Incontinentia Pigmenti
Pigmentation Disorders
Toe Phalanges
Dosage Compensation, Genetic
I-kappa B Kinase
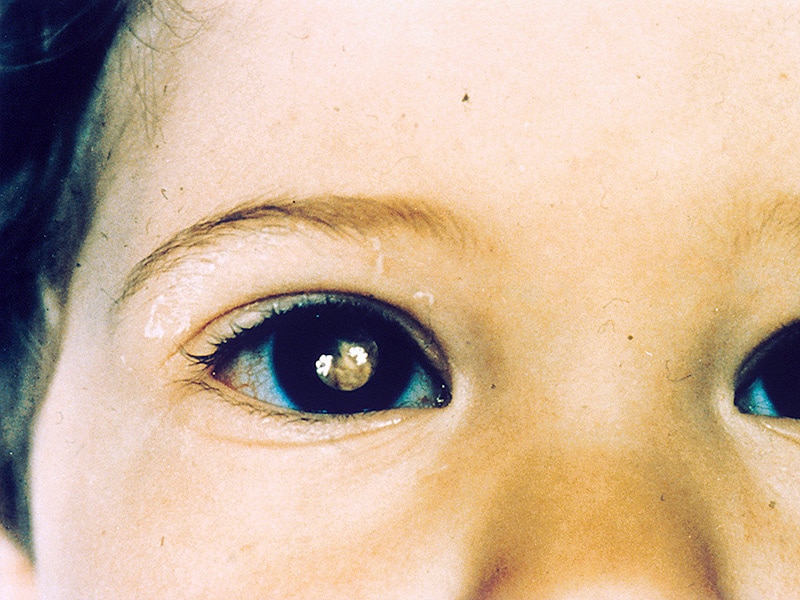
Macular vasculopathy and its evolution in incontinentia pigmenti. (1/55)
PURPOSE: To describe macular vasculopathy in incontinentia pigmenti. METHODS: Twelve baby girls with incontinentia pigmenti were examined under general anesthesia by fluorescein angiography of the macula. Nine eyes of 9 patients had sufficient detail to allow evaluation of capillary changes. Angiography was initiated as early as 3 months of age and was repeated in 7 eyes at 3- to 12-month intervals. Changes in capillary patterns were identified. RESULTS: Irregularly enlarged or distorted foveal avascular zones were noted in all 9 maculas. Sparseness of the perifoveolar capillary bed was a characteristic finding. Sequential macular angiography demonstrated non-progressive (stable) capillary closure in 2 eyes; progressive closure in another macula; progressive closure plus addition or reopening of macular capillaries in 3 eyes; and central retinal artery occlusion, with cherry-red spot formation, in 1 eye at 12 days of age. In addition, progressive tractional detachment of the macula occurred in 2 of these eyes, and progressive macular neovascularization occurred in 1 eye. CONCLUSIONS: Macular ischemia is characteristic of incontinentia pigmenti and is often progressive. It is the initiating event of a typical vasculopathy, characterized by capillary remodelling and, occasionally, by neovascularization and tractional detachment of the retina. (+info)Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. (2/55)
Rett syndrome (RTT) is a neurodevelopmental disorder characterized by loss of acquired skills after a period of normal development in infant girls. The responsible gene, encoding methyl-CpG binding protein 2 (MeCP2), was recently discovered. Here we explore the spectrum of phenotypes resulting from MECP2 mutations. Both nonsense (R168X and R255X) and missense (R106W and R306C) mutations have been found, with multiple recurrences. R168X mutations were identified in six unrelated sporadic cases, as well as in two affected sisters and their normal mother. The missense mutations were de novo and affect conserved domains of MeCP2. All of the nucleotide substitutions involve C-->T transitions at CpG hotspots. A single nucleotide deletion, at codon 137, that creates a L138X stop codon within the methyl-binding domain was found in an individual with features of RTT and incontinentia pigmenti. An 806delG deletion causing a V288X stop in the transcription-repression domain was identified in a woman with motor-coordination problems, mild learning disability, and skewed X inactivation; in her sister and daughter, who were affected with classic RTT; and in her hemizygous son, who died from congenital encephalopathy. Thus, some males with RTT-causing MECP2 mutations may survive to birth, and female heterozygotes with favorably skewed X-inactivation patterns may have little or no involvement. Therefore, MECP2 mutations are not limited to RTT and may be implicated in a much broader phenotypic spectrum. (+info)Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. (3/55)
IKK gamma/NEMO is the essential regulatory subunit of the I kappa B kinase (IKK), encoded by an X-linked gene in mice and humans. It is required for NF-kappa B activation and resistance to TNF-induced apoptosis. Female mice heterozygous for Ikk gamma/Nemo deficiency develop a unique dermatopathy characterized by keratinocyte hyperproliferation, skin inflammation, hyperkeratosis, and increased apoptosis. Although Ikk gamma+/- females eventually recover, Ikk gamma- males die in utero. These symptoms and inheritance pattern are very similar to those of incontinentia pigmenti (IP), a human genodermatosis, synthenic with the IKK gamma/NEMO locus. Indeed, biopsies and cells from IP patients exhibit defective IKK gamma/NEMO expression but normal expression of IKK catalytic subunits. This unique self-limiting disease, the first to be genetically linked to the IKK signaling pathway, is dependent on X-chromosome inactivation. We propose that the IKK gamma/NEMO-deficient cells trigger an inflammatory reaction that eventually leads to their death. (+info)NEMO/IKK gamma-deficient mice model incontinentia pigmenti. (4/55)
Disruption of the X-linked gene encoding NF-kappa B essential modulator (NEMO) produces male embryonic lethality, completely blocks NF-kappa B activation by proinflammatory cytokines, and interferes with the generation and/or persistence of lymphocytes. Heterozygous female mice develop patchy skin lesions with massive granulocyte infiltration and hyperproliferation and increased apoptosis of keratinocytes. Diseased animals present severe growth retardation and early mortality. Surviving mice recover almost completely, presumably through clearing the skin of NEMO-deficient keratinocytes. Male lethality and strikingly similar skin lesions in heterozygous females are hallmarks of the human genetic disorder incontinentia pigmenti (IP). Together with the recent discovery that mutations in the human NEMO gene cause IP, our results indicate that we have created a mouse model for that disease. (+info)A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). (5/55)
Hypohidrotic ectodermal dysplasia (HED), a congenital disorder of teeth, hair, and eccrine sweat glands, is usually inherited as an X-linked recessive trait, although rarer autosomal dominant and recessive forms exist. We have studied males from four families with HED and immunodeficiency (HED-ID), in which the disorder segregates as an X-linked recessive trait. Affected males manifest dysgammaglobulinemia and, despite therapy, have significant morbidity and mortality from recurrent infections. Recently, mutations in IKK-gamma (NEMO) have been shown to cause familial incontinentia pigmenti (IP). Unlike HED-ID, IP affects females and, with few exceptions, causes male prenatal lethality. IKK-gamma is required for the activation of the transcription factor known as "nuclear factor kappa B" and plays an important role in T and B cell function. We hypothesize that "milder" mutations at this locus may cause HED-ID. In all four families, sequence analysis reveals exon 10 mutations affecting the carboxy-terminal end of the IKK-gamma protein, a domain believed to connect the IKK signalsome complex to upstream activators. The findings define a new X-linked recessive immunodeficiency syndrome, distinct from other types of HED and immunodeficiency syndromes. The data provide further evidence that the development of ectodermal appendages is mediated through a tumor necrosis factor/tumor necrosis factor receptor-like signaling pathway, with the IKK signalsome complex playing a significant role. (+info)A pregnancy following PGD for X-linked dominant [correction of X-linked autosomal dominant] incontinentia pigmenti (Bloch-Sulzberger syndrome): case report. (6/55)
Incontinentia Pigmenti (Bloch-Sulzberger syndrome) is a rare multisystem, ectodermal disorder associated with dermatological, dental and ocular features, and in <10% of cases, severe neurological deficit. Pedigree review suggests X-linked dominance with lethality in affected males. Presentation in female carriers is variable. Following genetic counselling, a mildly affected female carrier diagnosed in infancy with a de novo mutation was referred for preimplantation sexing, unusually selecting for male gender, with an acceptance of either normality or early miscarriage in an affected male. Following standard in-vitro fertilization and embryo biopsy, fluorescence in situ hybridization (FISH) unambiguously identified two male and two female embryos. A single 8-cell, grade 4 male embryo was replaced. A positive pregnancy test was reported 2 weeks after embryo transfer, although ultrasonography failed to demonstrate a viable pregnancy. Post abortive fetal tissue karyotyping diagnosed a male fetus with trisomy 16. This is an unusual report of preimplantation genetic diagnosis (PGD) being used for selection of males in an X-linked autosomal dominant disorder and demonstrates the value of PGD where amniocentesis or chorion villus sampling followed by abortion is not acceptable to the patient. This case also demonstrates the importance of follow-up prenatal diagnosis. (+info)Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). (7/55)
Familial incontinentia pigmenti (IP [MIM 308310]), or Bloch-Sulzberger syndrome, is an X-linked dominant and male-lethal disorder. We recently demonstrated that mutations in NEMO (IKK-gamma), which encodes a critical component of the NF-kappaB signaling pathway, were responsible for IP. Virtually all mutations eliminate the production of NEMO, causing the typical skewing of X inactivation in female individuals and lethality in male individuals, possibly through enhanced sensitivity to apoptosis. Most mutations also give rise to classic signs of IP, but, in this report, we describe two mutations in families with atypical phenotypes. Remarkably, each family included a male individual with unusual signs, including postnatal survival and either immune dysfunction or hematopoietic disturbance. We found two duplication mutations in these families, at a cytosine tract in exon 10 of NEMO, both of which remove the zinc (Zn) finger at the C-terminus of the protein. Two deletion mutations were also identified in the same tract in additional families. However, only the duplication mutations allowed male individuals to survive, and affected female individuals with duplication mutations demonstrated random or slight skewing of X inactivation. Similarly, NF-kappaB activation was diminished in the presence of duplication mutations and was completely absent in cells with deletion mutations. These results strongly indicate that male individuals can also suffer from IP caused by NEMO mutations, and we therefore urge a reevaluation of the diagnostic criteria. (+info)A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. (8/55)
Incontinentia pigmenti (IP) is an X-linked dominant disorder characterized by abnormal skin pigmentation, retinal detachment, anodontia, alopecia, nail dystrophy and central nervous system defects. This disorder segregates as a male lethal disorder and causes skewed X-inactivation in female patients. IP is caused by mutations in a gene called NEMO, which encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappaB pathway. Here we report the identification of 277 mutations in 357 unrelated IP patients. An identical genomic deletion within NEMO accounted for 90% of the identified mutations. The remaining mutations were small duplications, substitutions and deletions. Nearly all NEMO mutations caused frameshift and premature protein truncation, which are predicted to eliminate NEMO function and cause cell lethality. Examination of families transmitting the recurrent deletion revealed that the rearrangement occurred in the paternal germline in most cases, indicating that it arises predominantly by intrachromosomal misalignment during meiosis. Expression analysis of human and mouse NEMO/Nemo showed that the gene becomes active early during embryogenesis and is expressed ubiquitously. These data confirm the involvement of NEMO in IP and will help elucidate the mechanism underlying the manifestation of this disorder and the in vivo function of NEMO. Based on these and other recent findings, we propose a model to explain the pathogenesis of this complex disorder. (+info)Incontinentia Pigmenti (IP) is a rare genetic disorder that primarily affects the skin, hair, and teeth. It is usually apparent at birth or in early infancy. The condition is characterized by four stages of skin changes:
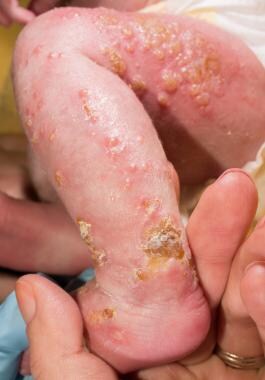
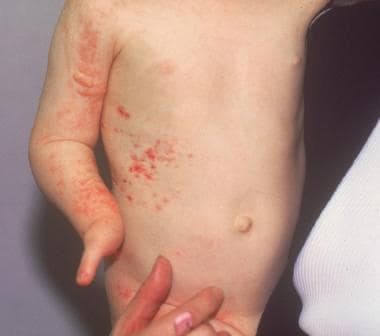
1. Vesiculobullous stage: This stage appears shortly after birth and is characterized by blisters and inflammation on the skin.
2. Verrucous stage: In this stage, which occurs around 6 months of age, the blisters turn into wart-like growths.
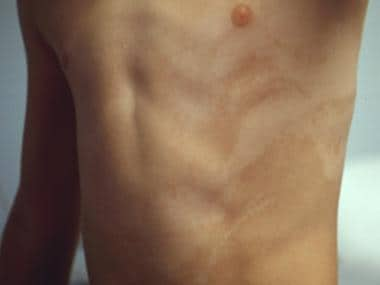
3. Hyperpigmented stage: This stage appears between ages 1 to 6 years and is characterized by swirling patterns of darkened skin.
4. Hypopigmented stage: In this final stage, which occurs in adolescence or early adulthood, the skin becomes paler in areas where the hyperpigmentation occurred.
Incontinentia Pigmenti is caused by mutations in the IKBKG gene and is inherited in an X-linked dominant pattern, meaning that females are more likely to be affected than males. The condition can also affect other organs, including the eyes, nails, hair, teeth, and central nervous system. Treatment typically focuses on managing symptoms and preventing complications.
Pigmentation disorders are conditions that affect the production or distribution of melanin, the pigment responsible for the color of skin, hair, and eyes. These disorders can cause changes in the color of the skin, resulting in areas that are darker (hyperpigmentation) or lighter (hypopigmentation) than normal. Examples of pigmentation disorders include melasma, age spots, albinism, and vitiligo. The causes, symptoms, and treatments for these conditions can vary widely, so it is important to consult a healthcare provider for an accurate diagnosis and treatment plan.
A toe phalanx is a bone in the toe, specifically referring to one of the 14 small bones that make up the digits of the foot, excluding the sesamoid bones. Each toe has three phalanges, except for the big toe, which only has two. These bones help form the basic structure of the toes and allow for their movement and flexibility. The term "phalanx" comes from Greek, meaning "a row of soldiers standing together in close order," which is fitting given how these bones are arranged in a line within each toe.
Tooth abnormalities refer to any variations or irregularities in the size, shape, number, structure, or development of teeth that deviate from the typical or normal anatomy. These abnormalities can occur in primary (deciduous) or permanent teeth and can be caused by genetic factors, environmental influences, systemic diseases, or localized dental conditions during tooth formation.
Some examples of tooth abnormalities include:
1. Microdontia - teeth that are smaller than normal in size.
2. Macrodontia - teeth that are larger than normal in size.
3. Peg-shaped teeth - teeth with a narrow, conical shape.
4. Talon cusps - additional cusps or points on the biting surface of a tooth.
5. Dens invaginatus - an abnormal development where the tooth crown has an extra fold or pouch that can trap bacteria and cause dental problems.
6. Taurodontism - teeth with large pulp chambers and short roots.
7. Supernumerary teeth - having more teeth than the typical number (20 primary and 32 permanent teeth).
8. Hypodontia - missing one or more teeth due to a failure of development.
9. Germination - two adjacent teeth fused together, usually occurring in the front teeth.
10. Fusion - two separate teeth that have grown together during development.
Tooth abnormalities may not always require treatment unless they cause functional, aesthetic, or dental health issues. A dentist can diagnose and manage tooth abnormalities through various treatments, such as fillings, extractions, orthodontic care, or restorative procedures.
Genetic dosage compensation is a process that evens out the effects of genes on an organism's phenotype (observable traits), even when there are differences in the number of copies of those genes present. This is especially important in cases where sex chromosomes are involved, as males and females often have different numbers of sex chromosomes.
In many species, including humans, females have two X chromosomes, while males have one X and one Y chromosome. To compensate for the difference in dosage, one of the female's X chromosomes is randomly inactivated during early embryonic development, resulting in each cell having only one active X chromosome, regardless of sex. This process ensures that both males and females have similar levels of gene expression from their X chromosomes and helps to prevent an imbalance in gene dosage between the sexes.
Defects in dosage compensation can lead to various genetic disorders, such as Turner syndrome (where a female has only one X chromosome) or Klinefelter syndrome (where a male has two or more X chromosomes). These conditions can result in developmental abnormalities and health issues due to the imbalance in gene dosage.
I-kappa B kinase (IKK) is a protein complex that plays a crucial role in the activation of NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells), a transcription factor involved in the regulation of immune response, inflammation, cell survival, and proliferation.
The IKK complex is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ (also known as NEMO). Upon stimulation by various signals such as cytokines, pathogens, or stress, the IKK complex becomes activated and phosphorylates I-kappa B (IkB), an inhibitor protein that keeps NF-kB in an inactive state in the cytoplasm.
Once IkB is phosphorylated by the IKK complex, it undergoes ubiquitination and degradation, leading to the release and nuclear translocation of NF-kB, where it can bind to specific DNA sequences and regulate gene expression. Dysregulation of IKK activity has been implicated in various pathological conditions, including chronic inflammation, autoimmune diseases, and cancer.
The X chromosome is one of the two types of sex-determining chromosomes in humans (the other being the Y chromosome). It's one of the 23 pairs of chromosomes that make up a person's genetic material. Females typically have two copies of the X chromosome (XX), while males usually have one X and one Y chromosome (XY).
The X chromosome contains hundreds of genes that are responsible for the production of various proteins, many of which are essential for normal bodily functions. Some of the critical roles of the X chromosome include:
1. Sex Determination: The presence or absence of the Y chromosome determines whether an individual is male or female. If there is no Y chromosome, the individual will typically develop as a female.
2. Genetic Disorders: Since females have two copies of the X chromosome, they are less likely to be affected by X-linked genetic disorders than males. Males, having only one X chromosome, will express any recessive X-linked traits they inherit.
3. Dosage Compensation: To compensate for the difference in gene dosage between males and females, a process called X-inactivation occurs during female embryonic development. One of the two X chromosomes is randomly inactivated in each cell, resulting in a single functional copy per cell.
The X chromosome plays a crucial role in human genetics and development, contributing to various traits and characteristics, including sex determination and dosage compensation.
 Incontinentia pigmenti
Incontinentia pigmenti
Incontinentia pigmenti achromians
Setleis syndrome
X-linked dominant inheritance
List of OMIM disorder codes
IKBKG
Marion Sulzberger
High-arched palate
PHF10
TMLHE
Phakomatosis
CLIC2
Linear and whorled nevoid hypermelanosis
David L. Nelson
Lucy Edwards
Genodermatosis
Rhabdomyosarcoma
Koebner phenomenon
Heterochromia iridum
Hypohidrotic ectodermal dysplasia with immune deficiency
Systemic disease
Dens evaginatus
NEMO deficiency syndrome
Acheiria
Epileptic spasms
Talon cusp
Genetic disorder
List of skin conditions
Sex linkage
Hypohidrosis
Incontinentia pigmenti - Wikipedia
 Incontinentia pigmenti : MedlinePlus Medical Encyclopedia
Incontinentia pigmenti : MedlinePlus Medical Encyclopedia
 Incontinentia Pigmenti: Background, Pathophysiology, Etiology
Incontinentia Pigmenti: Background, Pathophysiology, Etiology
Neurologic Manifestations of Incontinentia Pigmenti: Background, Pathophysiology, Epidemiology
 Incontinentia pigmenti achromians | health.am
Incontinentia pigmenti achromians | health.am
Incontinentia pigmenti
 X-inactivation and marker studies in three families with incontinentia pigmenti: implications for counselling and gene...
X-inactivation and marker studies in three families with incontinentia pigmenti: implications for counselling and gene...
Incontinentia Pigmenti: Background, Pathophysiology, Etiology
 Incontinentia Pigmenti International Foundation - Genetic Support Network Victoria (GSNV)
Incontinentia Pigmenti International Foundation - Genetic Support Network Victoria (GSNV)
 Incontinentia Pigmenti - Indian Journal of Dermatology, Venereology and Leprology
Incontinentia Pigmenti - Indian Journal of Dermatology, Venereology and Leprology
Incontinentia Pigmenti Medication
 Incontinentia Pigmenti in a 3-Year-Old Female: A Case Report
Incontinentia Pigmenti in a 3-Year-Old Female: A Case Report
Incontinentia Pigmenti: Background, Pathophysiology, Epidemiology
 Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of...
Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of...
 Multimodal Retinal Imaging in Incontinentia Pigmenti Including Optical Coherence Tomography Angiography: Findings From an Older...
Multimodal Retinal Imaging in Incontinentia Pigmenti Including Optical Coherence Tomography Angiography: Findings From an Older...
 Thieme E-Journals - American Journal of Perinatology Reports / Full Text
Thieme E-Journals - American Journal of Perinatology Reports / Full Text
 Enterovirus Surveillance --- United States, 1970--2005
Enterovirus Surveillance --- United States, 1970--2005
 Brown Patches On Skin: Causes & Home Remedies
Brown Patches On Skin: Causes & Home Remedies
 Caribbean | Page 6 | West Indian Medical Journal
Caribbean | Page 6 | West Indian Medical Journal
 Related Articles | Annals Singapore
Related Articles | Annals Singapore
 Molecular basis of hypohidrotic ectodermal dysplasia: an update | Journal of Applied Genetics
Molecular basis of hypohidrotic ectodermal dysplasia: an update | Journal of Applied Genetics
 IJMS | Free Full-Text | IKKγ/NEMO Localization into Multivesicular Bodies
IJMS | Free Full-Text | IKKγ/NEMO Localization into Multivesicular Bodies
Daniel METZGER | IGBMC
 Real Housewife Jenna Lyons Says Her Teeth and Hair Are Fake
Real Housewife Jenna Lyons Says Her Teeth and Hair Are Fake
![IKK gamma (IKBKG) Mouse Monoclonal Antibody [Clone ID: OTI12B11] - TA812491 | OriGene](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAYAAAAf8/9hAAABdklEQVQ4jZWQvUtbYRSHn3PvzTUZ0vgBYulSqENxCC6CitCAOImDYKAVvODmIKVYSv8Gh2yK6OJXW0H8B8TFOKggpdA0tEOH0qEmoCDN0Dah7+mSyjXvVeNve897nodzjhCRybdDbW7NZEHTgjgIn2qxP9tbk+/PAHiZmSC3vwMgjfDU6uBTRJeA1oavCqrPN4t+1VEZM7n8MwAn3BGsD4wj+i4CBkgistqdMsvh4qUguz2QUGUxaqpw+h4Yr8XTmCVI/GIYuH8TDOA5Gn/Uqh2WwCiPb4P/p+ve3wtLIEi1WYHn6rktEE6aFZQrzg9LsBEcHkNTkuLHsm/fAEFdJQDOo6h6fh5883ZVTbstANamj76oSz/InoUq+eNT7/X3ijMbLnuNfW+mjr7yKhOkO01Pb2ctZVScDyWn+vnMG1XVBcC9UQCIGN0olGSkUPJ/16f0QSN3sgVzT2aAkforHkmFcuUGvMg8FGH+Nug6gYirK0DyLoJ/0Pptg30xMl4AAAAASUVORK5CYII=) IKK gamma (IKBKG) Mouse Monoclonal Antibody [Clone ID: OTI12B11] - TA812491 | OriGene
IKK gamma (IKBKG) Mouse Monoclonal Antibody [Clone ID: OTI12B11] - TA812491 | OriGene
 Heterochromia: Types, Causes and Information
Heterochromia: Types, Causes and Information
 The 'Real Housewives of New York' Have Officially Killed the Term 'Girl's Girl'
The 'Real Housewives of New York' Have Officially Killed the Term 'Girl's Girl'
 RHONY star Jenna Lyons loves these beauty picks for women over 50
RHONY star Jenna Lyons loves these beauty picks for women over 50
 Mechanism of Human Tooth Eruption: Review Article Including a New Theory for Future Studies on the Eruption Process
Mechanism of Human Tooth Eruption: Review Article Including a New Theory for Future Studies on the Eruption Process
Identification of a 650 kb duplication at the X chromosome breakpoint in a patient with 46,X,t(X;8)(q28;q12) and non-syndromic...
Case of incontinentia pigmenti2
- Garrod reported the first probable case of incontinentia pigmenti in 1906 and described it as a peculiar pigmentation of the skin in an infant. (medscape.com)
- A Case of Incontinentia Pigmenti with Multiple Brain Infarction. (neo-med.org)
Stages of incontinentia pigmenti2
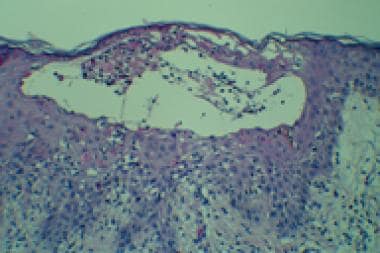
- The peripheral eosinophilia seen in the early stages of incontinentia pigmenti may result from the production of eotaxin, an eosinophil-selective cytokine, during the inflammatory cascade that results from a loss of NEMO/IKK -gamma activity. (medscape.com)
- Characteristic skin lesions compatible with the early, vesicular and/or verrucous stages of incontinentia pigmenti are present at birth or develop in the first few weeks of life in approximately 90% of patients. (medscape.com)
Hypohidrotic ectoderma3
- Incontinentia pigmenti has also been found to be allelic with hypohidrotic ectodermal dysplasia with severe immunodeficiency (EDAID), an X-linked immunodeficiency syndrome with developmental and immunologic defects in males. (medscape.com)
- Incontinentia pigmenti in a surviving male is accompanied by hypohidrotic ectodermal dysplasia and recurrent infection. (medscape.com)
- Mutations in this gene result in incontinentia pigmenti, hypohidrotic ectodermal dysplasia, and several other types of immunodeficiencies. (origene.com)
Retinal detachment1
- Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. (jamanetwork.com)
NEMO15
- Incontinentia pigmenti is caused by a mutation in the IKBKG gene, which encodes the NEMO protein, which serves to protect cells against TNF-alpha-induced apoptosis. (wikipedia.org)
- Incontinentia pigmenti is caused by mutations in the NEMO/IKK -gamma gene, which is located on chromosome Xq28. (medscape.com)
- The pathophysiology underlying the CNS manifestations in incontinentia pigmenti are unknown, but inflammation resulting from loss of NEMO/IKK -gamma activity may contribute to the development of vascular occlusive events. (medscape.com)
- Females with hypomorphic mutations in NEMO/IKK -gamma may have few clinical manifestations of incontinentia pigmenti. (medscape.com)
- A single mutation in NEMO/IKK -gamma involving the deletion of exons 4 through 10 accounts for most (80%) incontinentia pigmenti mutations. (medscape.com)
- In 2000, the International Incontinentia Pigmenti Consortium reported that incontinentia pigmenti is caused by a genomic rearrangement of the gene for NEMO, or nuclear factor kappa B essential modulator (IKBKG-IKK gamma). (medscape.com)
- Incontinentia pigmenti may rarely occur in males with Klinefelter syndrome (XXY syndrome) or as a result of somatic mosaicism or hypomorphic (less deleterious) mutations in the NEMO gene. (medscape.com)
- Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. (jamanetwork.com)
- Skin lesion development in a mouse model of incontinentia pigmenti is triggered by NEMO deficiency in epidermal keratinocytes and requires TNF signaling. (igbmc.fr)
- NEMO gene mutations in Chinese patients with incontinentia pigmenti. (lu.se)
- A male infant with anhidrotic ectodermal dysplasia/immunodeficiency accompanied by incontinentia pigmenti and a mutation in the NEMO pathway. (lu.se)
- A genetic cause for neonatal encephalopathy: incontinentia pigmenti with NEMO mutation. (lu.se)
- Identification of TRAF6-dependent NEMO polyubiquitination sites through analysis of a new NEMO mutation causing incontinentia pigmenti. (lu.se)
- Incontinentia pigmenti in a newborn with a novel nonsense mutation in the NEMO gene. (lu.se)
- A new mutation in exon 7 of NEMO gene: late skewed X-chromosome inactivation in an incontinentia pigmenti female patient with immunodeficiency. (lu.se)
Hypomelanosis2
- This has now been given its own name: 'Hypomelanosis of Ito' (incontinentia pigmenti achromians). (wikipedia.org)
- however, the disease with linkage to band Xp11.21 represents what has been referred to as incontinentia pigmenti type 1 or hypomelanosis of Ito. (medscape.com)
Achromians1
- Incontinentia pigmenti achromians is a congenital disorder that causes unusual and sometimes bizarre patterns of hypopigmented (diminished pigment) skin. (health.am)
Vesicular3
- This mechanism is believed to produce the cutaneous manifestations of the vesicular stage of incontinentia pigmenti. (medscape.com)
- Activation of eosinophils with subsequent release of cellular proteases may trigger the development of the vesicular stage of incontinentia pigmenti. (medscape.com)
- Osorio F, Magina S, Nogueira A, Azevedo F. Incontinentia Pigmenti with vesicular stage in utero. (medscape.com)
Mutation2
- Incontinentia pigmenti (IP), or Bloch-Sulzberger syndrome, is an X-linked dominant disease mainly of females that is lethal in males, caused by a mutation in the IKBKG gene. (logicalimages.com)
- Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. (medscape.com)
Manifestations4
- Incontinentia pigmenti is an X-linked dominant neurocutaneous syndrome with cutaneous, neurologic, ophthalmologic, and dental manifestations. (medscape.com)
- Macey-Dare LV, Goodman JR. Incontinentia pigmenti: seven cases with dental manifestations. (medscape.com)
- Retinal and other manifestations of incontinentia pigmenti (Bloch-Sulzberger syndrome). (jamanetwork.com)
- Incontinentia pigmenti is a rare X-linked dominant condition characterized by cutaneous, neural, ocular and dental manifestations. (uwi.edu)
Pigmentation1
- This article discusses what was formerly referred to as incontinentia pigmenti type 2, also known as Bloch-Sulzberger syndrome, a rare, X-linked, dominantly inherited disorder of skin pigmentation that is often associated with ocular, dental, and central nervous system abnormalities. (medscape.com)
Skewed X-inactivation1
- therefore, females with incontinentia pigmenti have an extremely skewed X-inactivation pattern. (medscape.com)
Dermatology1
- See also the Medscape Reference Dermatology article Incontinentia Pigmenti . (medscape.com)
Gene2
- The gene for incontinentia pigmenti is assigned to Xq28. (jamanetwork.com)
- Xq28 appears to be an unstable region of the human genome and genomic rearrangements are recognised as major causes of two single gene defects, haemophilia A and incontinentia pigmenti, which map within Xq28. (bmj.com)
Immunodeficiency1
- Incontinentia pigmenti can also cause immunodeficiency in women and this may not manifest in the neonatal period. (medscape.com)
Infant3
- A 7-month-old female infant with incontinentia pigmenti. (medscape.com)
- In the Journal of AAPOS , researchers described the first case report of the use of oral fluorescein and Optos noncontact ultra-widefield (UWF™) fundus and angiographic imaging in an office setting on a non-sedated infant with incontinentia pigmenti. (optos.com)
- Patel CK, Fung THM, Muqit MMK, Mordant DJ, Geh V. Non-contact ultra-widefield retinal imaging and fundus fluorescein angiography of an infant with incontinentia pigmenti without sedation in an ophthalmic office setting. (optos.com)
Newborn1
- Extensive cerebral infarction in the newborn due to incontinentia pigmenti. (medscape.com)
Genetic disorder5
- Incontinentia pigmenti (IP) is a rare X-linked dominant genetic disorder that affects the skin, hair, teeth, nails and central nervous system. (wikipedia.org)
- Incontinentia Pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare X-linked genetic disorder characterized by a distinctive cutaneous manifestation along with variable systemic involvement. (heraldopenaccess.us)
- Incontinentia Pigmenti (IP) is a rare X-linked dominant genetic disorder with an estimated incidence of 1 in 40,000 live births. (heraldopenaccess.us)
- Lyons has a genetic disorder called incontinentia pigmenti, which affects the color of her skin, as well as her teeth and hair. (yahoo.com)
- Lyons created Loveseen's high-quality, reusable lashes to meet a personal need: She has a rare genetic disorder - incontinentia pigmenti - that left her without natural lashes of her own. (yahoo.com)
Syndrome4
- Incontinentia pigmenti is an X-linked dominant, male lethal syndrome. (medscape.com)
- Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). (jamanetwork.com)
- Also known as Bloch-Sulzberger syndrome, incontinentia pigmenti is an inherited condition that gradually affects the skin and other bodily systems over time. (newbeauty.com)
- Incontinentia Pigmenti: Incontinentia pigmenti is also known as, 'Bloch-Sulzberger syndrome,' and affects a person's hair, teeth, nails and central nervous system. (disabled-world.com)
Anomalies1
- Minic S, Trpinac D, Gabriel H, Gencik M, Obradovic M. Dental and oral anomalies in incontinentia pigmenti: a systematic review. (medscape.com)
Verrucous1
- The proliferation of surviving IKK-positive cells may result in the production of the verrucous lesions seen in stage 2 of incontinentia pigmenti. (medscape.com)
Consortium1
- The International Incontinentia Pigmenti (IP) Consortium. (jamanetwork.com)
Abnormalities2
- Incontinentia pigmenti is an X-linked dominant genodermatosis characterized by abnormalities of the tissues and organs derived from the ectoderm and neuroectoderm and represents a type of ectodermal dysplasia. (medscape.com)
- Minic S, Novotny GE, Trpinac D, Obradovic M. Clinical features of incontinentia pigmenti with emphasis on oral and dental abnormalities. (medscape.com)
Ocular1
- Ocular findings in incontinentia pigmenti. (jamanetwork.com)
Lesions3
- In female incontinentia pigmenti patients, lyonization results in functional mosaicism of X-linked genes, which is manifested by the blaschkoid distribution of cutaneous lesions. (medscape.com)
- Nenci et al found that TNF signaling is necessary for development of the skin lesions in incontinentia pigmenti. (medscape.com)
- Bodak N, Hadj-Rabia S, Hamel-Teillac D, de Prost Y, Bodemer C. Late recurrence of inflammatory first-stage lesions in incontinentia pigmenti: an unusual phenomenon and a fascinating pathologic mechanism. (medscape.com)
Dominant3
- Familial incontinentia pigmenti (IP) is an X-linked dominant disorder with an extremely variable clinical presentation. (nih.gov)
- Incontinentia pigmenti (IP) is an X-linked dominant genodermatosis primarily affecting female children. (qxmd.com)
- Incontinentia pigmenti is transmitted as an X-linked dominant trait and is believed to be lethal in boys. (tripod.com)
Findings1
- The skin findings in incontinentia pigmenti represent changes in the epidermal cells. (medscape.com)
Probable1
- Alikhan A, Lee AD, Swing D, Carroll C, Yosipovitch G. Vaccination as a probable cause of incontinentia pigmenti reactivation. (medscape.com)
Clinical2
- Clinical study of 40 cases of incontinentia pigmenti. (jamanetwork.com)
- Clinical and molecular analysis of NF-kappaB essential modulator in Chinese incontinentia pigmenti patients. (lu.se)
Infants1
- Late-onset incontinentia pigmenti is occasionally reported in older infants. (medscape.com)
Patient2
- Jamnadas B, Agarwal R, Caddy CM. A rare case of SCC in a young patient with incontinentia pigmenti. (medscape.com)
- Multiple subungual squamous cell carcinomas in a patient with incontinentia pigmenti. (medscape.com)
Females1
- More than 95% of reported cases of incontinentia pigmenti occur in females. (medscape.com)
Patients2
- Cephalometric skeletal evaluation of patients with Incontinentia Pigmenti. (medscape.com)
- Incontinentia pigmenti appears to be more common among white patients, but it has also been reported in blacks and Asians. (medscape.com)
Skin2
- Incontinentia pigmenti (IP) is a rare skin condition passed down through families. (medlineplus.gov)
- Normal X chromosomes are active in unaffected skin, and mutated X chromosomes are active in skin affected with incontinentia pigmenti. (medscape.com)
Unusual1
- Nail dystrophy, an unusual presentation of incontinentia pigmenti. (medscape.com)
Incidence1
- No incidence or prevalence data are available on incontinentia pigmenti in the US population. (medscape.com)
Rare1
- The Incontinentia Pigmenti Genetic Biobank: study design and cohort profile to facilitate research into a rare disease worldwide. (lu.se)
Stage2
- Incontinentia pigmenti: an extensive second episode of a "first-stage" vesicobullous eruption. (medscape.com)
- Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. (qxmd.com)
Study1
- Montes CM, Maize JC, Guerry-Force ML. Incontinentia pigmenti with painful subungual tumors: a two-generation study. (medscape.com)
Journal1
- Retinal function in incontinentia pigmenti: a long-term electrophysiological follow-up , Scandinavian Journal of Optometry and Visual Science 13: (2) pp. 15-20. (doktori.hu)
Analysis1
- Incontinentia pigmenti: a world statistical analysis. (jamanetwork.com)