Leber Congenital Amaurosis
Optic Atrophy, Hereditary, Leber
Blindness
Optic Atrophies, Hereditary
Eye Proteins
Retinal Degeneration
Photoreceptor Connecting Cilium
Retinitis Pigmentosa
Retinal Dystrophies
Amaurosis Fugax
Photoreceptor Cells, Vertebrate
Night Vision
Eye Diseases, Hereditary
Cone Opsins
Pedigree
Carrier Proteins
Mutation
Retinal Cone Photoreceptor Cells
Guanylate Cyclase
Retina
Retinal Rod Photoreceptor Cells
Vision, Ocular
Retinaldehyde
Dependovirus
Nystagmus, Congenital
Genetic Therapy
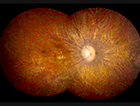
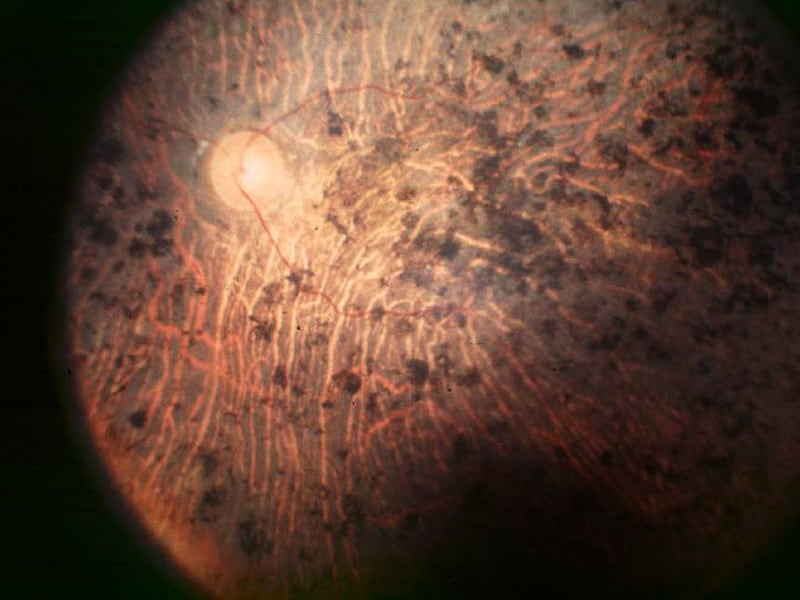
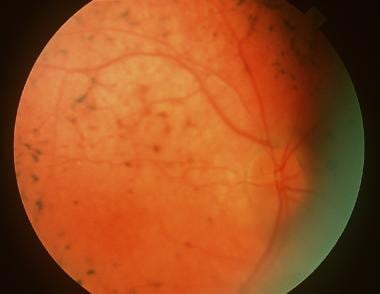
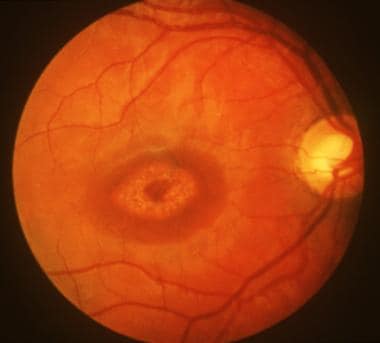
Fundus Oculi
Retinal Diseases
Tomography, Optical Coherence
Codon, Nonsense
Dark Adaptation
Phenotype
Polymorphism, Single-Stranded Conformational
Visual Acuity
Pigment Epithelium of Eye
Exons
Genetic Vectors
Disease Models, Animal
Proteins
Genotype
Visual Fields
Receptors, Cell Surface
Mutation, Missense
Molecular Sequence Data
Fluorescent Antibody Technique, Indirect
Nerve Tissue Proteins
Alcohol Oxidoreductases
Reflex, Pupillary
Genetic Testing
Membrane Proteins
Mice, Knockout
Polymerase Chain Reaction
Homeodomain Proteins
Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. (1/108)
(+info)Comprehensive SNP-chip for retinitis pigmentosa-Leber congenital amaurosis diagnosis: new mutations and detection of mutational founder effects. (2/108)
(+info)Bax-induced apoptosis in Leber's congenital amaurosis: a dual role in rod and cone degeneration. (3/108)
(+info)The Jeremiah Metzger Lecture: gene therapy for inherited disorders: from Christmas disease to Leber's amaurosis. (4/108)
This paper will focus on recent developments in the field of gene therapy for inherited disorders. From a historical perspective, this Metzger lecture is a follow-on to one presented by Dr. William Kelley in 1987, entitled "Current Status of Human Gene Therapy" (Transactions Am Clin. Climatol. Assoc. 99:152-169) (1). In 1987, gene transfer studies in human subjects were yet to be undertaken; the first clinical studies, infusion of genetically modified autologous T cells into two young girls with ADA-SCID, would not take place until 1990 (2). Today's lecture will summarize progress since that time in one area, that of in vivo gene transfer for genetic disease. I will describe progress in two areas, gene therapy for the bleeding disorder hemophilia B, and for a subset of retinal degenerative disorders termed Leber's congenital amaurosis, due to mutations in the gene encoding retinal pigment epithelium-specific 65 kilodalton protein (RPE65). This lecture will demonstrate the interconnected nature of progress in these two areas, as careful delineation of the obstacles in hemophilia led to the realization that success could be achieved in Leber's. (+info)Recent breakthroughs in gene therapy for inherited retinal degeneration. (5/108)
Gene therapy for inherited retinal degeneration has made major advances toward the ultimate goal of reversing blindness in human patients. With significant advances in recombinant viral vector design, safety and efficacy profiles have greatly improved. Although these recent advances have been applied to many different retinal diseases, one retinal degenerative disease, Leber congenital amaurosis, appears to have the greatest potential for reversing blindness. In pre-clinical animal studies, gene therapy for Leber congenital amaurosis has demonstrated visual recovery. Recently, in landmark clinical trials, preliminary results have indicated safety and efficacy for the use of gene therapy in Leber congenital amaurosis, thus laying the foundation for continued use of gene therapy in other forms of inherited blinding disease. (+info)Which Leber congenital amaurosis patients are eligible for gene therapy trials? (6/108)
(+info)Gene therapy for Leber's congenital amaurosis is safe and effective through 1.5 years after vector administration. (7/108)
(+info)Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis. (8/108)
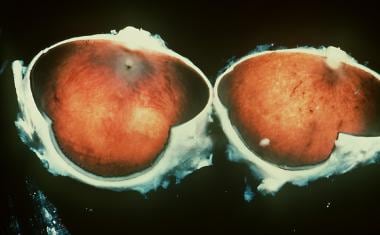
(+info)Leber Congenital Amaurosis (LCA) is a group of inherited retinal degenerative disorders that affect the development and function of the retina, a light-sensitive tissue at the back of the eye. It is characterized by severe visual impairment or blindness from birth or early infancy.
The condition is caused by mutations in various genes involved in the normal functioning of photoreceptor cells (rods and cones) in the retina, which are responsible for capturing light and transmitting visual signals to the brain. As a result, the photoreceptors fail to develop properly or degenerate over time, leading to vision loss.
Symptoms of LCA may include roving eye movements (nystagmus), lack of fixation, decreased or absent response to light, and abnormal pupillary reflexes. Some individuals with LCA may also have other ocular abnormalities such as keratoconus, cataracts, or glaucoma.
LCA is typically inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition. Currently, there is no cure for LCA, but various treatments such as gene therapy and assistive devices may help improve visual function and quality of life for affected individuals.
Cis-trans isomeres are molecules that have the same molecular formula and skeletal structure, but differ in the arrangement of their atoms around a double bond. In a cis isomer, the two larger groups or atoms are on the same side of the double bond, while in a trans isomer, they are on opposite sides.
Cis-trans isomerases are enzymes that catalyze the interconversion between cis and trans isomers of various molecules, such as fatty acids, steroids, and retinals. These enzymes play important roles in various biological processes, including membrane fluidity, vision, and the biosynthesis of hormones and other signaling molecules.
Examples of cis-trans isomerases include:
* Fatty acid desaturases, which introduce double bonds into fatty acids and can convert trans isomers to cis isomers
* Retinal isomerases, which interconvert the cis and trans isomers of retinal, a molecule involved in vision
* Steroid isomerases, which catalyze the interconversion of various steroids, including cholesterol and its derivatives.
Hereditary Optic Atrophy, Leber type (LOA) is a mitochondrial DNA-associated inherited condition that primarily affects the optic nerve and leads to vision loss. It is characterized by the degeneration of retinal ganglion cells and their axons, which make up the optic nerve. This results in bilateral, painless, and progressive visual deterioration, typically beginning in young adulthood (14-35 years).
Leber's hereditary optic atrophy is caused by mutations in the mitochondrial DNA (mtDNA) gene MT-ND4 or MT-ND6. The condition follows a maternal pattern of inheritance, meaning that it is passed down through the mother's lineage.
The onset of LOA usually occurs in one eye first, followed by the second eye within weeks to months. Central vision is initially affected, leading to blurriness and loss of visual acuity. Color vision may also be impaired. The progression of the condition generally stabilizes after a few months, but complete recovery of vision is unlikely.
Currently, there is no cure for Leber's hereditary optic atrophy. Treatment focuses on managing symptoms and providing visual rehabilitation to help affected individuals adapt to their visual impairment.
Blindness is a condition of complete or near-complete vision loss. It can be caused by various factors such as eye diseases, injuries, or birth defects. Total blindness means that a person cannot see anything at all, while near-complete blindness refers to having only light perception or the ability to perceive the direction of light, but not able to discern shapes or forms. Legal blindness is a term used to define a certain level of visual impairment that qualifies an individual for government assistance and benefits; it usually means best corrected visual acuity of 20/200 or worse in the better eye, or a visual field no greater than 20 degrees in diameter.
Hereditary optic atrophies (HOAs) are a group of genetic disorders that cause degeneration of the optic nerve, leading to vision loss. The optic nerve is responsible for transmitting visual information from the eye to the brain. In HOAs, this nerve degenerates over time, resulting in decreased visual acuity, color vision deficits, and sometimes visual field defects.
There are several types of HOAs, including dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), autosomal recessive optic atrophy (AROA), and Wolfram syndrome. Each type has a different inheritance pattern and is caused by mutations in different genes.
DOA is the most common form of HOA and is characterized by progressive vision loss that typically begins in childhood or early adulthood. It is inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the disease-causing mutation from an affected parent.
LHON is a mitochondrial disorder that primarily affects males and is characterized by sudden, severe vision loss that typically occurs in young adulthood. It is caused by mutations in the mitochondrial DNA and is inherited maternally.
AROA is a rare form of HOA that is inherited in an autosomal recessive manner, meaning that both copies of the gene must be mutated to cause the disease. It typically presents in infancy or early childhood with progressive vision loss.
Wolfram syndrome is a rare genetic disorder that affects multiple organs, including the eyes, ears, and endocrine system. It is characterized by diabetes insipidus, diabetes mellitus, optic atrophy, and hearing loss. It is inherited in an autosomal recessive manner.
There is currently no cure for HOAs, but treatments such as low-vision aids and rehabilitation may help to manage the symptoms. Research is ongoing to develop new therapies for these disorders.
Eye proteins, also known as ocular proteins, are specific proteins that are found within the eye and play crucial roles in maintaining proper eye function and health. These proteins can be found in various parts of the eye, including the cornea, iris, lens, retina, and other structures. They perform a wide range of functions, such as:
1. Structural support: Proteins like collagen and elastin provide strength and flexibility to the eye's tissues, enabling them to maintain their shape and withstand mechanical stress.
2. Light absorption and transmission: Proteins like opsins and crystallins are involved in capturing and transmitting light signals within the eye, which is essential for vision.
3. Protection against damage: Some eye proteins, such as antioxidant enzymes and heat shock proteins, help protect the eye from oxidative stress, UV radiation, and other environmental factors that can cause damage.
4. Regulation of eye growth and development: Various growth factors and signaling molecules, which are protein-based, contribute to the proper growth, differentiation, and maintenance of eye tissues during embryonic development and throughout adulthood.
5. Immune defense: Proteins involved in the immune response, such as complement components and immunoglobulins, help protect the eye from infection and inflammation.
6. Maintenance of transparency: Crystallin proteins in the lens maintain its transparency, allowing light to pass through unobstructed for clear vision.
7. Neuroprotection: Certain eye proteins, like brain-derived neurotrophic factor (BDNF), support the survival and function of neurons within the retina, helping to preserve vision.
Dysfunction or damage to these eye proteins can contribute to various eye disorders and diseases, such as cataracts, age-related macular degeneration, glaucoma, diabetic retinopathy, and others.
Retinal degeneration is a broad term that refers to the progressive loss of photoreceptor cells (rods and cones) in the retina, which are responsible for converting light into electrical signals that are sent to the brain. This process can lead to vision loss or blindness. There are many different types of retinal degeneration, including age-related macular degeneration, retinitis pigmentosa, and Stargardt's disease, among others. These conditions can have varying causes, such as genetic mutations, environmental factors, or a combination of both. Treatment options vary depending on the specific type and progression of the condition.
A photoreceptor connecting cilium, also known as the connecting cilium or the outer segment initial segment, is a specialized structure found in the eye's photoreceptor cells (rods and cones). It is a thin, non-motile cilium that connects the inner segment of the photoreceptor cell to the outer segment. The outer segment contains the visual pigments that absorb light and initiate the process of vision.
The connecting cilium plays a crucial role in the maintenance and function of the outer segment by providing a passageway for the transport of proteins, lipids, and other molecules from the inner segment to the outer segment. This process is essential for the renewal and turnover of the visual pigments and other components of the outer segment. The connecting cilium also helps maintain the structural integrity of the photoreceptor cells and their sensitivity to light.
Defects in the connecting cilium can lead to various retinal disorders, such as retinitis pigmentosa and Leber congenital amaurosis, which are characterized by progressive vision loss due to the degeneration of the photoreceptor cells.
Retinitis pigmentosa (RP) is a group of rare, genetic disorders that involve a breakdown and loss of cells in the retina - a light-sensitive tissue located at the back of the eye. The retina converts light into electrical signals which are then sent to the brain and interpreted as visual images.
In RP, the cells that detect light (rods and cones) degenerate more slowly than other cells in the retina, leading to a progressive loss of vision. Symptoms typically begin in childhood with night blindness (difficulty seeing in low light), followed by a gradual narrowing of the visual field (tunnel vision). Over time, this can lead to significant vision loss and even blindness.
The condition is usually inherited and there are several different genes that have been associated with RP. The diagnosis is typically made based on a combination of genetic testing, family history, and clinical examination. Currently, there is no cure for RP, but researchers are actively working to develop new treatments that may help slow or stop the progression of the disease.
Retinal dystrophies are a group of genetic eye disorders that primarily affect the retina, a light-sensitive layer at the back of the eye. These conditions are characterized by progressive degeneration and death of photoreceptor cells (rods and cones) in the retina, leading to vision loss.
The term "dystrophy" refers to a condition that results from the abnormal or defective development and function of tissues or organs. In the case of retinal dystrophies, the photoreceptor cells do not develop or function properly, resulting in visual impairment.
Retinal dystrophies can present at any age, from infancy to adulthood, and can have varying degrees of severity. Some common symptoms include night blindness, decreased visual acuity, loss of peripheral vision, light sensitivity, and color vision abnormalities.
Examples of retinal dystrophies include retinitis pigmentosa, Stargardt disease, Usher syndrome, and Leber congenital amaurosis, among others. These conditions are typically inherited and can be caused by mutations in various genes that play a role in the development and function of the retina.
There is currently no cure for retinal dystrophies, but research is ongoing to develop treatments that may slow or halt the progression of these conditions, such as gene therapy and stem cell transplantation.
Amaurosis fugax is a medical term that describes a temporary loss of vision in one eye, which is often described as a "shade or curtain falling over the field of vision." It's usually caused by a temporary interruption of blood flow to the retina or optic nerve. This condition is often associated with conditions such as giant cell arteritis, carotid artery stenosis, and cardiovascular disease.
It's important to note that Amaurosis fugax can be a warning sign for a more serious medical event, such as a stroke, so it's essential to seek medical attention promptly if you experience any symptoms of this condition.
Electroretinography (ERG) is a medical test used to evaluate the functioning of the retina, which is the light-sensitive tissue located at the back of the eye. The test measures the electrical responses of the retina to light stimulation.
During the procedure, a special contact lens or electrode is placed on the surface of the eye to record the electrical activity generated by the retina's light-sensitive cells (rods and cones) and other cells in the retina. The test typically involves presenting different levels of flashes of light to the eye while the electrical responses are recorded.
The resulting ERG waveform provides information about the overall health and function of the retina, including the condition of the photoreceptors, the integrity of the inner retinal layers, and the health of the retinal ganglion cells. This test is often used to diagnose and monitor various retinal disorders, such as retinitis pigmentosa, macular degeneration, and diabetic retinopathy.
Photoreceptor cells in vertebrates are specialized types of neurons located in the retina of the eye that are responsible for converting light stimuli into electrical signals. These cells are primarily responsible for the initial process of vision and have two main types: rods and cones.
Rods are more numerous and are responsible for low-light vision or scotopic vision, enabling us to see in dimly lit conditions. They do not contribute to color vision but provide information about the shape and movement of objects.
Cones, on the other hand, are less numerous and are responsible for color vision and high-acuity vision or photopic vision. There are three types of cones, each sensitive to different wavelengths of light: short (S), medium (M), and long (L) wavelengths, which correspond to blue, green, and red, respectively. The combination of signals from these three types of cones allows us to perceive a wide range of colors.
Both rods and cones contain photopigments that consist of a protein called opsin and a light-sensitive chromophore called retinal. When light hits the photopigment, it triggers a series of chemical reactions that ultimately lead to the generation of an electrical signal that is transmitted to the brain via the optic nerve. This process enables us to see and perceive our visual world.
Night vision refers to the ability to see in low light conditions, typically during night time. In a medical context, it often relates to the functionality of the eye and visual system. There are two types of night vision:
1. Scotopic vision: This is the primary type of night vision, enabled by the rod cells in our retina which are highly sensitive to light but lack color vision. During twilight or night conditions, when light levels are low, the rods take over from the cone cells (which are responsible for color and daytime vision) and provide us with limited vision, typically in shades of gray.
2. Mesopic vision: This is a state between photopic (daytime) and scotopic (night-time) vision, where both rod and cone cells contribute to vision. It allows for better color discrimination and visual acuity compared to scotopic vision alone.
In some cases, night vision can be impaired due to eye conditions such as cataracts, glaucoma, or retinal disorders. There are also medical devices called night vision goggles that amplify available light to enhance a person's ability to see in low-light environments.
Hereditary eye diseases refer to conditions that affect the eyes and are passed down from parents to their offspring through genetics. These diseases are caused by mutations or changes in an individual's DNA that are inherited from their parents. The mutations can occur in any of the genes associated with eye development, function, or health.
There are many different types of hereditary eye diseases, some of which include:
1. Retinitis Pigmentosa - a group of rare, genetic disorders that involve a breakdown and loss of cells in the retina.
2. Macular Degeneration - a progressive disease that damages the central portion of the retina, impairing vision.
3. Glaucoma - a group of eye conditions that damage the optic nerve, often caused by an increase in pressure inside the eye.
4. Cataracts - clouding of the lens inside the eye, which can lead to blurry vision and blindness.
5. Keratoconus - a progressive eye disease that causes the cornea to thin and bulge outward into a cone shape.
6. Color Blindness - a condition where an individual has difficulty distinguishing between certain colors.
7. Optic Neuropathy - damage to the optic nerve, which can result in vision loss.
The symptoms and severity of hereditary eye diseases can vary widely depending on the specific condition and the individual's genetic makeup. Some conditions may be present at birth or develop in early childhood, while others may not appear until later in life. Treatment options for these conditions may include medication, surgery, or lifestyle changes, and are often most effective when started early.
Cone opsins are a type of photopigment protein found in the cone cells of the retina, which are responsible for color vision. There are three types of cone opsins in humans, each sensitive to different wavelengths of light: short-wavelength (S) sensitive cone opsin (also known as blue cone opsin), medium-wavelength (M) sensitive cone opsin (also known as green cone opsin), and long-wavelength (L) sensitive cone opsin (also known as red cone opsin).
These cone opsins are activated by light, which triggers a chemical reaction that sends signals to the brain and enables us to perceive color. Differences in the genes that code for these cone opsins can result in variations in color perception and can contribute to individual differences in color vision. Certain genetic mutations can also lead to various forms of color blindness, including red-green color blindness and blue-yellow color blindness.
DNA Mutational Analysis is a laboratory test used to identify genetic variations or changes (mutations) in the DNA sequence of a gene. This type of analysis can be used to diagnose genetic disorders, predict the risk of developing certain diseases, determine the most effective treatment for cancer, or assess the likelihood of passing on an inherited condition to offspring.
The test involves extracting DNA from a patient's sample (such as blood, saliva, or tissue), amplifying specific regions of interest using polymerase chain reaction (PCR), and then sequencing those regions to determine the precise order of nucleotide bases in the DNA molecule. The resulting sequence is then compared to reference sequences to identify any variations or mutations that may be present.
DNA Mutational Analysis can detect a wide range of genetic changes, including single-nucleotide polymorphisms (SNPs), insertions, deletions, duplications, and rearrangements. The test is often used in conjunction with other diagnostic tests and clinical evaluations to provide a comprehensive assessment of a patient's genetic profile.
It is important to note that not all mutations are pathogenic or associated with disease, and the interpretation of DNA Mutational Analysis results requires careful consideration of the patient's medical history, family history, and other relevant factors.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Carrier proteins, also known as transport proteins, are a type of protein that facilitates the movement of molecules across cell membranes. They are responsible for the selective and active transport of ions, sugars, amino acids, and other molecules from one side of the membrane to the other, against their concentration gradient. This process requires energy, usually in the form of ATP (adenosine triphosphate).
Carrier proteins have a specific binding site for the molecule they transport, and undergo conformational changes upon binding, which allows them to move the molecule across the membrane. Once the molecule has been transported, the carrier protein returns to its original conformation, ready to bind and transport another molecule.
Carrier proteins play a crucial role in maintaining the balance of ions and other molecules inside and outside of cells, and are essential for many physiological processes, including nerve impulse transmission, muscle contraction, and nutrient uptake.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Retinal cone photoreceptor cells are specialized neurons located in the retina of the eye, responsible for visual phototransduction and color vision. They are one of the two types of photoreceptors, with the other being rods, which are more sensitive to low light levels. Cones are primarily responsible for high-acuity, color vision during daylight or bright-light conditions.
There are three types of cone cells, each containing different photopigments that absorb light at distinct wavelengths: short (S), medium (M), and long (L) wavelengths, which correspond to blue, green, and red light, respectively. The combination of signals from these three types of cones allows the human visual system to perceive a wide range of colors and discriminate between them. Cones are densely packed in the central region of the retina, known as the fovea, which provides the highest visual acuity.
Guanylate cyclase is an enzyme that catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which acts as a second messenger in various cellular signaling pathways. There are two main types of guanylate cyclases: soluble and membrane-bound. Soluble guanylate cyclase is activated by nitric oxide, while membrane-bound guanylate cyclase can be activated by natriuretic peptides. The increased levels of cGMP produced by guanylate cyclase can lead to a variety of cellular responses, including smooth muscle relaxation, neurotransmitter release, and regulation of ion channels. Dysregulation of guanylate cyclase activity has been implicated in several diseases, such as hypertension, heart failure, and cancer.
Consanguinity is a medical and genetic term that refers to the degree of genetic relationship between two individuals who share common ancestors. Consanguineous relationships exist when people are related by blood, through a common ancestor or siblings who have children together. The closer the relationship between the two individuals, the higher the degree of consanguinity.
The degree of consanguinity is typically expressed as a percentage or fraction, with higher values indicating a closer genetic relationship. For example, first-degree relatives, such as parents and children or full siblings, share approximately 50% of their genes and have a consanguinity coefficient of 0.25 (or 25%).
Consanguinity can increase the risk of certain genetic disorders and birth defects in offspring due to the increased likelihood of sharing harmful recessive genes. The risks depend on the degree of consanguinity, with closer relationships carrying higher risks. It is important for individuals who are planning to have children and have a history of consanguinity to consider genetic counseling and testing to assess their risk of passing on genetic disorders.
The retina is the innermost, light-sensitive layer of tissue in the eye of many vertebrates and some cephalopods. It receives light that has been focused by the cornea and lens, converts it into neural signals, and sends these to the brain via the optic nerve. The retina contains several types of photoreceptor cells including rods (which handle vision in low light) and cones (which are active in bright light and are capable of color vision).
In medical terms, any pathological changes or diseases affecting the retinal structure and function can lead to visual impairment or blindness. Examples include age-related macular degeneration, diabetic retinopathy, retinal detachment, and retinitis pigmentosa among others.
Retinal rod photoreceptor cells are specialized neurons in the retina of the eye that are primarily responsible for vision in low light conditions. They contain a light-sensitive pigment called rhodopsin, which undergoes a chemical change when struck by a single photon of light. This triggers a cascade of biochemical reactions that ultimately leads to the generation of electrical signals, which are then transmitted to the brain via the optic nerve.
Rod cells do not provide color vision or fine detail, but they allow us to detect motion and see in dim light. They are more sensitive to light than cone cells, which are responsible for color vision and detailed sight in bright light conditions. Rod cells are concentrated at the outer edges of the retina, forming a crescent-shaped region called the peripheral retina, with fewer rod cells located in the central region of the retina known as the fovea.
Ocular vision refers to the ability to process and interpret visual information that is received by the eyes. This includes the ability to see clearly and make sense of the shapes, colors, and movements of objects in the environment. The ocular system, which includes the eye and related structures such as the optic nerve and visual cortex of the brain, works together to enable vision.
There are several components of ocular vision, including:
* Visual acuity: the clarity or sharpness of vision
* Field of vision: the extent of the visual world that is visible at any given moment
* Color vision: the ability to distinguish different colors
* Depth perception: the ability to judge the distance of objects in three-dimensional space
* Contrast sensitivity: the ability to distinguish an object from its background based on differences in contrast
Disorders of ocular vision can include refractive errors such as nearsightedness or farsightedness, as well as more serious conditions such as cataracts, glaucoma, and macular degeneration. These conditions can affect one or more aspects of ocular vision and may require medical treatment to prevent further vision loss.
Retinaldehyde, also known as retinal, is a form of vitamin A that is essential for vision. It is the aldehyde form of retinol (vitamin A alcohol) and is involved in the visual cycle, where it plays a crucial role in the process of converting light into electrical signals that are sent to the brain.
When light hits the retina, it activates a protein called rhodopsin, which contains retinaldehyde as one of its components. This activation causes a chemical change in retinaldehyde, leading to the generation of an electrical signal that is transmitted to the brain via the optic nerve.
Retinaldehyde is also involved in other physiological processes, including the regulation of gene expression and cell growth and differentiation. It can be synthesized in the body from beta-carotene, a pigment found in fruits and vegetables, or obtained directly from animal sources such as liver, fish liver oil, and dairy products.
Intraocular injections are a type of medical procedure where medication is administered directly into the eye. This technique is often used to deliver drugs that treat various eye conditions, such as age-related macular degeneration, diabetic retinopathy, and endophthalmitis. The most common type of intraocular injection is an intravitreal injection, which involves injecting medication into the vitreous cavity, the space inside the eye filled with a clear gel-like substance called the vitreous humor. This procedure is typically performed by an ophthalmologist in a clinical setting and may be repeated at regular intervals depending on the condition being treated.
A dependovirus, also known as a dependent adenovirus or satellite adenovirus, is a type of virus that requires the presence of another virus, specifically an adenovirus, to replicate. Dependoviruses are small, non-enveloped viruses with a double-stranded DNA genome. They cannot complete their replication cycle without the help of an adenovirus, which provides necessary functions for the dependovirus to replicate.
Dependoviruses are clinically significant because they can cause disease in humans, particularly in individuals with weakened immune systems. In some cases, dependoviruses may also affect the severity and outcome of adenovirus infections. However, it is important to note that not all adenovirus infections are associated with dependovirus co-infections.
Congenital nystagmus is a type of involuntary eye movement that is present at birth or develops within the first few months of life. It is characterized by rhythmic oscillations or repetitive, rapid movements of the eyes in either horizontal, vertical, or rotatory directions. These movements can impair vision and may be associated with other ocular conditions such as albinism, congenital cataracts, or optic nerve hypoplasia. The exact cause of congenital nystagmus is not fully understood, but it is believed to result from abnormal development or dysfunction in the areas of the brain that control eye movements. In some cases, congenital nystagmus may be inherited as a genetic trait. Treatment options for congenital nystagmus include corrective lenses, prism glasses, surgery, and vision therapy, depending on the underlying cause and severity of the condition.
Genetic therapy, also known as gene therapy, is a medical intervention that involves the use of genetic material, such as DNA or RNA, to treat or prevent diseases. It works by introducing functional genes into cells to replace missing or faulty ones caused by genetic disorders or mutations. The introduced gene is incorporated into the recipient's genome, allowing for the production of a therapeutic protein that can help manage the disease symptoms or even cure the condition.
There are several approaches to genetic therapy, including:
1. Replacing a faulty gene with a healthy one
2. Inactivating or "silencing" a dysfunctional gene causing a disease
3. Introducing a new gene into the body to help fight off a disease, such as cancer
Genetic therapy holds great promise for treating various genetic disorders, including cystic fibrosis, muscular dystrophy, hemophilia, and certain types of cancer. However, it is still an evolving field with many challenges, such as efficient gene delivery, potential immune responses, and ensuring the safety and long-term effectiveness of the therapy.
"Fundus Oculi" is a medical term that refers to the back part of the interior of the eye, including the optic disc, macula, fovea, retinal vasculature, and peripheral retina. It is the area where light is focused and then transmitted to the brain via the optic nerve, forming visual images. Examinations of the fundus oculi are crucial for detecting various eye conditions such as diabetic retinopathy, macular degeneration, glaucoma, and other retinal diseases. The examination is typically performed using an ophthalmoscope or a specialized camera called a retinal camera.
Retinal diseases refer to a group of conditions that affect the retina, which is the light-sensitive tissue located at the back of the eye. The retina is responsible for converting light into electrical signals that are sent to the brain and interpreted as visual images. Retinal diseases can cause vision loss or even blindness, depending on their severity and location in the retina.
Some common retinal diseases include:
1. Age-related macular degeneration (AMD): A progressive disease that affects the central part of the retina called the macula, causing blurred or distorted vision.
2. Diabetic retinopathy: A complication of diabetes that can damage the blood vessels in the retina, leading to vision loss.
3. Retinal detachment: A serious condition where the retina becomes separated from its underlying tissue, requiring immediate medical attention.
4. Macular edema: Swelling or thickening of the macula due to fluid accumulation, which can cause blurred vision.
5. Retinitis pigmentosa: A group of inherited eye disorders that affect the retina's ability to respond to light, causing progressive vision loss.
6. Macular hole: A small break in the macula that can cause distorted or blurry vision.
7. Retinal vein occlusion: Blockage of the retinal veins that can lead to bleeding, swelling, and potential vision loss.
Treatment for retinal diseases varies depending on the specific condition and its severity. Some treatments include medication, laser therapy, surgery, or a combination of these options. Regular eye exams are essential for early detection and treatment of retinal diseases.
Recessive genes refer to the alleles (versions of a gene) that will only be expressed when an individual has two copies of that particular allele, one inherited from each parent. If an individual inherits one recessive allele and one dominant allele for a particular gene, the dominant allele will be expressed and the recessive allele will have no effect on the individual's phenotype (observable traits).
Recessive genes can still play a role in determining an individual's genetic makeup and can be passed down through generations even if they are not expressed. If two carriers of a recessive gene have children, there is a 25% chance that their offspring will inherit two copies of the recessive allele and exhibit the associated recessive trait.
Examples of genetic disorders caused by recessive genes include cystic fibrosis, sickle cell anemia, and albinism.
Optical coherence tomography (OCT) is a non-invasive imaging technique that uses low-coherence light to capture high-resolution cross-sectional images of biological tissues, particularly the retina and other ocular structures. OCT works by measuring the echo time delay of light scattered back from different depths within the tissue, creating a detailed map of the tissue's structure. This technique is widely used in ophthalmology to diagnose and monitor various eye conditions such as macular degeneration, diabetic retinopathy, and glaucoma.
A pupil, in medical terms, refers to the circular opening in the center of the iris (the colored part of the eye) that allows light to enter and reach the retina. The size of the pupil can change involuntarily in response to light intensity and emotional state, as well as voluntarily through certain eye exercises or with the use of eye drops. Pupillary reactions are important in clinical examinations as they can provide valuable information about the nervous system's functioning, particularly the brainstem and cranial nerves II and III.
A nonsense codon is a sequence of three nucleotides in DNA or RNA that does not code for an amino acid. Instead, it signals the end of the protein-coding region of a gene and triggers the termination of translation, the process by which the genetic code is translated into a protein.
In DNA, the nonsense codons are UAA, UAG, and UGA, which are also known as "stop codons." When these codons are encountered during translation, they cause the release of the newly synthesized polypeptide chain from the ribosome, bringing the process of protein synthesis to a halt.
Nonsense mutations are changes in the DNA sequence that result in the appearance of a nonsense codon where an amino acid-coding codon used to be. These types of mutations can lead to premature termination of translation and the production of truncated, nonfunctional proteins, which can cause genetic diseases or contribute to cancer development.
Dark adaptation is the process by which the eyes adjust to low levels of light. This process allows the eyes to become more sensitive to light and see better in the dark. It involves the dilation of the pupils, as well as chemical changes in the rods and cones (photoreceptor cells) of the retina. These changes allow the eye to detect even small amounts of light and improve visual acuity in low-light conditions. Dark adaptation typically takes several minutes to occur fully, but can be faster or slower depending on various factors such as age, prior exposure to light, and certain medical conditions. It is an important process for maintaining good vision in a variety of lighting conditions.
A homozygote is an individual who has inherited the same allele (version of a gene) from both parents and therefore possesses two identical copies of that allele at a specific genetic locus. This can result in either having two dominant alleles (homozygous dominant) or two recessive alleles (homozygous recessive). In contrast, a heterozygote has inherited different alleles from each parent for a particular gene.
The term "homozygote" is used in genetics to describe the genetic makeup of an individual at a specific locus on their chromosomes. Homozygosity can play a significant role in determining an individual's phenotype (observable traits), as having two identical alleles can strengthen the expression of certain characteristics compared to having just one dominant and one recessive allele.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Single-Stranded Conformational Polymorphism (SSCP) is not a medical condition but rather a laboratory technique used in molecular biology and genetics. It refers to the phenomenon where a single-stranded DNA or RNA molecule can adopt different conformations or shapes based on its nucleotide sequence, even if the difference in the sequence is as small as a single base pair change. This property is used in SSCP analysis to detect mutations or variations in DNA or RNA sequences.
In SSCP analysis, the denatured single-stranded DNA or RNA sample is subjected to electrophoresis on a non-denaturing polyacrylamide gel. The different conformations of the single-stranded molecules migrate at different rates in the gel, creating multiple bands that can be visualized by staining or other detection methods. The presence of additional bands or shifts in band patterns can indicate the presence of a sequence variant or mutation.
SSCP analysis is often used as a screening tool for genetic diseases, cancer, and infectious diseases to identify genetic variations associated with these conditions. However, it has largely been replaced by more sensitive and accurate methods such as next-generation sequencing.
Visual acuity is a measure of the sharpness or clarity of vision. It is usually tested by reading an eye chart from a specific distance, such as 20 feet (6 meters). The standard eye chart used for this purpose is called the Snellen chart, which contains rows of letters that decrease in size as you read down the chart.
Visual acuity is typically expressed as a fraction, with the numerator representing the testing distance and the denominator indicating the smallest line of type that can be read clearly. For example, if a person can read the line on the eye chart that corresponds to a visual acuity of 20/20, it means they have normal vision at 20 feet. If their visual acuity is 20/40, it means they must be as close as 20 feet to see what someone with normal vision can see at 40 feet.
It's important to note that visual acuity is just one aspect of overall vision and does not necessarily reflect other important factors such as peripheral vision, depth perception, color vision, or contrast sensitivity.
The pigment epithelium of the eye, also known as the retinal pigment epithelium (RPE), is a layer of cells located between the photoreceptor cells of the retina and the choroid, which is the vascular layer of the eye. The RPE plays a crucial role in maintaining the health and function of the photoreceptors by providing them with nutrients, removing waste products, and helping to regulate the light that enters the eye.
The RPE cells contain pigment granules that absorb excess light, preventing it from scattering within the eye and improving visual acuity. They also help to create a barrier between the retina and the choroid, which is important for maintaining the proper functioning of the photoreceptors. Additionally, the RPE plays a role in the regeneration of visual pigments in the photoreceptor cells, allowing us to see in different light conditions.
Damage to the RPE can lead to various eye diseases and conditions, including age-related macular degeneration (AMD), which is a leading cause of vision loss in older adults.
Exons are the coding regions of DNA that remain in the mature, processed mRNA after the removal of non-coding intronic sequences during RNA splicing. These exons contain the information necessary to encode proteins, as they specify the sequence of amino acids within a polypeptide chain. The arrangement and order of exons can vary between different genes and even between different versions of the same gene (alternative splicing), allowing for the generation of multiple protein isoforms from a single gene. This complexity in exon structure and usage significantly contributes to the diversity and functionality of the proteome.
A genetic vector is a vehicle, often a plasmid or a virus, that is used to introduce foreign DNA into a host cell as part of genetic engineering or gene therapy techniques. The vector contains the desired gene or genes, along with regulatory elements such as promoters and enhancers, which are needed for the expression of the gene in the target cells.
The choice of vector depends on several factors, including the size of the DNA to be inserted, the type of cell to be targeted, and the efficiency of uptake and expression required. Commonly used vectors include plasmids, adenoviruses, retroviruses, and lentiviruses.
Plasmids are small circular DNA molecules that can replicate independently in bacteria. They are often used as cloning vectors to amplify and manipulate DNA fragments. Adenoviruses are double-stranded DNA viruses that infect a wide range of host cells, including human cells. They are commonly used as gene therapy vectors because they can efficiently transfer genes into both dividing and non-dividing cells.
Retroviruses and lentiviruses are RNA viruses that integrate their genetic material into the host cell's genome. This allows for stable expression of the transgene over time. Lentiviruses, a subclass of retroviruses, have the advantage of being able to infect non-dividing cells, making them useful for gene therapy applications in post-mitotic tissues such as neurons and muscle cells.
Overall, genetic vectors play a crucial role in modern molecular biology and medicine, enabling researchers to study gene function, develop new therapies, and modify organisms for various purposes.
Animal disease models are specialized animals, typically rodents such as mice or rats, that have been genetically engineered or exposed to certain conditions to develop symptoms and physiological changes similar to those seen in human diseases. These models are used in medical research to study the pathophysiology of diseases, identify potential therapeutic targets, test drug efficacy and safety, and understand disease mechanisms.
The genetic modifications can include knockout or knock-in mutations, transgenic expression of specific genes, or RNA interference techniques. The animals may also be exposed to environmental factors such as chemicals, radiation, or infectious agents to induce the disease state.
Examples of animal disease models include:
1. Mouse models of cancer: Genetically engineered mice that develop various types of tumors, allowing researchers to study cancer initiation, progression, and metastasis.
2. Alzheimer's disease models: Transgenic mice expressing mutant human genes associated with Alzheimer's disease, which exhibit amyloid plaque formation and cognitive decline.
3. Diabetes models: Obese and diabetic mouse strains like the NOD (non-obese diabetic) or db/db mice, used to study the development of type 1 and type 2 diabetes, respectively.
4. Cardiovascular disease models: Atherosclerosis-prone mice, such as ApoE-deficient or LDLR-deficient mice, that develop plaque buildup in their arteries when fed a high-fat diet.
5. Inflammatory bowel disease models: Mice with genetic mutations affecting intestinal barrier function and immune response, such as IL-10 knockout or SAMP1/YitFc mice, which develop colitis.
Animal disease models are essential tools in preclinical research, but it is important to recognize their limitations. Differences between species can affect the translatability of results from animal studies to human patients. Therefore, researchers must carefully consider the choice of model and interpret findings cautiously when applying them to human diseases.
Proteins are complex, large molecules that play critical roles in the body's functions. They are made up of amino acids, which are organic compounds that are the building blocks of proteins. Proteins are required for the structure, function, and regulation of the body's tissues and organs. They are essential for the growth, repair, and maintenance of body tissues, and they play a crucial role in many biological processes, including metabolism, immune response, and cellular signaling. Proteins can be classified into different types based on their structure and function, such as enzymes, hormones, antibodies, and structural proteins. They are found in various foods, especially animal-derived products like meat, dairy, and eggs, as well as plant-based sources like beans, nuts, and grains.
Genotype, in genetics, refers to the complete heritable genetic makeup of an individual organism, including all of its genes. It is the set of instructions contained in an organism's DNA for the development and function of that organism. The genotype is the basis for an individual's inherited traits, and it can be contrasted with an individual's phenotype, which refers to the observable physical or biochemical characteristics of an organism that result from the expression of its genes in combination with environmental influences.
It is important to note that an individual's genotype is not necessarily identical to their genetic sequence. Some genes have multiple forms called alleles, and an individual may inherit different alleles for a given gene from each parent. The combination of alleles that an individual inherits for a particular gene is known as their genotype for that gene.
Understanding an individual's genotype can provide important information about their susceptibility to certain diseases, their response to drugs and other treatments, and their risk of passing on inherited genetic disorders to their offspring.
Visual fields refer to the total area in which objects can be seen while keeping the eyes focused on a central point. It is the entire area that can be observed using peripheral (side) vision while the eye gazes at a fixed point. A visual field test is used to detect blind spots or gaps (scotomas) in a person's vision, which could indicate various medical conditions such as glaucoma, retinal damage, optic nerve disease, brain tumors, or strokes. The test measures both the central and peripheral vision and maps the entire area that can be seen when focusing on a single point.
Cell surface receptors, also known as membrane receptors, are proteins located on the cell membrane that bind to specific molecules outside the cell, known as ligands. These receptors play a crucial role in signal transduction, which is the process of converting an extracellular signal into an intracellular response.
Cell surface receptors can be classified into several categories based on their structure and mechanism of action, including:
1. Ion channel receptors: These receptors contain a pore that opens to allow ions to flow across the cell membrane when they bind to their ligands. This ion flux can directly activate or inhibit various cellular processes.
2. G protein-coupled receptors (GPCRs): These receptors consist of seven transmembrane domains and are associated with heterotrimeric G proteins that modulate intracellular signaling pathways upon ligand binding.
3. Enzyme-linked receptors: These receptors possess an intrinsic enzymatic activity or are linked to an enzyme, which becomes activated when the receptor binds to its ligand. This activation can lead to the initiation of various signaling cascades within the cell.
4. Receptor tyrosine kinases (RTKs): These receptors contain intracellular tyrosine kinase domains that become activated upon ligand binding, leading to the phosphorylation and activation of downstream signaling molecules.
5. Integrins: These receptors are transmembrane proteins that mediate cell-cell or cell-matrix interactions by binding to extracellular matrix proteins or counter-receptors on adjacent cells. They play essential roles in cell adhesion, migration, and survival.
Cell surface receptors are involved in various physiological processes, including neurotransmission, hormone signaling, immune response, and cell growth and differentiation. Dysregulation of these receptors can contribute to the development of numerous diseases, such as cancer, diabetes, and neurological disorders.
A missense mutation is a type of point mutation in which a single nucleotide change results in the substitution of a different amino acid in the protein that is encoded by the affected gene. This occurs when the altered codon (a sequence of three nucleotides that corresponds to a specific amino acid) specifies a different amino acid than the original one. The function and/or stability of the resulting protein may be affected, depending on the type and location of the missense mutation. Missense mutations can have various effects, ranging from benign to severe, depending on the importance of the changed amino acid for the protein's structure or function.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
The Fluorescent Antibody Technique (FAT), Indirect is a type of immunofluorescence assay used to detect the presence of specific antigens in a sample. In this method, the sample is first incubated with a primary antibody that binds to the target antigen. After washing to remove unbound primary antibodies, a secondary fluorescently labeled antibody is added, which recognizes and binds to the primary antibody. This indirect labeling approach allows for amplification of the signal, making it more sensitive than direct methods. The sample is then examined under a fluorescence microscope to visualize the location and amount of antigen based on the emitted light from the fluorescent secondary antibody. It's commonly used in diagnostic laboratories for detection of various bacteria, viruses, and other antigens in clinical specimens.
Nerve tissue proteins are specialized proteins found in the nervous system that provide structural and functional support to nerve cells, also known as neurons. These proteins include:
1. Neurofilaments: These are type IV intermediate filaments that provide structural support to neurons and help maintain their shape and size. They are composed of three subunits - NFL (light), NFM (medium), and NFH (heavy).
2. Neuronal Cytoskeletal Proteins: These include tubulins, actins, and spectrins that provide structural support to the neuronal cytoskeleton and help maintain its integrity.
3. Neurotransmitter Receptors: These are specialized proteins located on the postsynaptic membrane of neurons that bind neurotransmitters released by presynaptic neurons, triggering a response in the target cell.
4. Ion Channels: These are transmembrane proteins that regulate the flow of ions across the neuronal membrane and play a crucial role in generating and transmitting electrical signals in neurons.
5. Signaling Proteins: These include enzymes, receptors, and adaptor proteins that mediate intracellular signaling pathways involved in neuronal development, differentiation, survival, and death.
6. Adhesion Proteins: These are cell surface proteins that mediate cell-cell and cell-matrix interactions, playing a crucial role in the formation and maintenance of neural circuits.
7. Extracellular Matrix Proteins: These include proteoglycans, laminins, and collagens that provide structural support to nerve tissue and regulate neuronal migration, differentiation, and survival.
Alcohol oxidoreductases are a class of enzymes that catalyze the oxidation of alcohols to aldehydes or ketones, while reducing nicotinamide adenine dinucleotide (NAD+) to NADH. These enzymes play an important role in the metabolism of alcohols and other organic compounds in living organisms.
The most well-known example of an alcohol oxidoreductase is alcohol dehydrogenase (ADH), which is responsible for the oxidation of ethanol to acetaldehyde in the liver during the metabolism of alcoholic beverages. Other examples include aldehyde dehydrogenases (ALDH) and sorbitol dehydrogenase (SDH).
These enzymes are important targets for the development of drugs used to treat alcohol use disorder, as inhibiting their activity can help to reduce the rate of ethanol metabolism and the severity of its effects on the body.
A pupillary reflex is a type of reflex that involves the constriction or dilation of the pupils in response to changes in light or near vision. It is mediated by the optic and oculomotor nerves. The pupillary reflex helps regulate the amount of light that enters the eye, improving visual acuity and protecting the retina from excessive light exposure.
In a clinical setting, the pupillary reflex is often assessed as part of a neurological examination. A normal pupillary reflex consists of both direct and consensual responses. The direct response occurs when light is shone into one eye and the pupil of that same eye constricts. The consensual response occurs when light is shone into one eye, causing the pupil of the other eye to also constrict.
Abnormalities in the pupillary reflex can indicate various neurological conditions, such as brainstem injuries or diseases affecting the optic or oculomotor nerves.
Genetic testing is a type of medical test that identifies changes in chromosomes, genes, or proteins. The results of a genetic test can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Genetic tests are performed on a sample of blood, hair, skin, amniotic fluid (the fluid that surrounds a fetus during pregnancy), or other tissue. For example, a physician may recommend genetic testing to help diagnose a genetic condition, confirm the presence of a gene mutation known to increase the risk of developing certain cancers, or determine the chance for a couple to have a child with a genetic disorder.
There are several types of genetic tests, including:
* Diagnostic testing: This type of test is used to identify or confirm a suspected genetic condition in an individual. It may be performed before birth (prenatal testing) or at any time during a person's life.
* Predictive testing: This type of test is used to determine the likelihood that a person will develop a genetic disorder. It is typically offered to individuals who have a family history of a genetic condition but do not show any symptoms themselves.
* Carrier testing: This type of test is used to determine whether a person carries a gene mutation for a genetic disorder. It is often offered to couples who are planning to have children and have a family history of a genetic condition or belong to a population that has an increased risk of certain genetic disorders.
* Preimplantation genetic testing: This type of test is used in conjunction with in vitro fertilization (IVF) to identify genetic changes in embryos before they are implanted in the uterus. It can help couples who have a family history of a genetic disorder or who are at risk of having a child with a genetic condition to conceive a child who is free of the genetic change in question.
* Pharmacogenetic testing: This type of test is used to determine how an individual's genes may affect their response to certain medications. It can help healthcare providers choose the most effective medication and dosage for a patient, reducing the risk of adverse drug reactions.
It is important to note that genetic testing should be performed under the guidance of a qualified healthcare professional who can interpret the results and provide appropriate counseling and support.
Membrane proteins are a type of protein that are embedded in the lipid bilayer of biological membranes, such as the plasma membrane of cells or the inner membrane of mitochondria. These proteins play crucial roles in various cellular processes, including:
1. Cell-cell recognition and signaling
2. Transport of molecules across the membrane (selective permeability)
3. Enzymatic reactions at the membrane surface
4. Energy transduction and conversion
5. Mechanosensation and signal transduction
Membrane proteins can be classified into two main categories: integral membrane proteins, which are permanently associated with the lipid bilayer, and peripheral membrane proteins, which are temporarily or loosely attached to the membrane surface. Integral membrane proteins can further be divided into three subcategories based on their topology:
1. Transmembrane proteins, which span the entire width of the lipid bilayer with one or more alpha-helices or beta-barrels.
2. Lipid-anchored proteins, which are covalently attached to lipids in the membrane via a glycosylphosphatidylinositol (GPI) anchor or other lipid modifications.
3. Monotopic proteins, which are partially embedded in the membrane and have one or more domains exposed to either side of the bilayer.
Membrane proteins are essential for maintaining cellular homeostasis and are targets for various therapeutic interventions, including drug development and gene therapy. However, their structural complexity and hydrophobicity make them challenging to study using traditional biochemical methods, requiring specialized techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single-particle cryo-electron microscopy (cryo-EM).
A "knockout" mouse is a genetically engineered mouse in which one or more genes have been deleted or "knocked out" using molecular biology techniques. This allows researchers to study the function of specific genes and their role in various biological processes, as well as potential associations with human diseases. The mice are generated by introducing targeted DNA modifications into embryonic stem cells, which are then used to create a live animal. Knockout mice have been widely used in biomedical research to investigate gene function, disease mechanisms, and potential therapeutic targets.
Polymerase Chain Reaction (PCR) is a laboratory technique used to amplify specific regions of DNA. It enables the production of thousands to millions of copies of a particular DNA sequence in a rapid and efficient manner, making it an essential tool in various fields such as molecular biology, medical diagnostics, forensic science, and research.
The PCR process involves repeated cycles of heating and cooling to separate the DNA strands, allow primers (short sequences of single-stranded DNA) to attach to the target regions, and extend these primers using an enzyme called Taq polymerase, resulting in the exponential amplification of the desired DNA segment.
In a medical context, PCR is often used for detecting and quantifying specific pathogens (viruses, bacteria, fungi, or parasites) in clinical samples, identifying genetic mutations or polymorphisms associated with diseases, monitoring disease progression, and evaluating treatment effectiveness.
Homeodomain proteins are a group of transcription factors that play crucial roles in the development and differentiation of cells in animals and plants. They are characterized by the presence of a highly conserved DNA-binding domain called the homeodomain, which is typically about 60 amino acids long. The homeodomain consists of three helices, with the third helix responsible for recognizing and binding to specific DNA sequences.
Homeodomain proteins are involved in regulating gene expression during embryonic development, tissue maintenance, and organismal growth. They can act as activators or repressors of transcription, depending on the context and the presence of cofactors. Mutations in homeodomain proteins have been associated with various human diseases, including cancer, congenital abnormalities, and neurological disorders.
Some examples of homeodomain proteins include PAX6, which is essential for eye development, HOX genes, which are involved in body patterning, and NANOG, which plays a role in maintaining pluripotency in stem cells.
 Leber congenital amaurosis
Leber congenital amaurosis
Chromosome 19
Akbar Khan (disability activist)
Congenital blindness
LCA5
Roche
Kim Umback
RPE65
Retina
RPGRIP1
Retinal dehydrogenase
Jean Bennett
Katherine A. High
CRB1
RDH8
Gene therapy
CRX (gene)
GUCY2D
Adeno-associated virus
RDH12
Kody Keplinger
Gene therapy of the human retina
NcRNA therapy
Retinitis pigmentosa
Retinal gene therapy using lentiviral vectors
AIPL1
Bestrophin 1
Letticia Martinez
Visual impairment
NUB1
Leber congenital amaurosis - Wikipedia
 Leber congenital amaurosis: MedlinePlus Genetics
Leber congenital amaurosis: MedlinePlus Genetics
 Leber Congenital Amaurosis News, Research - Page 2
Leber Congenital Amaurosis News, Research - Page 2
 Leber Congenital Amaurosis - American Academy of Ophthalmology
Leber Congenital Amaurosis - American Academy of Ophthalmology
 Leber's congenital amaurosis
Leber's congenital amaurosis
 Longitudinal clinical course of three Japanese patients with Leber congenital amaurosis/early-onset retinal dystrophy with...
Longitudinal clinical course of three Japanese patients with Leber congenital amaurosis/early-onset retinal dystrophy with...
 Gene Therapy for Inherited Blindness: Hannah's Leber Congenital Amaurosis Story | The Children's Hospital of Philadelphia
Gene Therapy for Inherited Blindness: Hannah's Leber Congenital Amaurosis Story | The Children's Hospital of Philadelphia
 Leber Congenital Amaurosis (LCA) Pipeline Report, 2023- Planned Drugs by Phase, Mechanism of Action, Route of Administration,...
Leber Congenital Amaurosis (LCA) Pipeline Report, 2023- Planned Drugs by Phase, Mechanism of Action, Route of Administration,...
 New Treatment for Leber's Congenital Amaurosis (LCA) | WonderBaby.org
New Treatment for Leber's Congenital Amaurosis (LCA) | WonderBaby.org
Peripherin mutations cause a distinct form of recessive Leber congenital amaurosis and dominant phenotypes in asymptomatic...
 Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital...
Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital...
 Coat's like vasculopathy in leber congenital amaurosis secondary to homozygous mutations in CRB1: a case report and discussion...
Coat's like vasculopathy in leber congenital amaurosis secondary to homozygous mutations in CRB1: a case report and discussion...
Leber's Congenital Amaurosis - PEDGAZ
 Leber Congenital Amaurosis - Oligofastx
Leber Congenital Amaurosis - Oligofastx
 Leber Congenital Amaurosis Definition - CorneaCare
Leber Congenital Amaurosis Definition - CorneaCare
 Leber congenital amaurosis - Global Genes
Leber congenital amaurosis - Global Genes
 Leber's congenital amaurosis - Athens Eye Hospital
Leber's congenital amaurosis - Athens Eye Hospital
 Leber's Congenital Amaurosis - Tactile Vision Graphics
Leber's Congenital Amaurosis - Tactile Vision Graphics
 Leber Congenital Amaurosis Precision Panel - Middle East
Leber Congenital Amaurosis Precision Panel - Middle East
 Gene Set - Leber congenital amaurosis Retina GSE3249
Gene Set - Leber congenital amaurosis Retina GSE3249
 Clinical and molecular analysis of leber congenital amaurosis.
Clinical and molecular analysis of leber congenital amaurosis.
 UCSD Leber Congenital Amaurosis Clinical Trials for 2023 - San Diego
UCSD Leber Congenital Amaurosis Clinical Trials for 2023 - San Diego
The Natural History of Leber Congenital Amaurosis and Cone-Rod Dystrophy Associated with Variants in the GUCY2D Gene<...
 Leber congenital amaurosis with early-onset deafness (Concept Id: C4693498)
- MedGen - NCBI
Leber congenital amaurosis with early-onset deafness (Concept Id: C4693498)
- MedGen - NCBI
 Leber congenital amaurosis (LCA)/Early onset severe retinal dystrophy (EOSRD): for professionals - Gene Vision
Leber congenital amaurosis (LCA)/Early onset severe retinal dystrophy (EOSRD): for professionals - Gene Vision
 OCT guided micro-focal ERG system with multiple stimulation wavelengths for characterization of ocular health | Scientific...
OCT guided micro-focal ERG system with multiple stimulation wavelengths for characterization of ocular health | Scientific...
 New gene therapy approach could help prevent blindness in children with Leber congenital amaurosis - Chores4Kids
New gene therapy approach could help prevent blindness in children with Leber congenital amaurosis - Chores4Kids
 Retinitis Pigmentosa: Practice Essentials, Background, Pathophysiology
Retinitis Pigmentosa: Practice Essentials, Background, Pathophysiology
 Can Blindness Be Cured?
Can Blindness Be Cured?
Retina - Wikipedia
Retinitis pigmentosa9
- LCA was originally described as a variety retinitis pigmentosa by Theodor Leber in 1869. (wikipedia.org)
- Mutations in RPE65 cause the childhood blindness disorder known as Leber congenital amaurosis (LCA), as well as autosomal recessive retinitis pigmentosa (RP). (molvis.org)
- It should be differentiated from other forms or chorioretinal dystrophies (juvenile retinitis pigmentosa or congenital stationary night blindness), cortical blindness or maturation delay and metabolic disorders. (nih.gov)
- Ocugen's OCU400, an investigational modifier gene therapy intended to treat inherited retinal diseases, has demonstrated improvement or stabilization of vision among patients with retinitis pigmentosa (RP) and leber congenital amaurosis (LCA) treated in a phase 1/2 clinical trial (NCT05203939). (cgtlive.com)
- Variations in the human Crumbs homolog-1 (CRB1) gene lead to an array of retinal dystrophies including early-onset of retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA) in children. (nin.nl)
- In eye diseases like retinitis pigmentosa, age-related macular degeneration and Leber congenital amaurosis, things are going on in the cellular level that occur before anything is detected by way of a test or observation. (guldenophthalmics.com)
- Common retinal diseases such as age-related macular degeneration, glaucoma, and retinitis pigmentosa are covered, as are rare disorders such as Leber congenital amaurosis and Usher syndrome. (cshlpress.com)
- Mutations in that gene are responsible for early onset blindness from Leber congenital amaurosis, some forms of retinitis pigmentosa , and other conditions. (aao.org)
- RPE65 gene mutations have been identified in two IRDs: Leber congenital amaurosis and retinitis pigmentosa. (medscape.com)
Types of Leber congenital amaurosis4
- At least 20 genetic types of Leber congenital amaurosis have been described. (medlineplus.gov)
- Different types of Leber congenital amaurosis can present with different symptoms. (guidedogs.org.uk)
- Some types of Leber congenital amaurosis remain stable, while others worsen with time, leading to degeneration in eyesight. (guidedogs.org.uk)
- How many types of Leber congenital amaurosis are there? (replicadb4.com)
Autosomal recessive2
- Leber's congenital amaurosis is an autosomal recessive disorder, characterized by the onset of blindness before the age of 6 months, a variable fundus aspect and an absent or extremely pathological ERG. (nih.gov)
- Leber congenital amaurosis (LCA) represents a leading cause of autosomal recessive blindness in children worldwide, affecting between 1 in 30,000 to 1 in 81,000 newborns annually. (entokey.com)
Mutations7
- citation needed] On 19 December 2017, the U.S. Food and Drug Administration approved voretigene neparvovec-rzyl (Luxturna), an adeno-associated virus vector-based gene therapy for children and adults with biallelic RPE65 gene mutations responsible for retinal dystrophy, including Leber congenital amaurosis. (wikipedia.org)
- Leber congenital amaurosis can result from variants (also known as mutations) in at least 20 genes, all of which are necessary for function of the retina and normal vision. (medlineplus.gov)
- August 1, 2012 - Four independent research teams have described mutations in a gene known for its role in energy metabolism as the cause of the 18th recognized form of Leber congenital amaurosis (LCA), the most common type of childhood blindness. (medscape.com)
- Two groups of investigators have reported independently on their initial observations from Phase I clinical trials of gene transfer for Leber congenital amaurosis (LCA) caused by mutations in the RPE65 gene. (nih.gov)
- Mutations in the KCNJ13 gene that encodes the inwardly rectifying potassium channel Kir7.1 cause snowflake vitreoretinal degeneration (SVD) and leber congenital amaurosis (LCA). (mcw.edu)
- 12. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. (nih.gov)
- NMNAT1 mutations cause Leber congenital amaurosis. (opusgtx.com)
RPE657
- Variants in the CEP290 , CRB1 , GUCY2D , and RPE65 genes are the most common causes of Leber congenital amaurosis, while variants in the other genes generally account for a smaller percentage of cases. (medlineplus.gov)
- RPE65 -related Leber congenital amaurosis / early-onset severe retinal dystrophy ( RPE65 -LCA/EOSRD) is a severe inherited retinal degeneration (IRD) with a typical presentation between birth and age five years. (nih.gov)
- About 10% of people with Leber congenital amaurosis (LCA), an inherited disorder that causes vision loss starting in childhood, have an altered form of the gene RPE65 . (nih.gov)
- Biallelic pathogenic variants in RPE65 cause early-onset severe retinal dystrophy (EOSRD) or Leber congenital amaurosis (LCA). (hindawi.com)
- 8. Gene therapy rescues cone structure and function in the 3-month-old rd12 mouse: a model for midcourse RPE65 leber congenital amaurosis. (nih.gov)
- 9. Lentiviral gene transfer of RPE65 rescues survival and function of cones in a mouse model of Leber congenital amaurosis. (nih.gov)
- In this study, we investigated the expression of the gene encoding β-galactosidase (Glb)-1-like protein 3 (Glb1l3), a member of the glycosyl hydrolase 35 family, during retinal degeneration in the retinal pigment epithelium (RPE)-specific 65-kDa protein knockout ( Rpe65 −/− ) mouse model of Leber congenital amaurosis (LCA). (molvis.org)
Causes Leber congenital3
- What causes Leber congenital amaurosis? (guidedogs.org.uk)
- A defect in a number of genes causes Leber congenital amaurosis. (guidedogs.org.uk)
- A Novel KCNJ13 Nonsense Mutation and Loss of Kir7.1 Channel Function Causes Leber Congenital Amaurosis (LCA16). (mcw.edu)
Leber's Congenita4
- Leber's congenital amaurosis associated with familial juvenile nephronophthisis and cone-shaped epiphyses of the hands (the Saldino-Mainzer syndrome). (nih.gov)
- Leber's Congenital Amaurosis and Gene Therapy. (igenomix.it)
- Long-term effect of gene therapy on Leber's congenital amaurosis. (nih.gov)
- 10. Bax-induced apoptosis in Leber's congenital amaurosis: a dual role in rod and cone degeneration. (nih.gov)
Dystrophy4
- Leber congenital amaurosis is a severe retinal dystrophy and is sometimes known as Leber hereditary optic neuropathy or Leber optic atrophy. (guidedogs.org.uk)
- Two other pieces of evidence were accepted after further analysis of these Brazilian families: (i) p.Phe83Leu and p.Gly187Glu segregate with childhood retinal dystrophy within families, and (ii) their prevalence in Leber congenital amaurosis (LCA)/early-onset retinal dystrophy (EORD) patients can be considered higher than in other inherited retinal dystrophy patients. (nih.gov)
- Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. (igenomix.it)
- GUCY2D -associated Leber congenital amaurosis is a severe early-onset retinal dystrophy. (entokey.com)
Retina9
- Leber congenital amaurosis (LCA) is an eye disorder that primarily affects the retina. (nih.gov)
- Variants in any of the genes associated with Leber congenital amaurosis disrupt the development and function of the retina, resulting in early vision loss. (medlineplus.gov)
- 600 genes differentially expressed during Leber congenital amaurosis_Retina_GSE3249 disease perturbation from the GEO Signatures of Differentially Expressed Genes for Diseases dataset. (maayanlab.cloud)
- The Leber congenital amaurosis gene product AIPL1 is localized exclusively in rod photoreceptors of the adult human retina. (replicadb4.com)
- Leber congenital amaurosis (LCA) primarily affects the retina, the specialized tissue at the back of the eye that detects light and color. (replicadb4.com)
- Leber Congenital Amaurosis (LCA) is a genetic eye disease that affects the retina. (guldenophthalmics.com)
- The search led them first to Emory University, where physicians found a genetic mutation for Leber congenital amaurosis (LCA), a rare vision disorder that affects the retina. (savannahbee.com)
- La enfermedad se cree que es causada por el desarrollo anormal de las CÉLULAS FOTORRECEPTORAS en la RETINA o por la degeneración extremadamente prematura de las células retinianas. (bvsalud.org)
- Not to be confused with LEBER HEREDITARY OPTIC NEUROPATHY, the disease is thought to be caused by abnormal development of PHOTORECEPTOR CELLS in the RETINA, or by the extremely premature degeneration of retinal cells. (bvsalud.org)
Genetic4
- If your child does have Leber congenital amaurosis, then genetic testing may be used to identify which gene is responsible for the condition. (guidedogs.org.uk)
- Leber congenital amaurosis: Current genetic basis, scope for genetic testing and personalized medicine. (igenomix.it)
- The diagnosis of Leber congenital amaurosis was suggested, and a genetic CRB1 sequencing for the patient and her two younger siblings, who also had severe vision loss, was done, upon which the diagnosis of Leber congenital amaurosis associated with exudative retinal detachment due to coat's like vasculopathy was made. (biomedcentral.com)
- Genetic therapies that are currently approved by the FDA are available for people who have Leber congenital amaurosis , a rare inherited condition that leads to blindness. (nih.gov)
Dystrophies4
- Leber Congenital Amaurosis (LCA) belongs to the spectrum of early-onset retinal dystrophies. (igenomix.it)
- MIM# 204000) is the earliest and most severe form of all hereditary retinal dystrophies, responsible for congenital blindness [ 1 ]. (biomedcentral.com)
- Leber congenital amaurosis (LCA) and other early-onset severe retinal dystrophies (EOSRD) have a prevalence of approximately 1 : 50,000 worldwide [ 1 ]. (hindawi.com)
- Leber congenital amaurosis (LCA) is the earliest and most severe form of inherited retinal dystrophies, characterized by blindness or severe visual impairment from birth. (molvis.org)
Early-onset1
- Leber congenital amaurosis (LCA) is an inherited retinal disease that causes early-onset severe visual impairment. (nih.gov)
Diagnosis2
- The role of molecular diagnosis in Leber congenital amaurosis. (nih.gov)
- The Igenomix Leber Congenital Amaurosis Precision Panel can be used to make a n accurate and directed diagnosis as well as a differential diagnosis of blindness ultimately leading to a better management and prognosis of the disease. (igenomix.it)
Disorder4
- Leber congenital amaurosis, also known as LCA, is an eye disorder that is present from birth (congenital). (medlineplus.gov)
- In about 30 percent of all people with Leber congenital amaurosis, the cause of the disorder is unknown, though research is ongoing. (medlineplus.gov)
- When Leber congenital amaurosis is caused by varaints in the CRX or IMPDH1 genes, the disorder has an autosomal dominant pattern of inheritance. (medlineplus.gov)
- Leber congenital amaurosis (LCA) is a rare type of inherited eye disorder that causes severe vision loss at birth. (replicadb4.com)
Variants2
- A Mini-Review: Leber Congenital Amaurosis: Identification of Disease-Causing Variants and Personalised Therapies. (igenomix.it)
- To describe the natural history of Leber congenital amaurosis (LCA) associated with GUCY2D variants ( GUCY2D -LCA) in a cohort of children and adults, in preparation for trials of novel therapies. (entokey.com)
Hereditary optic1
- It should not be confused with Leber's hereditary optic neuropathy, which is a different disease also described by Theodor Leber. (wikipedia.org)
Childhood blindness1
- CEP290-associated Leber congenital amaurosis type 10 (LCA10) is a retinal disease resulting in childhood blindness. (ucl.ac.uk)
Mutation2
- This is the first report of the occurrence of coat's like vasculopathy in a patient diagnosed with Leber congenital amaurosis caused by a CRB1 mutation. (biomedcentral.com)
- In this unique case we are reporting the incidence of coat's like vasculopathy in a patient diagnosed with Leber congenital amaurosis caused by CRB1 gene mutation, and its management. (biomedcentral.com)
Symptoms3
- When Do Symptoms of Leber congenital amaurosis Begin? (nih.gov)
- The symptoms of Leber congenital amaurosis can be similar to some other diseases, including Loken-Senior syndrome and Joubert syndrome. (guidedogs.org.uk)
- What are the symptoms of Leber congenital amaurosis? (replicadb4.com)
Multiple systemic2
- Two sib cases of Leber congenital amaurosis with cerebellar vermis hypoplasia and multiple systemic anomalies. (nih.gov)
- Leber congenital amaurosis is reported to be associated with multiple systemic and ocular findings, none of which is coat's like vasculopathy. (biomedcentral.com)
Disorders1
- Children with possible congenital Leber amaurosis should not only have a thorough ophthalmological examination, but should also be seen by a paediatrician experienced in metabolic disorders. (nih.gov)
Vision5
- Leber congenital amaurosis is also associated with other vision problems, including an increased sensitivity to light (photophobia), involuntary movements of the eyes (nystagmus), and extreme farsightedness ( hyperopia ). (medlineplus.gov)
- Leber Congenital Amaurosis (LCA): Potential for Improvement of Vision. (igenomix.it)
- 5. Gene Therapy Fully Restores Vision to the All-Cone Nrl(-/-) Gucy2e(-/-) Mouse Model of Leber Congenital Amaurosis-1. (nih.gov)
- 11. Pharmacological Amelioration of Cone Survival and Vision in a Mouse Model for Leber Congenital Amaurosis. (nih.gov)
- La présente étude, conduite en 2005, évaluait les causes et les principales localisations anatomiques de la cécité et des pertes de vision sévères dans une école pour enfants aveugles de la province d'Ispahan en République islamique d'Iran. (who.int)
Disease1
- Leber congenital amaurosis (LCA) is a rare inherited eye disease that appears at birth or in the first few months of life. (wikipedia.org)
Affects1
- Leber congenital amaurosis leads to poor visual function because it affects the optic nerve at the back of the eye. (guidedogs.org.uk)
Advances1
- 17. Gene therapy for Leber congenital amaurosis: advances and future directions. (nih.gov)
Rare1
- In very rare cases, delayed development and intellectual disability have been reported in people with the features of Leber congenital amaurosis. (medlineplus.gov)
Newborns1
- Leber congenital amaurosis occurs in 2 to 3 per 100,000 newborns. (medlineplus.gov)
Type2
Common1
- How common is Leber congenital amaurosis? (replicadb4.com)
Children1
- Providing children with opportunities to play, hear, touch, understand and other early educational interventions may prevent developmental delays in children with Leber congenital amaurosis. (medlineplus.gov)
Condition1
- Leber congenital amaurosis (LCA) is an inherited retinal condition. (guidedogs.org.uk)