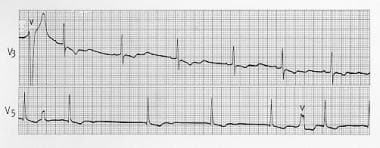
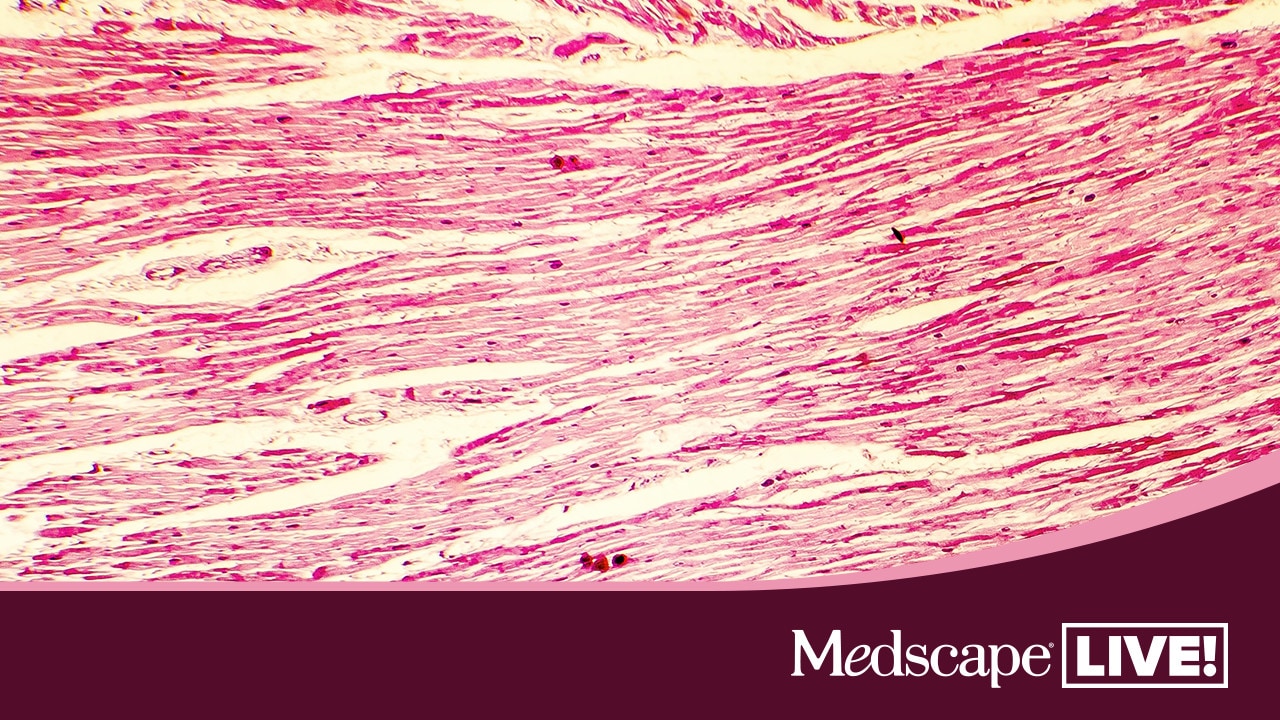
Hypokalemic Periodic Paralysis
Paralyses, Familial Periodic
Hypokalemia
Paralysis, Hyperkalemic Periodic
NAV1.4 Voltage-Gated Sodium Channel
Thyrotoxicosis
Paralysis
Sodium Channels
Andersen Syndrome
Potassium
Muscle, Skeletal
Hyperkalemia
Calcium Channels
Mutation
Bartter Syndrome
Acidosis, Renal Tubular
Myotonia
Channelopathies
Effects of mutations causing hypokalaemic periodic paralysis on the skeletal muscle L-type Ca2+ channel expressed in Xenopus laevis oocytes. (1/68)
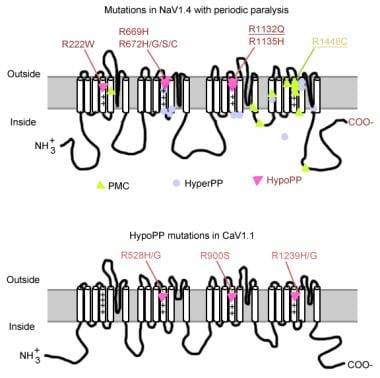
1. A truncated form of the rabbit alpha1S Ca2+ channel subunit (alpha1SDeltaC) was expressed with the beta1b, alpha2delta and gamma auxiliary subunits in Xenopus laevis oocytes. After 5-7 days, skeletal muscle L-type currents were measured (469 +/- 48 nA in 10 mM Ba2+). All three of the auxiliary subunits were necessary to record significant L-type current. A rapidly inactivating, dihydropyridine-insensitive endogenous Ba2+ current was observed in oocytes expressing the auxiliary subunits without an exogenous alpha subunit. Expression of full-length alpha1S gave 10-fold smaller currents than the truncated form. 2. Three missense mutations causing hypokalaemic periodic paralysis (R528H in domain II S4 of the alpha1S subunit; R1239H and R1239G in domain IV S4) were introduced into alpha1SDeltaC and expressed in oocytes. L-type current was separated from the endogenous current by nimodipine subtraction. All three of the mutations reduced L-type current amplitude ( approximately 40 % for R528H, approximately 60-70 % for R1239H and R1239G). 3. The disease mutations altered the activation properties of L-type current. R528H shifted the G(V) curve approximately 5 mV to the left and modestly reduced the voltage dependence of the activation time constant, tauact. R1239H and R1239G shifted the G(V) curve approximately 5-10 mV to the right and dramatically slowed tauact at depolarized test potentials. 4. The voltage dependence of steady-state inactivation was not significantly altered by any of the disease mutations. 5. Wild-type and mutant L-type currents were also measured in the presence of (-)-Bay K8644, which boosted the amplitude approximately 5- to 7-fold. The effects of the mutations on the position of the G(V) curve and the voltage dependence of tauact were essentially the same as in the absence of agonist. Bay K-enhanced tail currents were slowed by R528H and accelerated by R1239H and R1239G. 6. We conclude that the domain IV mutations R1239H and R1239G have similar effects on the gating properties of the skeletal muscle L-type Ca2+ channel expressed in Xenopus oocytes, while the domain II mutation R528H has distinct effects. This result implies that the location of the substitutions is more important than their degree of conservation in determining their biophysical consequences. (+info)Hypokalaemic paralysis. (2/68)
Hypokalaemic paralysis is a relatively uncommon but potentially life-threatening clinical syndrome. If recognised and treated appropriately, patients recover without any clinical sequellae. The syndrome of hypokalaemic paralysis represents a heterogeneous group of disorders characterised clinically by hypokalaemia and acute systemic weakness. Most cases are due to familial or primary hypokalaemic periodic paralysis; sporadic cases are associated with numerous other conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies and gastrointestinal potassium losses. The age of onset, race, family history, medications, and underlying disease states can help in identifying the cause of hypokalaemic paralysis. Initial therapy of the patient with hypokalaemic paralysis includes potassium replacement and search for underlying aetiology. Further management depends on the aetiology of hypokalaemia, severity of symptoms, and duration of disease. This review presents the differential diagnosis for hypokalaemic paralysis and discusses management of the syndrome. (+info)Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. (3/68)
The pathomechanism of familial hypokalemic periodic paralysis (HypoPP) is a mystery, despite knowledge of the underlying dominant point mutations in the dihydropyridine receptor (DHPR) voltage sensor. In five HypoPP families without DHPR gene defects, we identified two mutations, Arg-672-->His and -->Gly, in the voltage sensor of domain 2 of a different protein: the skeletal muscle sodium channel alpha subunit, known to be responsible for hereditary muscle diseases associated with myotonia. Excised skeletal muscle fibers from a patient heterozygous for Arg-672-->Gly displayed depolarization and weakness in low-potassium extracellular solution. Slowing and smaller size of action potentials were suggestive of excitability of the wild-type channel population only. Heterologous expression of the two sodium channel mutations revealed a 10-mV left shift of the steady-state fast inactivation curve enhancing inactivation and a sodium current density that was reduced even at potentials at which inactivation was removed. Decreased current and small action potentials suggested a low channel protein density. The alterations are decisive for the pathogenesis of episodic muscle weakness by reducing the number of excitable sodium channels particularly at sustained membrane depolarization. The results prove that SCN4A, the gene encoding the sodium channel alpha subunit of skeletal muscle is responsible for HypoPP-2 which does not differ clinically from DHPR-HypoPP. HypoPP-2 represents a disease caused by enhanced channel inactivation and current reduction showing no myotonia. (+info)Clinical-molecular study of a family with essential tremor, late onset seizures and periodic paralysis. (4/68)
We report the clinical features of, and the molecular study performed on, a Spanish family with essential tremor (ET), late onset epilepsy and autosomal dominant hypokalemic periodic paralysis (hypoPP). The presence of hypoPP in this kindred suggested an ion channel as a candidate gene for ET. Our study identified an Arg528His CACNL1A3 mutation in patients with hypoPP, and excluded this mutation as the cause of tremor or epilepsy in this kindred. (+info)The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. (5/68)
Missense mutations of the human skeletal muscle voltage-gated Na channel (hSkM1) underlie a variety of diseases, including hyperkalemic periodic paralysis (HyperPP), paramyotonia congenita, and potassium-aggravated myotonia. Another disorder of sarcolemmal excitability, hypokalemic periodic paralysis (HypoPP), which is usually caused by missense mutations of the S4 voltage sensors of the L-type Ca channel, was associated recently in one family with a mutation in the outermost arginine of the IIS4 voltage sensor (R669H) of hSkM1 (Bulman et al., 1999). Intriguingly, an arginine-to-histidine mutation at the homologous position in the L-type Ca(2+) channel (R528H) is a common cause of HypoPP. We have studied the gating properties of the hSkM1-R669H mutant Na channel experimentally in human embryonic kidney cells and found that it has no significant effects on activation or fast inactivation but does cause an enhancement of slow inactivation. R669H channels exhibit an approximately 10 mV hyperpolarized shift in the voltage dependence of slow inactivation and a twofold to fivefold prolongation of recovery after prolonged depolarization. In contrast, slow inactivation is often disrupted in HyperPP-associated Na channel mutants. These results demonstrate that, in R669H-associated HypoPP, enhanced slow inactivation does not preclude, and may contribute to, prolonged attacks of weakness and add support to previous evidence implicating the IIS4 voltage sensor in slow-inactivation gating. (+info)Identification of mutations including de novo mutations in Korean patients with hypokalaemic periodic paralysis. (6/68)
BACKGROUND: Hypokalaemic periodic paralysis (hypoPP) is an autosomal dominant disorder involving the abnormal function of ion channels and it is characterized by paralysis attacks of varying severity, accompanied by a fall in blood potassium levels. Linkage analysis showed that the candidate locus responsible for hypoPP was localized to chromosome 1q31-32, and this locus encoded the muscle dihydropyridine-sensitive calcium channel alpha(1)-subunit (CACNA1S). So far, three different mutations in CACNA1S gene have been identified in patients with hypoPP: Arg528His, Arg1239His and Arg1239Gly in Caucasian patients. However, there are few reports about the mutations of CACNA1S gene in other races. METHODS: In this study, four Korean families with five hypoPP patients were screened for mutations of CACNA1S gene with polymerase chain reaction-based restriction analysis and single-strand conformation polymorphism analysis. To determine the mode of inheritance, haplotype analysis was done with three microsatellite markers (D1S1726, CACNL1A3, and D1S1723). RESULTS: Arg528His mutation was detected in three families, and one family had no known mutations. Moreover, for the first time, we detected de novo Arg528His mutations in two out of three families with hypoPP. Haplotype analysis using three microsatellite markers (D1S1726, CACNL1A3, and D1S1723) suggested the occurrence of de novo Arg528His mutations in two of the three families with Arg528His mutation. CONCLUSIONS: Arg528His mutations of CACNA1S, including de novo Arg528His mutations, were found in Korean patients with hypoPP. These results imply that de novo mutation, in addition to non-penetrance, is one of the genetic mechanisms that can explain the previous clinical observation that hypoPP occurs sporadically without family history. (+info)Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. (7/68)
Hypokalaemic periodic paralysis (hypoPP) is an autosomal dominant muscle disorder characterized by episodic attacks of muscle weakness associated with a decrease in blood potassium levels. Mutations in the gene encoding the skeletal muscle voltage-gated calcium channel alpha-1 subunit (CACNL1A3) account for the majority of cases. Recently, mutations in the gene coding for the skeletal muscle voltage-gated sodium channel alpha subunit (SCN4A) have been reported in a small number of hypoPP families. In order to determine the relative frequency of the CANCL1A3 and SCN4A mutations in a large population of hypoPP patients, and to specify the clinical and pathological features associated with each of them, we searched for mutations in 58 independent hypoPP index cases. We detected the causative mutation in 45 cases: 40 were linked to the CACNL1A3 gene and five to the SCN4A gene. One mutation has not been described before. Some remarkable clinical features were observed in a large hypoPP family carrying an SCN4A mutation: a complete penetrance in men and women, an early age at onset, postcritic myalgias and an increased number and severity of attacks induced by acetazolamide. A muscle biopsy, performed in two members of this family, revealed a peculiar myopathy characterized by tubular aggregates. In contrast, vacuoles were predominant in muscles from hypoPP patients carrying CACNL1A3 mutations. Our findings point to the usefulness of a molecular characterization of hypoPP patients in clinical practice. They also provide new clues for understanding the mechanisms behind functional and structural alterations of the skeletal muscle in hypoPP. (+info)Viral vector-mediated expression of K+ channels regulates electrical excitability in skeletal muscle. (8/68)
Modification of K+ currents by exogenous gene expression may lead to therapeutic interventions in skeletal muscle diseases characterized by alterations in electrical excitability. In order to study the specific effects of increasing outward K+ currents, we expressed a modified voltage-dependent K+ channel in primary cultured rat skeletal muscle cells. The rat Kv1.4 channel was expressed as an N-terminal fusion protein containing a bioluminescent marker (green fluorescent protein). Transgene expression was carried out using the helper-dependent herpes simplex 1 amplicon system. Transduced myoballs, identified using fluorescein optics and studied electrophysiologically with single-cell patch clamp, exhibited a greater than two-fold increase in K+ conductance by 20-30 h after infection. This increase in K+ current led to a decrease in membrane resistance and a 10-fold increase in the current threshold for action potential generation. Electrical hyperexcitability induced by the Na+ channel toxin anemone toxin II (1 microM) was effectively counteracted by overexpression of Kv1.4 at 30-32 h after transduction. Thus, virally induced overexpression of a voltage-gated K+ channel in skeletal muscle has a powerful effect in reducing electrical excitability. (+info)Hypokalemic Periodic Paralysis (HPP) is a group of rare inherited disorders characterized by episodes of muscle weakness or paralysis, often associated with low potassium levels in the blood (hypokalemia). During an attack, muscles may become weak or fully paralyzed, typically affecting the legs and arms. The episodes can last from several hours to days. HPP is caused by genetic mutations that affect ion channels in muscle cells, leading to an imbalance of electrolytes and impaired muscle function. There are two main types: primary (or classic) HPP and secondary HPP. Primary HPP is further divided into thyrotoxic HPP and normokalemic HPP. Secondary HPP can be caused by various factors, such as medications or underlying medical conditions that cause hypokalemia.
Familial periodic paralysis is a group of rare genetic disorders characterized by episodes of muscle weakness or paralysis that recur over time. There are several types of familial periodic paralysis, including hypokalemic periodic paralysis, hyperkalemic periodic paralysis, and normokalemic periodic paralysis, each with its own specific genetic cause and pattern of symptoms.
In general, these disorders are caused by mutations in genes that regulate ion channels in muscle cells, leading to abnormalities in the flow of ions such as potassium in and out of the cells. This can result in changes in muscle excitability and contractility, causing episodes of weakness or paralysis.
The episodes of paralysis in familial periodic paralysis can vary in frequency, duration, and severity. They may be triggered by factors such as rest after exercise, cold or hot temperatures, emotional stress, alcohol consumption, or certain medications. During an episode, the affected muscles may become weak or completely paralyzed, often affecting the limbs but sometimes also involving the muscles of the face, throat, and trunk.
Familial periodic paralysis is typically inherited in an autosomal dominant pattern, meaning that a child has a 50% chance of inheriting the disorder if one parent is affected. However, some cases may arise from new mutations in the affected gene and occur in people with no family history of the disorder.
Treatment for familial periodic paralysis typically involves avoiding triggers and managing symptoms during episodes. In some cases, medications such as potassium-binding agents or diuretics may be used to help prevent or reduce the severity of episodes. Lifestyle modifications, such as a low-carbohydrate or high-sodium diet, may also be recommended in some cases.
Hypokalemia is a medical condition characterized by abnormally low potassium levels in the blood, specifically when the concentration falls below 3.5 milliequivalents per liter (mEq/L). Potassium is an essential electrolyte that helps regulate heart function, nerve signals, and muscle contractions.
Hypokalemia can result from various factors, including inadequate potassium intake, increased potassium loss through the urine or gastrointestinal tract, or shifts of potassium between body compartments. Common causes include diuretic use, vomiting, diarrhea, certain medications, kidney diseases, and hormonal imbalances.
Mild hypokalemia may not cause noticeable symptoms but can still affect the proper functioning of muscles and nerves. More severe cases can lead to muscle weakness, fatigue, cramps, paralysis, heart rhythm abnormalities, and in rare instances, respiratory failure or cardiac arrest. Treatment typically involves addressing the underlying cause and replenishing potassium levels through oral or intravenous (IV) supplementation, depending on the severity of the condition.
Hyperkalemic periodic paralysis (HypPK) is a rare genetic disorder characterized by episodes of muscle weakness or paralysis due to an abnormality in the ion channels that regulate the movement of potassium into and out of muscle fibers. This results in an excessive accumulation of potassium in the blood (hyperkalemia) during attacks, which can interfere with the ability of nerve cells to communicate with muscles and cause temporary muscle weakness or paralysis.
HypPK is caused by mutations in the SCN4A gene, which encodes a sodium channel protein found in skeletal muscle. These genetic changes disrupt the normal functioning of the ion channels, leading to an imbalance in the flow of ions across the muscle cell membrane and affecting muscle excitability.
Episodes of paralysis in HypPK typically begin in childhood or adolescence and can last from several hours to days. Triggers for attacks may include rest following exercise, cold or hot weather, stress, alcohol consumption, infection, or certain medications. Between episodes, individuals with HypPK usually have normal muscle strength and function.
Management of HypPK includes avoiding triggers, maintaining a low-potassium diet, and using medications to prevent or treat attacks. Medications such as thiazide diuretics, acetazolamide, or dichlorphenamide can help lower potassium levels in the blood and reduce the frequency and severity of episodes. In some cases, intravenous glucose and insulin may be used to drive potassium into cells during an attack, thereby reducing its concentration in the blood and alleviating symptoms.
NAV1.4, also known as SCN4A, is a gene that encodes for the α subunit of the voltage-gated sodium channel in humans. This channel, specifically located in the skeletal muscle, is responsible for the rapid influx of sodium ions during the initiation and propagation of action potentials, which are critical for muscle contraction.
The NAV1.4 Voltage-Gated Sodium Channel plays a crucial role in the functioning of skeletal muscles. Mutations in this gene can lead to various neuromuscular disorders such as hyperkalemic periodic paralysis, paramyotonia congenita, and potassium-aggravated myotonia, which are characterized by muscle stiffness, cramps, and episodes of weakness or paralysis.
Thyrotoxicosis is a medical condition that results from an excess of thyroid hormones in the body, leading to an overactive metabolic state. It can be caused by various factors such as Graves' disease, toxic adenoma, Plummer's disease, or excessive intake of thyroid hormone medication. Symptoms may include rapid heart rate, weight loss, heat intolerance, tremors, and increased sweating, among others. Thyrotoxicosis is not a diagnosis itself but a manifestation of various underlying thyroid disorders. Proper diagnosis and management are crucial to prevent complications and improve quality of life.
Paralysis is a loss of muscle function in part or all of your body. It can be localized, affecting only one specific area, or generalized, impacting multiple areas or even the entire body. Paralysis often occurs when something goes wrong with the way messages pass between your brain and muscles. In most cases, paralysis is caused by damage to the nervous system, especially the spinal cord. Other causes include stroke, trauma, infections, and various neurological disorders.
It's important to note that paralysis doesn't always mean a total loss of movement or feeling. Sometimes, it may just cause weakness or numbness in the affected area. The severity and extent of paralysis depend on the underlying cause and the location of the damage in the nervous system.
Sodium channels are specialized protein structures that are embedded in the membranes of excitable cells, such as nerve and muscle cells. They play a crucial role in the generation and transmission of electrical signals in these cells. Sodium channels are responsible for the rapid influx of sodium ions into the cell during the initial phase of an action potential, which is the electrical signal that travels along the membrane of a neuron or muscle fiber. This sudden influx of sodium ions causes the membrane potential to rapidly reverse, leading to the depolarization of the cell. After the action potential, the sodium channels close and become inactivated, preventing further entry of sodium ions and helping to restore the resting membrane potential.
Sodium channels are composed of a large alpha subunit and one or two smaller beta subunits. The alpha subunit forms the ion-conducting pore, while the beta subunits play a role in modulating the function and stability of the channel. Mutations in sodium channel genes have been associated with various inherited diseases, including certain forms of epilepsy, cardiac arrhythmias, and muscle disorders.
Andersen Syndrome is a rare genetic disorder characterized by the presence of three major features:
1. Periodic episodes of muscle weakness (periodic paralysis)
2. Potassium-sensitive ventricular arrhythmias
3. Physical deformities of the face and skeleton
The periodic paralysis in Andersen Syndrome is typically less severe than other forms of periodic paralysis, and it can be triggered by factors such as cold, emotional stress, or infection. The potassium-sensitive ventricular arrhythmias can be life-threatening and may require treatment with medications or an implantable cardioverter-defibrillator (ICD).
The physical deformities associated with Andersen Syndrome can include a short stature, low-set ears, a broad nose, widely spaced eyes, a cleft palate, and skeletal abnormalities such as scoliosis or clubfoot. These features may vary in severity among individuals with the disorder.
Andersen Syndrome is caused by mutations in the gene for the protein called the inward rectifier potassium channel (Kir2.1), which is involved in regulating the flow of potassium ions across cell membranes. This gene is located on chromosome 17 and is designated KCNJ2. The disorder is inherited in an autosomal dominant manner, meaning that a person has a 50% chance of inheriting the mutated gene from an affected parent. However, some cases of Andersen Syndrome are due to new (de novo) mutations and occur in people with no family history of the disorder.
Potassium is a essential mineral and an important electrolyte that is widely distributed in the human body. The majority of potassium in the body (approximately 98%) is found within cells, with the remaining 2% present in blood serum and other bodily fluids. Potassium plays a crucial role in various physiological processes, including:
1. Regulation of fluid balance and maintenance of normal blood pressure through its effects on vascular tone and sodium excretion.
2. Facilitation of nerve impulse transmission and muscle contraction by participating in the generation and propagation of action potentials.
3. Protein synthesis, enzyme activation, and glycogen metabolism.
4. Regulation of acid-base balance through its role in buffering systems.
The normal serum potassium concentration ranges from 3.5 to 5.0 mEq/L (milliequivalents per liter) or mmol/L (millimoles per liter). Potassium levels outside this range can have significant clinical consequences, with both hypokalemia (low potassium levels) and hyperkalemia (high potassium levels) potentially leading to serious complications such as cardiac arrhythmias, muscle weakness, and respiratory failure.
Potassium is primarily obtained through the diet, with rich sources including fruits (e.g., bananas, oranges, and apricots), vegetables (e.g., leafy greens, potatoes, and tomatoes), legumes, nuts, dairy products, and meat. In cases of deficiency or increased needs, potassium supplements may be recommended under the guidance of a healthcare professional.
Skeletal muscle, also known as striated or voluntary muscle, is a type of muscle that is attached to bones by tendons or aponeuroses and functions to produce movements and support the posture of the body. It is composed of long, multinucleated fibers that are arranged in parallel bundles and are characterized by alternating light and dark bands, giving them a striped appearance under a microscope. Skeletal muscle is under voluntary control, meaning that it is consciously activated through signals from the nervous system. It is responsible for activities such as walking, running, jumping, and lifting objects.
Hyperkalemia is a medical condition characterized by an elevated level of potassium (K+) in the blood serum, specifically when the concentration exceeds 5.0-5.5 mEq/L (milliequivalents per liter). Potassium is a crucial intracellular ion that plays a significant role in various physiological processes, including nerve impulse transmission, muscle contraction, and heart rhythm regulation.
Mild to moderate hyperkalemia might not cause noticeable symptoms but can still have harmful effects on the body, particularly on the cardiovascular system. Severe cases of hyperkalemia (potassium levels > 6.5 mEq/L) can lead to potentially life-threatening arrhythmias and heart failure.
Hyperkalemia may result from various factors, such as kidney dysfunction, hormonal imbalances, medication side effects, trauma, or excessive potassium intake. Prompt identification and management of hyperkalemia are essential to prevent severe complications and ensure proper treatment.
Calcium channels are specialized proteins that span the membrane of cells and allow calcium ions (Ca²+) to flow in and out of the cell. They are crucial for many physiological processes, including muscle contraction, neurotransmitter release, hormone secretion, and gene expression.
There are several types of calcium channels, classified based on their biophysical and pharmacological properties. The most well-known are:
1. Voltage-gated calcium channels (VGCCs): These channels are activated by changes in the membrane potential. They are further divided into several subtypes, including L-type, P/Q-type, N-type, R-type, and T-type. VGCCs play a critical role in excitation-contraction coupling in muscle cells and neurotransmitter release in neurons.
2. Receptor-operated calcium channels (ROCCs): These channels are activated by the binding of an extracellular ligand, such as a hormone or neurotransmitter, to a specific receptor on the cell surface. ROCCs are involved in various physiological processes, including smooth muscle contraction and platelet activation.
3. Store-operated calcium channels (SOCCs): These channels are activated by the depletion of intracellular calcium stores, such as those found in the endoplasmic reticulum. SOCCs play a critical role in maintaining calcium homeostasis and signaling within cells.
Dysregulation of calcium channel function has been implicated in various diseases, including hypertension, arrhythmias, migraine, epilepsy, and neurodegenerative disorders. Therefore, calcium channels are an important target for drug development and therapy.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Bartter syndrome is a rare genetic disorder that affects the kidneys' ability to reabsorb sodium and chloride, leading to an imbalance of electrolytes in the body. This condition is characterized by hypokalemia (low potassium levels), metabolic alkalosis (high pH levels in the blood), and normal or low blood pressure. It can also result in increased urine production, excessive thirst, and growth retardation in children. There are two major types of Bartter syndrome, based on the genes affected: type I caused by mutations in the SLC12A1 gene, and type II caused by mutations in the KCNJ1 gene. Type III is caused by mutations in the CLCNKB gene, while type IV is caused by mutations in the BSND or CLCNKB genes. Treatment typically involves supplementation of electrolytes, such as potassium and magnesium, as well as nonsteroidal anti-inflammatory drugs (NSAIDs) to help reduce sodium loss in the urine.
Renal tubular acidosis (RTA) is a medical condition that occurs when the kidneys are unable to properly excrete acid into the urine, leading to an accumulation of acid in the bloodstream. This results in a state of metabolic acidosis.
There are several types of RTA, but renal tubular acidosis type 1 (also known as distal RTA) is characterized by a defect in the ability of the distal tubules to acidify the urine, leading to an inability to lower the pH of the urine below 5.5, even in the face of metabolic acidosis. This results in a persistently alkaline urine, which can lead to calcium phosphate stones and bone demineralization.
Type 1 RTA is often caused by inherited genetic defects, but it can also be acquired due to various kidney diseases, drugs, or autoimmune disorders. Symptoms of type 1 RTA may include fatigue, weakness, muscle cramps, decreased appetite, and vomiting. Treatment typically involves alkali therapy to correct the acidosis and prevent complications.
Myotonia is a condition characterized by the delayed relaxation of a muscle after voluntary contraction or electrical stimulation, resulting in stiffness or difficulty with relaxing the muscles. It's often associated with certain neuromuscular disorders such as myotonic dystrophy and myotonia congenita. The prolonged muscle contraction can cause stiffness, especially after periods of rest, and may improve with repeated contractions (warm-up phenomenon).
Channelopathies are genetic disorders that are caused by mutations in the genes that encode for ion channels. Ion channels are specialized proteins that regulate the flow of ions, such as sodium, potassium, and calcium, across cell membranes. These ion channels play a crucial role in various physiological processes, including the generation and transmission of electrical signals in the body.
Channelopathies can affect various organs and systems in the body, depending on the type of ion channel that is affected. For example, mutations in sodium channel genes can cause neuromuscular disorders such as epilepsy, migraine, and periodic paralysis. Mutations in potassium channel genes can cause cardiac arrhythmias, while mutations in calcium channel genes can cause neurological disorders such as episodic ataxia and hemiplegic migraine.
The symptoms of channelopathies can vary widely depending on the specific disorder and the severity of the mutation. Treatment typically involves managing the symptoms and may include medications, lifestyle modifications, or in some cases, surgery.
Alkalosis is a medical condition that refers to an excess of bases or a decrease in the concentration of hydrogen ions (H+) in the blood, leading to a higher than normal pH level. The normal range for blood pH is typically between 7.35 and 7.45. A pH above 7.45 indicates alkalosis.
Alkalosis can be caused by several factors, including:
1. Metabolic alkalosis: This type of alkalosis occurs due to an excess of bicarbonate (HCO3-) in the body, which can result from conditions such as excessive vomiting, hyperventilation, or the use of certain medications like diuretics.
2. Respiratory alkalosis: This form of alkalosis is caused by a decrease in carbon dioxide (CO2) levels in the blood due to hyperventilation or other conditions that affect breathing, such as high altitude, anxiety, or lung disease.
Symptoms of alkalosis can vary depending on its severity and underlying cause. Mild alkalosis may not produce any noticeable symptoms, while severe cases can lead to muscle twitching, cramps, tremors, confusion, and even seizures. Treatment for alkalosis typically involves addressing the underlying cause and restoring the body's normal pH balance through medications or other interventions as necessary.
 Hypokalemic periodic paralysis
Hypokalemic periodic paralysis
Thyrotoxic periodic paralysis
List of OMIM disorder codes
Cav1.1
Hypokalemic sensory overstimulation
Hyperkalemic periodic paralysis
Nav1.4
Kir2.6
Inward-rectifier potassium channel
Periodic paralysis
Sleep paralysis
KCNE3
Wolff-Parkinson-White syndrome
Conversion disorder
Signs and symptoms of Graves' disease
Hypokalemia
ZBED1
Mary Broadfoot Walker
Elizabeth Barrett Browning
Diclofenamide
Andersen-Tawil syndrome
Ankle jerk reflex
List of MeSH codes (C05)
Long QT syndrome
HPP
Renal tubular acidosis
List of MeSH codes (C10)
Familial hemiplegic migraine
List of MeSH codes (C18)
List of MeSH codes (C16)
Hypokalemic periodic paralysis - Wikipedia
JCI - A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis
 Hypokalemic periodic paralysis Information | Mount Sinai - New York
Hypokalemic periodic paralysis Information | Mount Sinai - New York
 CACNA1S gene: MedlinePlus Genetics
CACNA1S gene: MedlinePlus Genetics
 Keveyis for the Treatment of Primary Hyperkalemic and Hypokalemic Periodic Paralysis - Clinical Trials Arena
Keveyis for the Treatment of Primary Hyperkalemic and Hypokalemic Periodic Paralysis - Clinical Trials Arena
 Hypokalemic - Surveys Archives - Periodic Paralysis International
Hypokalemic - Surveys Archives - Periodic Paralysis International
 A mutation in the KCNE3 potassium channel gene is associated with susceptibility to thyrotoxic hypokalemic periodic paralysis
A mutation in the KCNE3 potassium channel gene is associated with susceptibility to thyrotoxic hypokalemic periodic paralysis
 Hypokalemic Periodic Paralysis Due To Distal Renal Tubular Acidosis
Hypokalemic Periodic Paralysis Due To Distal Renal Tubular Acidosis
 Conn's syndrome, subclinical Cushing's syndrome and thyrotoxicosis presenting as hypokalemic periodic paralysis: A case report ...
Conn's syndrome, subclinical Cushing's syndrome and thyrotoxicosis presenting as hypokalemic periodic paralysis: A case report ...
Health Library | Rutgers Cancer Institute of New Jersey
 Pediatric Hypokalemia: Background, Pathophysiology, Etiology
Pediatric Hypokalemia: Background, Pathophysiology, Etiology
![Ginjaar HB[au] - Search Results - PubMed](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAMAAAAoLQ9TAAAARVBMVEVHcEwoU45gYmYAUpQAUpRPYGVgYmZLXnJgYmYAUZUAUpRJXnIAUpQAUpRgYmYAUpRgYmZgYmZhYmYAUpQAUpQAUpRgYmaDiPJuAAAAFXRSTlMADOJ+6QewGO8/uTRqtH7GdFJ11p1bCL3TAAAAZUlEQVQYlV2PVw7AIAxDTeney7n/UcsoldX3E+VJOAboEi7MBpHWMs1ADlG8u7UYWauwyZFeRQVPOhG2o+aiwhByJxUx91Jxhje3iJSqGfHuLKI0+0TpXvY1twCOPlFh5pa/++MB0vIOBm+1zaoAAAAASUVORK5CYII=) Ginjaar HB[au] - Search Results - PubMed
Ginjaar HB[au] - Search Results - PubMed
Periodic Paralyses Follow-up: Prognosis, Patient Education
 Thyrotoxic Hypokalaemic Periodic Paralysis (TPP) in Turkey: Report of a Case and Review of the Literature
Thyrotoxic Hypokalaemic Periodic Paralysis (TPP) in Turkey: Report of a Case and Review of the Literature
![Thyrotoxic hypokalemic periodic paralysis, an endocrine emergency: clinical and genetic features in 25 patients]. | Arq Bras...](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAYAAAAf8/9hAAACpklEQVQ4jaXTe2iNcRzH8ffz7Jzncp4ZZyZjZ6bmEvPPxrDIJSnJZZJIbn8Qk1CI2pQ7JX8h5fKHW4o/3EuaWnJvrUNqiFZssTWcs83z7DnPc87XH88Ziv98//z1+7369v38vvCfpQBY0w6LeHb2RCHl+fg9vaiGxuyq0ZQMzef0tSeoOSqGEQaR4Go4QghAPBvxHRzbBddjUGGUhXMmULN0KhVlxQDMLC+iZt9VEt+6MPNMCIwAAEj7aSrGxFg5r5KVCyoZGM2lx3Y5dbGB6jnlLJs7nvIxMZbvOE9TvAUjagXtAygVu2Tk3P3ieb6IiPjpjBw9Vy+F02uFYWulaEadnLn2WEREJCOyetdFYdRGMSfXSQ4ABVV7Jo4rYtWCiVyvf8nizWe4cuMZdkYwBlh863a4fa+JR/EWyscWs37pFAxD4/7T96hB/2lKhuYDcORsPW+aWzHz+6FrIRDB1MMY0Vwe3I9Td+IuALMmjyIjZAGBkiFRACKmBqb+77hyDQw9DED8TRukvCygKpQWFwCQyUb0zxIwtQD40NoZPAVQdY3S2EAAkj9cfo/3b0DJ+h8/fwdVCYA8S6d0WNBBbHB/6LJJeWmUPkiB3pQPPQ7RPDMAviQglBMAtuNSs/cqyW6HO8fXcaB2CZYWwk78IOWlcb72UDjA4tihFRzcOg+Ato4EaigIkUjVbmHEBimbf1Diza0iIvKupV2qN52W/MrtsuPoDUl22dJXl2+9EHP8NtEn1YnSB0i6F6fbwYronKxdwupFkwCwbZdIRKe9s5sLN59z6W4jr962oZs6OVrk91dGhEg/A8f1WbPzPI3Nnzi8ZT7NLe2cuPyQWw2vSXQmwdAwLIM/xvP3NmYyGXptl+GxAlo7kvi2S9gyCIfVX0sEwTb+BBBpI5QgousUAAAAAElFTkSuQmCC) Thyrotoxic hypokalemic periodic paralysis, an endocrine emergency: clinical and genetic features in 25 patients]. | Arq Bras...
Thyrotoxic hypokalemic periodic paralysis, an endocrine emergency: clinical and genetic features in 25 patients]. | Arq Bras...
 Case Report: Recurrent hypokalemic periodic paralysis associated with distal renal tubular acidosis (type 1) and hypothyroidism...
Case Report: Recurrent hypokalemic periodic paralysis associated with distal renal tubular acidosis (type 1) and hypothyroidism...
 Hypokalemia | Diagnosaurus
Hypokalemia | Diagnosaurus
 Best FOAM Podcasts (2023)
Best FOAM Podcasts (2023)
 Vol 66, Issue 2: 2017 | West Indian Medical Journal
Vol 66, Issue 2: 2017 | West Indian Medical Journal
 Paralysis: Definition and Patient Education
Paralysis: Definition and Patient Education
 Vol. 11 No. 2 (2023)
| Bangladesh Critical Care Journal
Vol. 11 No. 2 (2023)
| Bangladesh Critical Care Journal
Perioperative Management of Neuromuscular Disorders: Practice Essentials, Problem, Management
 ICGNMD Publications | International Centre for Genomic Medicine in Neuromuscular Diseases - UCL - University College London
ICGNMD Publications | International Centre for Genomic Medicine in Neuromuscular Diseases - UCL - University College London
 Familial Periodic Paralysis - Pediatrics - MSD Manual Professional Edition
Familial Periodic Paralysis - Pediatrics - MSD Manual Professional Edition
Orphanet: Search a disease
Dr.Debmalya Sanyal :.
 Division of Nephrology, Hypertension & Apheresis - Research output
- Research Profiles at Washington University School of...
Division of Nephrology, Hypertension & Apheresis - Research output
- Research Profiles at Washington University School of...
 Klor-Con M: Package Insert - Drugs.com
Klor-Con M: Package Insert - Drugs.com
Abstracts | 0012 | SFE2006 | 197th Meeting of the Society for Endocrinology | Endocrine Abstracts
 An Unusual Malfunction of an Anesthetic Machine | Anesthesiology | American Society of Anesthesiologists
An Unusual Malfunction of an Anesthetic Machine | Anesthesiology | American Society of AnesthesiologistsHyperkalemic11
- citation needed] In contrast, hyperkalemic periodic paralysis refers to gain-of-function mutations in sodium channels that maintain muscle depolarisation and therefore are aggravated by high potassium ion concentrations. (wikipedia.org)
- Mutations of Na V 1.4 give rise to a heterogeneous group of muscle disorders, with gain-of-function defects causing myotonia or hyperkalemic periodic paralysis. (jci.org)
- HypoPP is one of a group of genetic disorders that includes hyperkalemic periodic paralysis and thyrotoxic periodic paralysis . (mountsinai.org)
- Keveyis (dichlorphenamide) is the first medicine indicated for the treatment of primary hyperkalemic and hypokalemic periodic paralysis, which are a group of rare hereditary disorders that cause occasional episodes of muscle weakness or paralysis. (clinicaltrialsarena.com)
- Keveyis 50mg tablet was approved by US Food and Drug Administration (FDA) for the treatment for primary hyperkalemic and hypokalemic periodic paralysis and related variants in August 2015. (clinicaltrialsarena.com)
- Common types of primary PP include hyperkalemic and hypokalemic. (clinicaltrialsarena.com)
- The approval of Keveyis by the FDA was based on results from two clinical trials, namely Study 1 and Study 2, conducted to evaluate the efficacy of the drug in patients with hyperkalemic and hypokalemic periodic paralysis. (clinicaltrialsarena.com)
- Sub-study 1 was conducted on patients with hypokalemic periodic paralysis and the other on patients with hyperkalemic periodic paralysis. (clinicaltrialsarena.com)
- One sub-study was on patients with hypokalemic PP and the other was in those with hyperkalemic PP, including patients with paramyotonia congenita, a disorder that affects muscles used for movement. (clinicaltrialsarena.com)
- The carbonic anhydrase inhibitor dichlorphenamide is indicated for long-term treatment of primary hyperkalemic periodic paralysis, primary hypokalemic periodic paralysis, and related variants. (medscape.com)
- Some patients may develop chronic muscle weakness later in life.Hyperkalemic periodic paralysis is characterized by a rise in potassium levels in the blood. (brainfacts.org)
Familial11
- Hypokalemic periodic paralysis (hypoKPP), also known as familial hypokalemic periodic paralysis (FHPP), is a rare, autosomal dominant channelopathy characterized by muscle weakness or paralysis when there is a fall in potassium levels in the blood. (wikipedia.org)
- the most common causes are Familial Hypokalemic Periodic Paralysis (FHypoKPP), an autosomal dominant disease, and Thyrotoxic Hypokalemic Periodic Paralysis (THypoKPP), secondary to thyrotoxicosis. (unifesp.br)
- Familial periodic paralyses information page. (epnet.com)
- Available at: https://www.ninds.nih.gov/Disorders/All-Disorders/Familial-Periodic-Paralyses-Information-Page. (epnet.com)
- Attacks are transient , self -limited, associated with hypokalemia and resemble those of familial hypokalemic periodic paralysis (FHPP), an autosomal dominant neurological channelopathy . (bvsalud.org)
- Familial periodic paralysis is a rare autosomal dominant condition with considerable variation in penetrance characterized by episodes of flaccid paralysis with loss of deep tendon reflexes and failure of muscle to respond to electrical stimulation. (msdmanuals.com)
- Each form of familial periodic paralysis involves a different gene and electrolyte channel. (msdmanuals.com)
- Although the hypokalemic form is the most common form of familial periodic paralysis, it is nonetheless quite rare, with a prevalence of 1/100,000. (msdmanuals.com)
- Familial periodic paralyses are a group of inherited neurological disorders caused by mutations in genes that regulate sodium and calcium channels in nerve cells. (brainfacts.org)
- The prognosis for the familial periodic paralyses varies. (brainfacts.org)
- The NINDS conducts and supports research on neuromuscular disorders such as the familial periodic paralyses. (brainfacts.org)
Thyrotoxic periodic4
- This entity is distinguished with thyroid function tests, and the diagnosis is instead called thyrotoxic periodic paralysis. (wikipedia.org)
- Chaudhry MA, Wayangankar S. Thyrotoxic Periodic Paralysis: A Concise Review of the Literature. (medscape.com)
- Thyrotoxic periodic paralysis (TPP) is a potentially life-threatening complication of Graves' disease (GD). (hindawi.com)
- Thyrotoxic periodic paralysis (TPP) is a rare but potentially life-threatening complication of thyrotoxicosis characterized by muscle paralysis and serum hypokalemia due to massive shifting of potassium into the intracellular space [ 1 , 2 ]. (hindawi.com)
HypoKPP1
- Background: Hypokalemic periodic paralysis (HypoKPP) is characterized by transient episodes of flaccid muscle weakness. (edu.pe)
Thyrotoxicosis4
- Symptoms of paralysis are similar in both diseases, distinguished by thyrotoxicosis present in THypoKPP. (unifesp.br)
- Since both diseases are similar, we tested the hypothesis that THypoKPP could carry the same mutations described in FHypoKPP, being the paralysis a genetically conditioned complication of thyrotoxicosis. (unifesp.br)
- Siddiqui FA, Sheikh A. Muscle paralysis in thyrotoxicosis. (medscape.com)
- Thyrotoxic hypokalemic periodic paralysis (THPP) is a medical emergency characterized by acute attacks of weakness, hypokalemia , and thyrotoxicosis that resolve with the treatment of hyperthyroidism . (bvsalud.org)
Autosomal1
- Hypokalemic periodic paralysis is a rare autosomal dominant hereditary disorder that is characterized by transient episodes of skeletal muscle weakness/paralysis when the serum potassium level decreases. (medscape.com)
HypoPP8
- Hypokalemic periodic paralysis (HypoPP) is an ion channelopathy of skeletal muscle characterized by attacks of muscle weakness associated with low serum K + . HypoPP results from a transient failure of muscle fiber excitability. (jci.org)
- To address the question of specificity for the allele encoding the Na V 1.4-R669H variant as a cause of HypoPP and to produce a model system in which to characterize functional defects of the mutant channel and susceptibility to paralysis, we generated knockin mice carrying the ortholog of the gene encoding the Na V 1.4-R669H variant (referred to herein as R669H mice). (jci.org)
- Hypokalemic periodic paralysis (hypoPP) is a disorder that causes occasional episodes of muscle weakness and sometimes a lower than normal level of potassium in the blood. (mountsinai.org)
- HypoPP is the most common form of periodic paralysis. (mountsinai.org)
- Unlike other forms of periodic paralysis, people with hypoPP have normal thyroid function. (mountsinai.org)
- In the hypokalemic form, 70% of affected people have a mutation in the alpha-subunit of the voltage-sensitive muscle calcium channel gene on chromosome 1q (HypoPP type I). In some families, the mutation is in the alpha-subunit of the sodium channel gene on chromosome 17 (HypoPP type II). (msdmanuals.com)
- A rare genetic disorder called hypokalemic periodic paralysis (hypoPP) causes sudden, profound muscle weakness in people who occasionally exhibit low levels of potassium in their blood, or hypokalemia. (lbl.gov)
- When a patient is hypokalemic, hypoPP affects the function of the muscles responsible for skeletal movement. (lbl.gov)
Flaccid paralysis3
- Flaccid paralysis causes your muscles to shrink and become flabby. (healthline.com)
- Potassium depletion may produce weakness, fatigue, disturbances or cardiac rhythm (primarily ectopic beats), prominent U-waves in the electrocardiogram, and in advanced cases, flaccid paralysis and/or impaired ability to concentrate urine. (drugs.com)
- Guillain-Barré Syndrome (GBS) is the most frequent cause of acute and sub-acute flaccid paralysis after polio eradication. (bvsalud.org)
Susceptibility1
- Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. (medscape.com)
Types of Periodic Paralysis1
- Arzel-Hezode M, McGoey S, Sternberg D, Vicart S, Eymard B, Fontaine B. Glucocorticoids may trigger attacks in several types of periodic paralysis. (medscape.com)
Skeletal muscle1
- Hypokalemic periodic paralysis (HPP) is a disorder that characterized by attacks of skeletal muscle paralysis depending on the changes in serum potassium levels, and can occur due to primary and secondary causes. (ejgm.org)
Hypokalemia1
- During an acute attack of TPP with marked hypokalemia, cautious potassium supplementation is of paramount importance as it prevents major cardiopulmonary complications [ 6 ] but, at the same time, one must bear in mind that overadministration of potassium could lead to rebound hyperkalemia and fatal dysrhythmia during recovery from paralysis [ 7 ]. (hindawi.com)
Weakness or paralysis3
- Attacks cause severe weakness or paralysis that usually lasts from hours to days. (wikipedia.org)
- citation needed] Mutations altering the usual structure and function of these channels therefore disrupt regulation of muscle contraction, leading to episodes of severe muscle weakness or paralysis. (wikipedia.org)
- Periodic paralysis (PP) is a heterogeneous group of hereditary muscle disorders characterised by episodes of muscle weakness or paralysis occurring at irregular intervals. (clinicaltrialsarena.com)
Mutations5
- Mutations in the following genes can cause hypokalemic periodic paralysis: An association with KCNE3 (voltage-gated potassium channel) has also been described, but is currently disputed, and excluded from the disease definition in OMIM. (wikipedia.org)
- Mutations in KCNJ2 lead to hypokalemic periodic paralysis with cardiac arrhythmias called Andersen-Tawil syndrome. (wikipedia.org)
- At least 11 mutations in the CACNA1S gene have been identified in people with hypokalemic periodic paralysis, a condition that causes episodes of extreme muscle weakness, usually in the arms and legs. (medlineplus.gov)
- Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. (medlineplus.gov)
- Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. (medscape.com)
Symptoms6
- Some people only develop symptoms of periodic paralysis due to hyperthyroidism (overactive thyroid). (wikipedia.org)
- Symptoms include attacks of muscle weakness or loss of muscle movement ( paralysis ) that come and go. (mountsinai.org)
- A treatment plan and outlook for the condition will depend on the underlying cause of paralysis, as well as symptoms experienced. (healthline.com)
- What are the symptoms of paralysis? (healthline.com)
- The symptoms of paralysis are usually easy to identify. (healthline.com)
- Treatment of the periodic paralyses focuses on preventing further attacks and relieving acute symptoms. (brainfacts.org)
20231
- 2023 ) Retigabine suppresses loss of force in mouse models of hypokalaemic periodic paralysis. (neurotree.org)
Attacks3
- Hypokalemic Periodic Paralyses comprise diverse diseases characterized by acute and reversible attacks of severe muscle weakness, associated with low serum potassium. (unifesp.br)
- Attacks are usually shorter, more frequent, and less severe than the hypokalemic form. (brainfacts.org)
- Avoiding carbohydrate-rich meals and strenuous exercise, and taking acetazolamide daily may prevent hypokalemic attacks. (brainfacts.org)
Sodium2
- Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemicperiodic paralysis. (medscape.com)
- In the disease state, a clear passage allows sodium to leak through, resulting in muscle paralysis. (lbl.gov)
Acetazolamide1
- Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. (medscape.com)
20202
- 2020 ) Hypokalaemic periodic paralysis with a charge-retaining substitution in the voltage sensor. (neurotree.org)
- 2020 ) A role for external Ca2+ in maintaining muscle contractility in periodic paralysis. (neurotree.org)
Myopathy1
- Muscle MRI in periodic paralysis shows myopathy is common and correlates with intramuscular fat accumulation. (ucl.ac.uk)
Disorders2
- Periodic paralyses (PP) are a rare group of disorders. (epnet.com)
- Neuromuscular disorders encompass a number of different disease processes, including myasthenia gravis (MG), Lambert-Eaton myasthenic syndrome (LEMS), Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and hypokalemic periodic paralysis. (medscape.com)
Muscle paralysis1
- X-ray crystallography of a membrane protein provided a structural understanding of how a single mutation can result in periodic muscle paralysis. (lbl.gov)
Gitelman1
- 2) Gitelman syndrome presenting as Hypokalaemic Periodic Paralysis. (calcuttayellowpages.com)
Chronic1
- Cienna has about 20 chronic conditions including hypokalemic periodic paralysis, mast cell activation syndrome, central hypoventilation syndrome, postural orthostatic tachycardia syndrome, narcolepsy with cataplexy, and others. (healthline.com)
Diagnosis3
- Review of the Diagnosis and Treatment of Periodic Paralysis. (medscape.com)
- If you're seeking treatment for paralysis, ask your doctor for more information about your specific diagnosis, treatment plan, and long-term outlook. (healthline.com)
- The clinicopathologic findings were consistent with a diagnosis of hypokalemic polymyopathy. (vin.com)
Genetic1
- Thyrotoxic hypokalemic periodic paralysis, an endocrine emergency: clinical and genetic features in 25 patients]. (bvsalud.org)
Affects1
- Localized paralysis affects only one part of your body, such as your face or hand. (healthline.com)
Patients2
- Patients who report muscle pain in association with their episodes are too often told that the periodic paralyses are not painful despite many authoritative reports to the contrary. (hkpp.org)
- Submitted by deb on Mon, 06/27/2011 - 00:31 In July of 1998 we conducted a survey of 64 self-reported clinically diagnosed periodic paralysis patients, all members of the HKPP Listserv. (hkpp.org)
Potassium levels1
- The two most common types of periodic paralyses are:Hypokalemic periodic paralysis is characterized by a fall in potassium levels in the blood. (brainfacts.org)
Treatment2
- Treatment for periodic paralysis. (epnet.com)
- [2] Treatment options for sleep paralysis have been poorly studied. (wikipedia.org)
Voltage sensor1
- Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. (medscape.com)
Severe1
- Weakness may be mild and limited to certain muscle groups, or more severe full-body paralysis. (wikipedia.org)