homocystinuric cell line and is associated with a B(6)-responsive phenotype. In this study, we have examined the kinetic properties of this mutant and demonstrate that the mutation affects the PLP but not the heme content. The approximately 13-fold diminution in activity because of the mutation corresponds to an approximately 7-fold decrease in the level of bound PLP. This may be explained by half of the sites activity associated with cystathionine beta-synthase. The addition of PLP results in partial but not full restoration of activity to wild type levels. Elimination of the C-terminal quarter of the mutant protein results in alleviation of the catalytic penalty imposed by the V168M mutation. The resulting truncated protein is very similar to the corresponding truncated enzyme with wild type sequence and is now able to bind the full complement of both heme and PLP cofactors. These results indicate that the V168M mutation per se does not affect binding of PLP directly and that interactions between the regulatory C terminus and the catalytic N terminus are important in modulating the cofactor content and therefore the activity of the full-length enzyme. These studies provide the first biochemical explanation for the B(6)-responsive phenotype associated with a cystathionine beta-synthase-impaired homocystinuric genotype. (+info)Reduction of false negative results in screening of newborns for homocystinuria. (3/142)
BACKGROUND: Mental retardation and other disabilities (including ectopia lentis, osteoporosis, and thromboembolism) in patients who have homocystinuria as a result of a deficiency of cystathionine beta-synthase can be prevented by the screening of newborns with measurement of blood methionine, followed by the early treatment of affected infants. Many infants with this disorder, however, are not identified by screening and have irreversible brain damage. METHODS: We reviewed the results of neonatal screening for homocystinuria over a period of 32 years in New England. Additional specimens were requested for repeated analysis when blood methionine measurements were at or above the established cutoff level. Homocystinuria due to cystathionine beta-synthase deficiency was confirmed by quantitative amino acid analyses. RESULTS: For the first 23.5 years of the review period, the blood methionine cutoff value was 2 mg per deciliter (134 micromol per liter). Among the 2.2 million infants screened during that period, 8 with homocystinuria were identified (1:275,000). In 1990, the cutoff value was reduced to 1 mg per deciliter (67 micromol per liter). Among the 1.1 million infants screened in the subsequent 8.5 years, 7 with the disorder were identified (1:157,000). During the latter period, the specimens were collected from six of the seven infants when they were two days of age or less; five of the six had blood methionine concentrations below 2 mg per deciliter. Use of the reduced cutoff level increased the false positive rate from 0.006 percent to 0.03 percent. CONCLUSIONS: A cutoff level for blood methionine of 1 mg per deciliter in neonatal screening tests for homocystinuria should identify affected infants who have only slightly elevated concentrations of methionine and reduce the frequency of false negative results. (+info)Relationship between homocysteine and superoxide dismutase in homocystinuria: possible relevance to cardiovascular risk. (4/142)
A modest homocysteine elevation is associated with an increased cardiovascular risk. Marked circulating homocysteine elevations occur in homocystinuria due to cystathionine beta-synthase (CbetaS) deficiency, a disorder associated with a greatly enhanced cardiovascular risk. Lowering homocysteine levels reduces this risk significantly. Because homocysteine-induced oxidative damage may contribute to vascular changes and extracellular superoxide dismutase (EC-SOD) is an important antioxidant in vascular tissue, we assessed EC-SOD and homocysteine in patients with homocystinuria. We measured circulating EC-SOD, total homocysteine (free plus bound), and methionine levels during the treatment of 21 patients with homocystinuria, 18 due to CbetaS deficiency, aged 8 to 59 years, and 3 with remethylating defects. We measured total homocysteine by immunoassay, EC-SOD by ELISA, and methionine by amino acid analysis and assessed interindividual and intraindividual relationships. There was a significant, positive relationship between EC-SOD and total homocysteine. For the interindividual assessment, levels were highly correlated, r=0.746, N=21, P<0.0001. This relationship was maintained after taking into account intraindividual patient variation (r=0.607, N=62, P<0.0001). In 2 newly diagnosed CbetaS-deficient patients, treatment that lowered the markedly elevated pretreatment homocysteine level (from 337 to 72 and from 298 to 50 micromol/L) reduced the associated elevated EC-SOD in each by 50%. EC-SOD and methionine levels were unrelated (r=0.148, n=39, P=0.368). The positive relationship between circulating EC-SOD and homocysteine could represent a protective antioxidant response to homocysteine-induced oxidative damage and contribute to reducing cardiovascular risk in homocystinuric patients. EC-SOD levels may be relevant to the pathogenesis of vascular disease in other patient groups. (+info)Homocysteine and cardiovascular disease: cause or effect? (5/142)
Both markedly and mildly elevated circulating homocysteine concentrations are associated with increased risk of vascular occlusion. Here we review possible mechanisms that mediate these effects. Inborn errors of homocysteine metabolism result in markedly elevated plasma homocysteine (200-300 micromol/L) and thromboembolic (mainly venous) disease: treatment to lower but not to normalize these concentrations prevents vascular events. Mild homocysteine elevation (>15 micromol/L) occurs in approximately 20-30% of patients with atherosclerotic disease. Usually, this is easily normalized with oral folate and ongoing trials are assessing the effect of folate treatment on outcomes. Although there is evidence of endothelial dysfunction with both markedly and mildly elevated homocysteine concentrations, the elevated homocysteine concentration in atherosclerotic patients is also associated with most standard vascular risk factors, and importantly, with early decline in renal function, which is common in atherosclerosis. Decline in renal function alone causes elevated plasma homocysteine (and cysteine). These observations suggest that mild hyperhomocysteinemia could often be an effect rather than a cause of atherosclerotic disease. Data on the common C677T methylenetetrahydrofolate reductase polymorphism supports this, in that, although homozygosity is a frequent cause of mild hyperhomocysteinemia when plasma folate is below median population concentrations, it appears not to increase cardiovascular risk. Indeed, there is recent evidence suggesting an acute antioxidant effect of folic acid independent of its effect on homocysteine concentrations. This antioxidant mechanism may oppose an oxidant effect of homocysteine and be relevant to treatment of patients with vascular disease, especially those with chronic renal insufficiency. Such patients have moderately elevated plasma homocysteine and greatly increased cardiovascular risk that is largely unexplained. (+info)The controversy over homocysteine and cardiovascular risk. (6/142)
Elevated plasma total homocysteine (tHcy) is a risk factor for occlusive cardiovascular disease (CVD). This concept is based on the observations of premature vascular disease in patients with homocystinuria, the relation between tHcy and both clinical CVD as well as preclinical atherosclerotic disease, the relation between tHcy in children and CVD in their parents or relatives, and reduction in CVD or surrogate endpoints after tHcy-lowering intervention with B vitamins. Plausible mechanisms include the in vivo interference with nitric oxide-dependent reactive vasodilatation. Some observations have raised questions about tHcy as a risk factor. 1) Some prospective studies showed a weak relation or no relation between tHcy and CVD. 2) Several traditional risk factors are associated with tHcy and may confound the relation between tHcy and CVD. 3) tHcy is related to renal function, and hyperhomocysteinemia may reflect early nephrosclerosis. 4) The C677T transition of the methylenetetrahydrofolate reductase gene causes a moderate increase in tHcy but no or only minor increased CVD risk. However, the strength of some of these arguments can be questioned because there is increasing evidence that tHcy is a proximate risk factor provoking the acute event, it strongly interacts with traditional risk factors, and it may predict CVD or death in patients with chronic renal failure. Furthermore, the studies of the C677T polymorphism lack statistical power, and the TT genotype may even modulate CVD risk independently of homocysteine. Thus, only placebo-controlled intervention studies with tHcy-lowering B vitamins and clinical endpoints can provide additional valid arguments for the debate over whether tHcy is a causal CVD risk factor. (+info)Homocystine, atherosclerosis and thrombosis: implications for oral contraceptive users.(7/142)
(+info)Mutations in the regulatory domain of cystathionine beta synthase can functionally suppress patient-derived mutations in cis. (8/142)
Human cystathionine beta--synthase (CBS) is an S-adenosylmethionine-regulated enzyme that plays a key role in the metabolism of homocysteine. Mutations in CBS are known to cause homocystinuria, an inborn error in metabolism. We previously developed a yeast functional assay for CBS and used it to characterize mutations found in homocystinuric patients. We discovered that many patient-derived mutations are functionally suppressed by deletion of the C-terminal 142 amino acids, which contain a 53 amino acid motif known as the CBS domain. This domain is found in a wide variety of proteins of diverse biological function. Here we have used a genetic screen to identify missense mutations in the C-terminal region of CBS that can suppress the most common patient mutation, I278T. Seven suppressor mutations were identified, four of which map to the CBS domain. When combined in cis with another pathogenic mutation, V168M, six of seven of the suppressor mutations rescued the yeast phenotype. Enzyme activity analyses indicate that the suppressors restore activity from <2% to 17--64% of the wild-type levels. Analysis of the suppressor mutations in the absence of the pathogenic mutation shows that six of the seven suppressor alleles have lost enzymatic responsiveness to S-adenosylmethionine. Using homology modeling, we show that the suppressor mutations appear to map on one face of the CBS domain. Our results indicate that subtle changes to the C-terminus of CBS can restore activity to mutant proteins and provide a rationale for screening for compounds that can activate mutant CBS alleles. (+info)Homocystinuria is a genetic disorder characterized by the accumulation of homocysteine and its metabolites in the body due to a deficiency in the enzyme cystathionine beta-synthase (CBS). This enzyme is responsible for converting homocysteine to cystathionine, which is a critical step in the metabolic pathway that breaks down methionine.
As a result of this deficiency, homocysteine levels in the blood increase and can lead to various health problems, including neurological impairment, ocular abnormalities (such as ectopia lentis or dislocation of the lens), skeletal abnormalities (such as Marfan-like features), and vascular complications.
Homocystinuria can be diagnosed through newborn screening or by measuring homocysteine levels in the blood or urine. Treatment typically involves a low-methionine diet, supplementation with vitamin B6 (pyridoxine), betaine, and/or methylcobalamin (a form of vitamin B12) to help reduce homocysteine levels and prevent complications associated with the disorder.
Cystathionine beta-synthase (CBS) is an enzyme that plays a crucial role in the metabolic pathway responsible for the production of the amino acid cysteine from homocysteine. CBS catalyzes the condensation of serine with homocysteine to form cystathionine, which is subsequently hydrolyzed to cysteine and alpha-ketobutyrate by another enzyme called cystathionine gamma-lyase.
CBS requires the cofactor pyridoxal 5'-phosphate (PLP) for its activity and is primarily located in the liver, where it helps regulate homocysteine levels in the body. Elevated levels of homocysteine have been linked to various health issues, including cardiovascular disease and neurological disorders.
In addition to its role in cysteine synthesis, CBS also contributes to the transsulfuration pathway, which is involved in the detoxification of methionine and the production of glutathione, an essential antioxidant in the body. Genetic mutations in the CBS gene can lead to conditions such as homocystinuria, a rare inherited metabolic disorder characterized by elevated levels of homocysteine and methionine in the blood and urine.
Ectopia lentis is a medical term that refers to the displacement or malpositioning of the lens in the eye. The lens, which is normally located behind the iris and held in place by tiny fibers called zonules, can become dislocated due to various reasons such as genetic disorders like Marfan syndrome, trauma, or other ocular diseases.
When the lens becomes displaced, it can cause a variety of symptoms including blurry vision, double vision, sensitivity to light, and distorted images. In some cases, ectopia lentis may be asymptomatic and only discovered during a routine eye examination. Treatment for ectopia lentis depends on the severity of the displacement and any associated symptoms. In mild cases, no treatment may be necessary, while in more severe cases, surgery may be required to reposition or remove the lens and replace it with an artificial one.
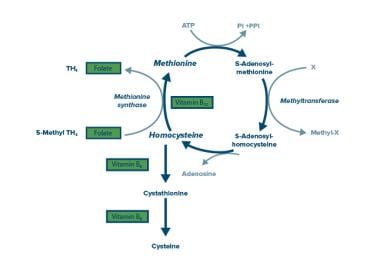
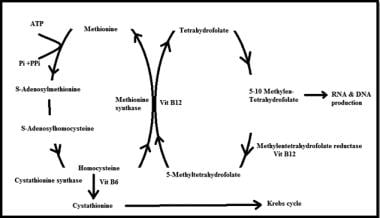
Homocysteine is an amino acid that is formed from the metabolism of another amino acid called methionine. It is not normally present in significant amounts in the diet, but it can be elevated in some people due to genetic factors or nutritional deficiencies (such as a lack of vitamin B12, folate, or betaine). Elevated levels of homocysteine in the blood have been linked to an increased risk of cardiovascular disease, including heart attack and stroke. Homocysteine can be converted back to methionine through a process that requires the presence of vitamin B12, folate, and betaine. It can also be converted to another amino acid called cystathionine through a reaction that requires the enzyme cystathionine beta-synthase and the cofactor vitamin B6.
Cystathionine is a non-proteinogenic amino acid, which means that it is not used in the synthesis of proteins. It is an intermediate in the biosynthetic pathway that converts the amino acid methionine to cysteine in the body. This process involves the removal of a sulfur atom from methionine, resulting in the formation of cystathionine. Further breakdown of cystathionine leads to the production of cysteine and another amino acid called alpha-ketobutyrate.
Cystathionine plays a crucial role in the metabolism of certain sulfur-containing amino acids, and its levels are regulated by an enzyme called cystathionine beta-synthase (CBS). Genetic defects or deficiencies in this enzyme can result in a disorder known as homocystinuria, which is characterized by the accumulation of homocysteine and methionine in the body and an increased risk of various health complications.
In summary, cystathionine is a biologically important amino acid that functions as an intermediate in the conversion of methionine to cysteine, and its levels are tightly regulated by enzymatic processes in the body.
Pyridoxine is the chemical name for Vitamin B6. According to the medical definition, Pyridoxine is a water-soluble vitamin that is part of the B-vitamin complex and is essential for the metabolism of proteins, carbohydrates, and fats. It plays a vital role in the regulation of homocysteine levels in the body, the formation of neurotransmitters such as serotonin and dopamine, and the synthesis of hemoglobin.
Pyridoxine can be found naturally in various foods, including whole grains, legumes, vegetables, nuts, seeds, meat, poultry, and fish. It is also available as a dietary supplement and may be prescribed by healthcare providers to treat or prevent certain medical conditions, such as vitamin B6 deficiency, anemia, seizures, and carpal tunnel syndrome.
Like other water-soluble vitamins, Pyridoxine cannot be stored in the body and must be replenished regularly through diet or supplementation. Excessive intake of Pyridoxine can lead to toxicity symptoms such as nerve damage, skin lesions, and light sensitivity.
Homocysteine is an amino acid that is formed in the body during the metabolism of another amino acid called methionine. It's an important intermediate in various biochemical reactions, including the synthesis of proteins, neurotransmitters, and other molecules. However, elevated levels of homocysteine in the blood (a condition known as hyperhomocysteinemia) have been linked to several health issues, such as cardiovascular disease, stroke, and cognitive decline.
Homocysteine can be converted back to methionine with the help of vitamin B12 and a cofactor called betaine, or it can be converted to another amino acid called cystathionine with the help of vitamin B6 and folate (vitamin B9). Imbalances in these vitamins and other factors can lead to an increase in homocysteine levels.
It is crucial to maintain normal homocysteine levels for overall health, as high levels may contribute to the development of various diseases. Regular monitoring and maintaining a balanced diet rich in folate, vitamin B6, and vitamin B12 can help regulate homocysteine levels and reduce the risk of related health issues.
Betaine, also known as trimethylglycine, is a naturally occurring compound that can be found in various foods such as beets, spinach, and whole grains. In the body, betaine functions as an osmolyte, helping to regulate water balance in cells, and as a methyl donor, contributing to various metabolic processes including the conversion of homocysteine to methionine.
In medical terms, betaine is also used as a dietary supplement and medication. Betaine hydrochloride is a form of betaine that is sometimes used as a supplement to help with digestion by providing additional stomach acid. Betaine anhydrous, on the other hand, is often used as a supplement for improving athletic performance and promoting liver health.
Betaine has also been studied for its potential role in protecting against various diseases, including cardiovascular disease, diabetes, and neurological disorders. However, more research is needed to fully understand its mechanisms of action and therapeutic potential.
Inborn errors of metabolism (IEM) refer to a group of genetic disorders caused by defects in enzymes or transporters that play a role in the body's metabolic processes. These disorders result in the accumulation or deficiency of specific chemicals within the body, which can lead to various clinical manifestations, such as developmental delay, intellectual disability, seizures, organ damage, and in some cases, death.
Examples of IEM include phenylketonuria (PKU), maple syrup urine disease (MSUD), galactosemia, and glycogen storage diseases, among many others. These disorders are typically inherited in an autosomal recessive manner, meaning that an affected individual has two copies of the mutated gene, one from each parent.
Early diagnosis and management of IEM are crucial to prevent or minimize complications and improve outcomes. Treatment options may include dietary modifications, supplementation with missing enzymes or cofactors, medication, and in some cases, stem cell transplantation or gene therapy.
Inborn errors of amino acid metabolism refer to genetic disorders that affect the body's ability to properly break down and process individual amino acids, which are the building blocks of proteins. These disorders can result in an accumulation of toxic levels of certain amino acids or their byproducts in the body, leading to a variety of symptoms and health complications.
There are many different types of inborn errors of amino acid metabolism, each affecting a specific amino acid or group of amino acids. Some examples include:
* Phenylketonuria (PKU): This disorder affects the breakdown of the amino acid phenylalanine, leading to its accumulation in the body and causing brain damage if left untreated.
* Maple syrup urine disease: This disorder affects the breakdown of the branched-chain amino acids leucine, isoleucine, and valine, leading to their accumulation in the body and causing neurological problems.
* Homocystinuria: This disorder affects the breakdown of the amino acid methionine, leading to its accumulation in the body and causing a range of symptoms including developmental delay, intellectual disability, and cardiovascular problems.
Treatment for inborn errors of amino acid metabolism typically involves dietary restrictions or supplementation to manage the levels of affected amino acids in the body. In some cases, medication or other therapies may also be necessary. Early diagnosis and treatment can help prevent or minimize the severity of symptoms and health complications associated with these disorders.
Megaloblastic anemia is a type of macrocytic anemia, which is characterized by the presence of large, structurally abnormal, and immature red blood cells called megaloblasts in the bone marrow. This condition arises due to impaired DNA synthesis during erythropoiesis (the process of red blood cell production), often as a result of deficiencies in vitamin B12 or folate, or from the use of certain medications that interfere with DNA synthesis.
The hallmark feature of megaloblastic anemia is the presence of megaloblasts in the bone marrow, which exhibit an asynchrony between nuclear and cytoplasmic maturation. This means that although the cytoplasm of these cells may appear well-developed, their nuclei remain underdeveloped and fragmented. As a result, the peripheral blood shows an increase in mean corpuscular volume (MCV), reflecting the larger size of the red blood cells.
Additional hematological findings include decreased reticulocyte counts, neutrophil hypersegmentation, and occasionally thrombocytopenia or leukopenia. Neurological symptoms may also be present due to the involvement of the nervous system in vitamin B12 deficiency.
Megaloblastic anemia is typically treated with supplementation of the deficient vitamin (B12 or folate), which helps restore normal erythropoiesis and alleviate symptoms over time.
Marfan syndrome is a genetic disorder that affects the body's connective tissue. Connective tissue helps to strengthen and support various structures in the body, including the skin, ligaments, blood vessels, and heart. In Marfan syndrome, the body produces an abnormal amount of a protein called fibrillin-1, which is a key component of connective tissue. This leads to problems with the formation and function of connective tissue throughout the body.
The most serious complications of Marfan syndrome typically involve the heart and blood vessels. The aorta, which is the large artery that carries blood away from the heart, can become weakened and stretched, leading to an increased risk of aortic dissection or rupture. Other common features of Marfan syndrome include long, thin fingers and toes; tall stature; a curved spine; and eye problems such as nearsightedness and lens dislocation.
Marfan syndrome is usually inherited in an autosomal dominant pattern, which means that a child has a 50% chance of inheriting the gene mutation from a parent who has the condition. However, about 25% of cases are the result of a new mutation and occur in people with no family history of the disorder. There is no cure for Marfan syndrome, but treatment can help to manage the symptoms and reduce the risk of complications.
Methionine is an essential amino acid, which means that it cannot be synthesized by the human body and must be obtained through the diet. It plays a crucial role in various biological processes, including:
1. Protein synthesis: Methionine is one of the building blocks of proteins, helping to create new proteins and maintain the structure and function of cells.
2. Methylation: Methionine serves as a methyl group donor in various biochemical reactions, which are essential for DNA synthesis, gene regulation, and neurotransmitter production.
3. Antioxidant defense: Methionine can be converted to cysteine, which is involved in the formation of glutathione, a potent antioxidant that helps protect cells from oxidative damage.
4. Homocysteine metabolism: Methionine is involved in the conversion of homocysteine back to methionine through a process called remethylation, which is essential for maintaining normal homocysteine levels and preventing cardiovascular disease.
5. Fat metabolism: Methionine helps facilitate the breakdown and metabolism of fats in the body.
Foods rich in methionine include meat, fish, dairy products, eggs, and some nuts and seeds.
I'm sorry for any confusion, but "Hydro-Lyases" is not a recognized medical term or category in biochemistry. It seems like there might be a misunderstanding or a typo in the term.
In biochemistry, "lyases" are enzymes that catalyze the removal of groups from substrates by means other than hydrolysis or oxidation, often forming a double bond or a ring-forming reaction. They are classified and named based on the type of bond they break.
If you meant to ask about a specific enzyme or reaction, could you please provide more context or clarify the term? I'd be happy to help further with accurate information.
Renal aminoacidurias are a group of inherited kidney disorders characterized by the abnormal excretion of amino acids in the urine (aminoaciduria). This condition results from defects in the renal tubular transport systems that are responsible for the reabsorption of amino acids from the filtrate in the kidneys.
There are several types of renal aminoacidurias, each associated with a specific genetic mutation affecting different transporter proteins in the proximal renal tubules. The most common type is cystinuria, which is caused by a defect in the transport system for four amino acids: cystine, ornithine, lysine, and arginine. Other types of renal aminoacidurias include Hartnup disorder, Lowe syndrome, and Dent disease, among others.
The clinical manifestations of renal aminoacidurias vary depending on the specific type and severity of the disorder. Some individuals may be asymptomatic or have only mild symptoms, while others may experience severe complications such as kidney stones, urinary tract infections, neurological symptoms, or growth retardation.
Treatment for renal aminoacidurias typically involves dietary modifications, increased fluid intake, and medications to reduce the risk of kidney stone formation and other complications. In some cases, surgery may be necessary to remove large kidney stones.
Livedo reticularis is a cutaneous manifestation characterized by a bluish-purple, netlike pattern of discoloration on the skin. It is caused by the abnormal dilation and constriction of blood vessels near the skin's surface, leading to impaired circulation in the affected areas.
The condition can be idiopathic (primary) or secondary to various underlying disorders such as autoimmune diseases, vasculitis, hematologic disorders, infections, or medications that affect the blood vessels. In some cases, livedo reticularis may be a sign of an underlying medical condition requiring further evaluation and treatment.
It is essential to differentiate livedo reticularis from other related conditions like livedo racemosa, which presents with more irregular and diffuse patterns, and is typically associated with vasculitis or severe systemic disorders. Additionally, livedo reticularis should not be confused with cutis marmorata, a physiological response to cold temperatures that resolves upon warming the affected area.
5-Methyltetrahydrofolate-Homocysteine S-Methyltransferase is also known as Methionine Synthase. It is a vital enzyme in the human body that plays a crucial role in methionine metabolism and homocysteine regulation.
The medical definition of 5-Methyltetrahydrofolate-Homocysteine S-Methyltransferase is as follows:
A enzyme (EC 2.1.1.13) that catalyzes the methylation of homocysteine to methionine, using 5-methyltetrahydrofolate as a methyl donor. This reaction also requires the cofactor vitamin B12 (cobalamin) as a coenzyme. The enzyme is located in the cytosol of cells and is essential for the synthesis of methionine, which is an important amino acid required for various biological processes such as protein synthesis, methylation reactions, and the formation of neurotransmitters.
Deficiency or dysfunction of this enzyme can lead to several health issues, including homocystinuria, a genetic disorder characterized by elevated levels of homocysteine in the blood, which can cause serious complications such as neurological damage, cardiovascular disease, and skeletal abnormalities.
Vitamin B12, also known as cobalamin, is a water-soluble vitamin that plays a crucial role in the synthesis of DNA, formation of red blood cells, and maintenance of the nervous system. It is involved in the metabolism of every cell in the body, particularly affecting DNA regulation and neurological function.
Vitamin B12 is unique among vitamins because it contains a metal ion, cobalt, from which its name is derived. This vitamin can be synthesized only by certain types of bacteria and is not produced by plants or animals. The major sources of vitamin B12 in the human diet include animal-derived foods such as meat, fish, poultry, eggs, and dairy products, as well as fortified plant-based milk alternatives and breakfast cereals.
Deficiency in vitamin B12 can lead to various health issues, including megaloblastic anemia, fatigue, neurological symptoms such as numbness and tingling in the extremities, memory loss, and depression. Since vitamin B12 is not readily available from plant-based sources, vegetarians and vegans are at a higher risk of deficiency and may require supplementation or fortified foods to meet their daily requirements.
Neonatal screening is a medical procedure in which specific tests are performed on newborn babies within the first few days of life to detect certain congenital or inherited disorders that are not otherwise clinically apparent at birth. These conditions, if left untreated, can lead to serious health problems, developmental delays, or even death.
The primary goal of neonatal screening is to identify affected infants early so that appropriate treatment and management can be initiated as soon as possible, thereby improving their overall prognosis and quality of life. Commonly screened conditions include phenylketonuria (PKU), congenital hypothyroidism, galactosemia, maple syrup urine disease, sickle cell disease, cystic fibrosis, and hearing loss, among others.
Neonatal screening typically involves collecting a small blood sample from the infant's heel (heel stick) or through a dried blood spot card, which is then analyzed using various biochemical, enzymatic, or genetic tests. In some cases, additional tests such as hearing screenings and pulse oximetry for critical congenital heart disease may also be performed.
It's important to note that neonatal screening is not a diagnostic tool but rather an initial step in identifying infants who may be at risk of certain conditions. Positive screening results should always be confirmed with additional diagnostic tests before any treatment decisions are made.
Methylmalonic acid (MMA) is an organic compound that is produced in the human body during the metabolism of certain amino acids, including methionine and threonine. It is a type of fatty acid that is intermediate in the breakdown of these amino acids in the liver and other tissues.
Under normal circumstances, MMA is quickly converted to succinic acid, which is then used in the Krebs cycle to generate energy in the form of ATP. However, when there are deficiencies or mutations in enzymes involved in this metabolic pathway, such as methylmalonyl-CoA mutase, MMA can accumulate in the body and cause methylmalonic acidemia, a rare genetic disorder that affects approximately 1 in every 50,000 to 100,000 individuals worldwide.
Elevated levels of MMA in the blood or urine can be indicative of various metabolic disorders, including methylmalonic acidemia, vitamin B12 deficiency, and renal insufficiency. Therefore, measuring MMA levels is often used as a diagnostic tool to help identify and manage these conditions.
 Homocystinuria
Homocystinuria
Renal vein thrombosis
David Haigh
Eric Arnott
List of OMIM disorder codes
Hyperhomocysteinemia
Cystathionine beta synthase
MTRR (gene)
Marfanoid
Arachnodactyly
Akhenaten
Deep vein thrombosis
Megavitamin-B6 syndrome
List of genetic disorders
Malar flush
Trimethylaminuria
MMADHC
MMACHC
Serine dehydratase
CBS domain
Vesanto Melina
Cystinuria
Transsulfuration pathway
Methylenetetrahydrofolate reductase
Methionine synthase) reductase
Nasal septum deviation
Archibald's sign
Charles Enrique Dent
Dolichostenomelia
Methylmalonic acidemia
Homocystinuria - Wikipedia
 Homocystinuria: MedlinePlus Genetics
Homocystinuria: MedlinePlus Genetics
 Homocystinuria/Homocysteinemia: Overview, Pathophysiology, Epidemiology
Homocystinuria/Homocysteinemia: Overview, Pathophysiology, Epidemiology
 Adult-onset homocystinuria arteriopathy mimics fibromuscular dysplasia
Adult-onset homocystinuria arteriopathy mimics fibromuscular dysplasia
 Homocystinuria, Newborn Bloodspot Screening - HSE.ie
Homocystinuria, Newborn Bloodspot Screening - HSE.ie
 Treatment of homocystinuria. | Archives of Disease in Childhood
Treatment of homocystinuria. | Archives of Disease in Childhood
The molecular basis of cystathionine beta-synthase deficiency in Dutch patients with homocystinuria: effect of CBS genotype on...
 Platelet and monocyte variables in homocystinuria due to cystathionine-beta-synthase deficiency
| Haematologica
Platelet and monocyte variables in homocystinuria due to cystathionine-beta-synthase deficiency
| Haematologica
Homocystinuria (HCU) - heel prick screening
- HSE.ie
 Methylmalonic acidemia with Homocystinuria in adults | Mayo Clinic Connect
Methylmalonic acidemia with Homocystinuria in adults | Mayo Clinic Connect
Methylmalonic Aciduria and Homocystinuria | Asper Biogene
Homocystinuria/Homocysteinemia: Overview, Pathophysiology, Epidemiology
 Homocystinuria - Paddle For Kids Corp
Homocystinuria - Paddle For Kids Corp
 Stats Homocystinuria - Homocystinuria Map | Diseasemaps
Stats Homocystinuria - Homocystinuria Map | Diseasemaps
HCU Support - HOMOCYSTINURIA SUPPORT SITE
 Homocystinuria: Video, Anatomy, Definition & Function | Osmosis
Homocystinuria: Video, Anatomy, Definition & Function | Osmosis
 Gene Therapy and Classical Homocystinuria | HCU
Gene Therapy and Classical Homocystinuria | HCU
 Homocystinuria - Children's Health Issues - MSD Manual Consumer Version
Homocystinuria - Children's Health Issues - MSD Manual Consumer Version
 Homocystinuria | Quick Medical Diagnosis & Treatment 2023 | AccessMedicine | McGraw Hill Medical
Homocystinuria | Quick Medical Diagnosis & Treatment 2023 | AccessMedicine | McGraw Hill Medical
 Enzyme replacement prevents neonatal death, liver damage, and osteoporosis in murine homocystinuria
Enzyme replacement prevents neonatal death, liver damage, and osteoporosis in murine homocystinuria
 Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
HCFC1 gene: MedlinePlus Genetics
Homocystinuria (HCU) | Syndromes: Rapid Recognition and Perioperative Implications, 2e | AccessAnesthesiology | McGraw Hill...
 1.7.99.5: 5,10-methylenetetrahydrofolate reductase (FADH2) - BRENDA Enzyme Database
1.7.99.5: 5,10-methylenetetrahydrofolate reductase (FADH2) - BRENDA Enzyme Database
 Synlogic Granted FDA Fast Track Designation for SYNB1353 for the Treatment of Homocystinuria (HCU) - Synlogic
Synlogic Granted FDA Fast Track Designation for SYNB1353 for the Treatment of Homocystinuria (HCU) - Synlogic
Amino Acid Metabolism Disorders: MedlinePlus
Osteoporosis Treatment & Management: Approach Considerations, Pharmacologic Therapy, Dietary Measures
Cystadane (betaine) dosing, indications, interactions, adverse effects, and more
 Cataplexy - Epidemiology Forecast - 2032
Cataplexy - Epidemiology Forecast - 2032
Cystathionine8
- Homocystinuria or HCU is an inherited disorder of the metabolism of the amino acid methionine due to a deficiency of cystathionine beta synthase or methionine synthase. (wikipedia.org)
- Bublil EM, Majtan T. Classical homocystinuria: From cystathionine beta-synthase deficiency to novel enzyme therapies. (medlineplus.gov)
- Homocystinuria due to cystathionine beta-synthase (CBS) deficiency, inherited as an autosomal recessive trait, is the most prevalent inborn error of methionine metabolism. (nih.gov)
- To gain insight into the mechanisms responsible for enhanced thromboxane (TX) A2 biosynthesis in homozygous homocystinuria due to cystathionine-beta-synthase deficiency (CBSD), we measured a series of platelet and monocyte variables in 9 homozygous and 8 obligate heterozygous CBSD patients and evaluated their relationships to thromboxane formation, as reflected by urinary excretion of its major metabolite, 11-dehydro-TXB2 (TXM). (haematologica.org)
- Deficiency of cystathionine synthase (homocystinuria I) leads to a failure of transsulfuration of precursors of cysteine, an important component of collagen. (mhmedical.com)
- Molecular basis of cystathionine beta-synthase deficiency in pyridoxine responsive and nonresponsive homocystinuria. (nih.gov)
- Delayed diagnosis of homocystinuria by major deficiency in cystathionine beta synthase]. (bvsalud.org)
- Diagnostic tardif d'homocystinurie par déficit majeur en cystathionine beta synthase. (bvsalud.org)
Methylmalonic acidemia7
- @lilli64 , I had to look up methylmalonic acidemia with homocystinuria as I had never heard of it before. (mayoclinic.org)
- Methylmalonic acidemia with homocystinuria is an inherited disorder in which the body is unable to properly process certain nutrients from food including amino acids, lipids and cholesterol. (mayoclinic.org)
- People with this disorder have a combination of features from two separate conditions: methylmalonic acidemia and homocystinuria. (mayoclinic.org)
- Several HCFC1 gene variants (also known as mutations) have been identified in people with methylmalonic acidemia with homocystinuria, cblX type, which is one form of a disorder that causes developmental delay, eye defects, neurological problems, and blood abnormalities. (medlineplus.gov)
- This combination of imbalances leads to the signs and symptoms of methylmalonic acidemia with homocystinuria. (medlineplus.gov)
- These individuals have delayed development and other neurological problems but do not show other features of methylmalonic acidemia with homocystinuria, cblX type (described above). (medlineplus.gov)
- 121 Mendelian pathogenic or likely pathogenic variants associated with 31 inherited diseases were detected, among these hearing loss, congenital hypothyroidism, methylmalonic acidemia, methylmalonic acidemia with homocystinuria, phenylketonuria(PKU) and benign hyperphenylalaninemia accounted for half of the carrier variants. (researchsquare.com)
Pyridoxine1
- 50% of patients with homocystinuria show pyridoxine responsiveness, including 13% who can be completely controlled with pyridoxine alone. (mhmedical.com)
Deficiency2
- Symptoms of homocystinuria can also be caused by a deficiency of vitamins B6, B12, or folate. (wikipedia.org)
- The new diseases are homocystinuria, maple syrup urine disease, tyrosinemia type 1, isovaleric acidemia, glutaric aciduria type I, long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency, and carnitine deficiency. (medscape.com)
Autosomal3
- Homocystinuria is an autosomal recessively inherited defect in the transsulfuration pathway (homocystinuria I) or methylation pathway (homocystinuria II and III). (medscape.com)
- Homocystinuria is an autosomal recessive condition resulting from the accumulation in blood of the essential amino acid methionine and one of its metabolic products homocysteine. (hse.ie)
- Familial homocystinuria is an autosomal recessive genetic disorder that first manifests early in life. (osmosis.org)
Inherited disorder1
- This medication is used to treat high levels of a certain important body chemical (homocysteine) due to an inherited disorder (homocystinuria). (medscape.com)
Phenylketonuria1
- Synlogic's pipeline includes its lead program in phenylketonuria (PKU), which has demonstrated proof of concept with plans to start a pivotal, Phase 3 study in the first half of 2023, and additional novel drug candidates designed to treat homocystinuria (HCU) and enteric hyperoxaluria. (synlogictx.com)
Diagnosis2
- Homocystinurias and defects of folate and methylation metabolism: practical approaches to diagnosis and treatment. (com.es)
- Knowledge on frequency, pathophysiology, clinical manifestation, diagnosis and treatment of homocystinurias, methylation defects and genetic disorders of B-vitamin metabolism. (com.es)
Homocysteine levels1
- SYNB1353 is a novel orally-administered, non-systemically absorbed drug candidate designed to consume methionine in the gastrointestinal tract thereby lowering homocysteine levels in patients with homocystinuria (HCU). (synlogictx.com)
Symptoms5
- The signs and symptoms of homocystinuria typically develop during childhood, although some mildly affected people may not show signs and symptoms until adulthood. (medlineplus.gov)
- Researchers have not determined how excess homocysteine and related compounds lead to the signs and symptoms of homocystinuria. (medlineplus.gov)
- Consequently all infants who present clinically in later life with signs and symptoms suggestive of homocystinuria, such as dislocation of lenses, osteoporosis and inappropriate tall stature should have the disorder excluded formally by measuring plasma levels of total homocysteine. (hse.ie)
- Children with homocystinuria are unable to break down (metabolize) the amino acid homocysteine, which, along with certain toxic by-products, builds up to cause a variety of symptoms. (msdmanuals.com)
- Symptoms of homocystinuria range from mild to severe. (msdmanuals.com)
20221
- CAMBRIDGE, Mass. , Aug. 23, 2022 (GLOBE NEWSWIRE) -- Synlogic, Inc. (Nasdaq: SYBX), a clinical-stage biotechnology company developing medicines for metabolic and immunological diseases through its proprietary approach to synthetic biology, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to SYNB1353 for the potential treatment of homocystinuria (HCU). (synlogictx.com)
Urine5
- Homocystinuria represents a group of hereditary metabolic disorders characterized by an accumulation of the amino acid homocysteine in the serum and an increased excretion of homocysteine in the urine. (wikipedia.org)
- The term homocystinuria describes an increased excretion of the thiol amino acid homocysteine in urine (and incidentally, also an increased concentration in plasma). (wikipedia.org)
- Homocystinuria is a disorder of methionine metabolism, leading to an abnormal accumulation of homocysteine and its metabolites (homocystine, homocysteine-cysteine complex, and others) in blood and urine. (medscape.com)
- In homocystinuria , "homocysteine" is a metabolite of the amino acid methionine, and "uria" means, a substance present in urine. (osmosis.org)
- So people with homocystinuria have large amounts of homocysteine in their urine, as well as other problems in the connective tissue, muscles, brain, heart, and blood vessels. (osmosis.org)
Metabolic2
- Clinical syndromes resulting from each of these metabolic abnormalities have been termed homocystinuria I, II, and III. (medscape.com)
- Prof. Kruger joins the Journal of Inherited Metabolic Disease Podcast to talk about homocystinuria, successful trials in gene therapy, why it costs so much to make viruses and what inspires him. (hcunetworkaustralia.org.au)
Clinical2
- The Patient-Expert Meetings bring together leaders in the field of homocystinuria research, clinical care, nutrition and advocacy with patients, families and caregivers. (hcunetworkaustralia.org.au)
- Seeking volunteers with homocystinuria to join a paid clinical research study. (rareshare.org)
Methionine1
- Two other interrelated pathways of methionine metabolism can produce accumulation of homocysteine and its metabolites: defective methylcobalamin synthesis (homocystinuria II) and abnormality in MTHFR (homocystinuria III). (mhmedical.com)
Genetic1
- Homocysteinemia may be due to a genetic predisposition to abnormal activity in the same pathways as homocystinuria. (medscape.com)
Genes2
- Variants (also called mutations) in the CBS , MTHFR , MTR , MTRR , and MMADHC genes cause homocystinuria. (medlineplus.gov)
- Rarely, homocystinuria can be caused by variants in several other genes. (medlineplus.gov)
MTHFR1
- An abnormal gene on chromosome 1 has been proposed as the cause of reduction in methylene tetrahydrofolate reductase ([MTHFR] or homocystinuria III). (mhmedical.com)
Abnormalities1
- Our results argue against abnormalities of platelet and monocyte function being responsible for the abnormally high in vivo TXA2 biosynthesis in homocystinuria due to CBSD. (haematologica.org)
Enzyme2
- I was diagnosed at the age of 2 that I had a condition called Homocystinuria, which is due to the absence of an enzyme in my body. (hcusupport.com)
- Homocystinuria is caused by a lack of the enzyme needed to metabolize homocysteine. (msdmanuals.com)
Intellectual disability1
- Less common forms of homocystinuria can cause intellectual disability, slower growth and weight gain (failure to thrive), seizures, and problems with movement. (medlineplus.gov)
Variants1
- Variants in the CBS gene cause classic homocystinuria. (medlineplus.gov)
Cardiovascular1
- 01). This intervention in Dutch patients significantly reduces the risk of cardiovascular disease and other sequelae of classical homocystinuria syndrome. (nih.gov)
Disease1
- Classical Homocystinuria Disease. (mhmedical.com)
Classical2
- Our Scientific Advisory Board's co-chair, Professor Warren Kruger is pioneering gene therapy for classical homocystinuria. (hcunetworkaustralia.org.au)
- Do you have classical homocystinuria? (rareshare.org)
Patients1
- Some basic information about what kinds of food Homocystinuria patients can eat is given in diet section. (hcusupport.com)
Mild1
- A mild elevation of plasma homocysteine may exist without homocystinuria. (medscape.com)
Treatment1
- Treatment of homocystinuria. (bmj.com)
People3
- Classic homocystinuria affects at least 1 in 200,000 to 335,000 people worldwide. (medlineplus.gov)
- Although people who carry one altered copy and one normal copy of the CBS gene do not have homocystinuria, they are more likely than people without a CBS variant to have shortages (deficiencies) of vitamin B12 and folic acid. (medlineplus.gov)
- In 1999, this site was developed in order to help people with HOMOCYSTINURIA( HCU ). (hcusupport.com)
Condition1
- Homocystinuria (HCU) is an inherited condition caused by an altered gene. (hse.ie)