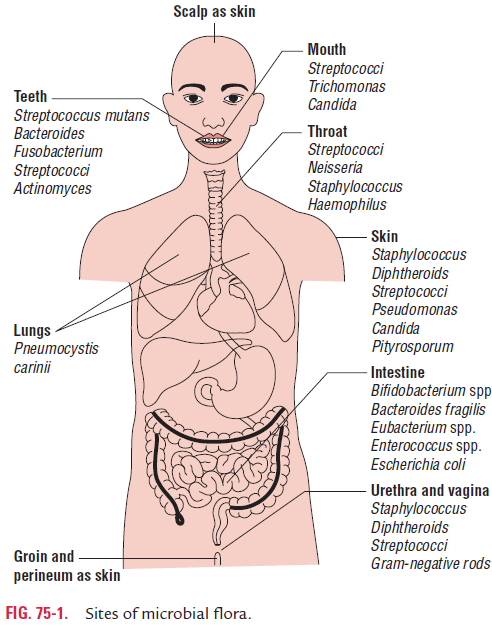
Malabsorption Syndromes
Sprue, Tropical
Celiac Disease
Jejunum
Lactose Intolerance
Missense mutations in SGLT1 cause glucose-galactose malabsorption by trafficking defects. (1/482)
Glucose-galactose malabsorption (GGM) is an autosomal recessive disorder caused by defects in the Na+/glucose cotransporter (SGLT1). Neonates present with severe diarrhea while on any diet containing glucose and/or galactose [1]. This study focuses on a patient of Swiss and Dominican descent. All 15 exons of SGLT1 were screened using single stranded conformational polymorphism analyses, and aberrant PCR products were sequenced. Two missense mutations, Gly318Arg and Ala468Val, were identified. SGLT1 mutants were expressed in Xenopus laevis oocytes for radiotracer uptake, electrophysiological experiments, and Western blotting. Uptakes of [14C]alpha-methyl-d-glucoside by the mutants were 5% or less than that of wild-type. Two-electrode voltage-clamp experiments confirmed the transport defects, as no noticeable sugar-induced current could be elicited from either mutant [2]. Western blots of cell protein showed levels of each SGLT1 mutant protein comparable to that of wild-type, and that both were core-glycosylated. Presteady-state current measurements indicated an absence of SGLT1 in the plasma membrane. We suggest that the compound heterozygote missense mutations G318R and A468V lead to GGM in this patient by defective trafficking of mutant proteins from the endoplasmic reticulum to the plasma membrane. (+info)Geography of intestinal permeability and absorption. (2/482)
BACKGROUND: Intestinal morphology and function vary geographically. AIMS: These functions were assessed in asymptomatic volunteers in European, North American, Middle Eastern, Asian, African, and Caribbean countries. METHODS: Five hour urine collections were obtained from each subject following ingestion of a 100 ml iso-osmolar test solution containing 3-0-methyl-D-glucose, D-xylose, L-rhamnose, and lactulose after an overnight fast, to assess active (3-0-methyl-D-glucose) and passive (D-xylose) carrier mediated, and non-mediated (L-rhamnose) absorption capacity, as well as intestinal permeability (lactulose:rhamnose ratio). RESULTS: A comparison of results for subjects from tropical countries (n=218) with those resident in the combined temperate and subtropical region (Europe, United States, Qatar) (n=224) showed significant differences. Residents in tropical areas had a higher mean lactulose:rhamnose ratio and lower mean five hour recoveries of 3-0-methyl-D-glucose, D-xylose, and L-rhamnose, indicating higher intestinal permeability and lower absorptive capacity. Investigation of visiting residents suggested that differences in intestinal permeability and absorptive capacity were related to the area of residence. Subjects from Texas and Qatar, although comprised of several ethnic groups and resident in a subtropical area, showed no significant difference from European subjects. CONCLUSIONS: There are clearly demarcated variations in intestinal permeability and absorptive capacity affecting asymptomatic residents of different geographical areas which correspond with the condition described as tropical enteropathy. Results suggest the importance of environmental factors. The parameters investigated may be relevant to the predisposition of the indigenous population and travellers to diarrhoeal illness and malnutrition. Intestinal function in patients from the tropics may be difficult to interpret, but should take into account the range of values found in the asymptomatic normal population. (+info)Primary hypomagnesemia caused by isolated magnesium malabsorption: atypical case in adult. (3/482)
Isolated magnesium malabsorption is a rare disorder, which bas been described in no more than 30 patients worldwide. Patients with this disorder typically present with convulsion and diarrhea in early infancy. Hypomagnesemia and hypocalcemia were found in a 35-year-old man with muscle cramps, who bad been diagnosed as primary hypoparathyroidism. Oral magnesium therapy corrected the low serum calcium, magnesium and parathyroid hormone levels. We report an atypical case of isolated magnesium malabsorption in an adult. (+info)Lymphatic absorption of structured triglycerides vs. physical mix in a rat model of fat malabsorption. (4/482)
Comparison was made between the intestinal absorption and lymphatic transport of a randomly interesterified fish oil and medium-chain triglyceride (MCT) structured triglycerides (STG) vs. the physical mix in rat small intestine following ischemia and reperfusion (I/R) injury. Under halothane anesthesia, the superior mesenteric artery (SMA) was occluded for 20 min and then reperfused in I/R rats. The SMA was isolated but not occluded in control rats. In both treatment groups, the mesenteric lymph duct was cannulated and a gastric tube was inserted. Each treatment group received 1 ml of the fish oil-MCT STG or physical mix (7 rats/group) through the gastric tube followed by an infusion of PBS at 3 ml/h for 8 h. Lymph was collected hourly for 8 h. Lymph triglyceride, cholesterol, and decanoic and eicosapentaenoic acids increased rapidly and maintained a significantly higher output (P < 0.01) with STG compared with physical mix in control rats over 8 h. After I/R, lymphatic triglyceride output decreased 50% compared with control. Gastric infusion of STG significantly improved lipid transport by having a twofold higher triglyceride, cholesterol, and decanoic and eicosapentaenoic acids output to lymph compared with its physical mix (P < 0.01). We conclude that STG is absorbed into lymph significantly better than physical mix by both the normal intestine and the intestine injured by I/R. (+info)Metabolic adaptations to dietary fat malabsorption in chylomicron-deficient mice. (5/482)
A mouse model of chylomicron deficiency was recently developed; these mice express a human apolipoprotein (apo) B transgene in the liver but do not synthesize any apoB in the intestine. Despite severe intestinal fat malabsorption, the mice maintain normal concentrations of plasma lipids and liver-derived apoB 100-containing lipoproteins. We investigated the metabolic mechanisms by which plasma lipid levels are kept normal. De novo lipogenesis (DNL) and cholesterogenesis were measured by mass isotopomer distribution analysis (MIDA). Plasma non-esterified fatty acid (NEFA) fluxes and hepatic re-esterification of labelled plasma NEFA were also measured. Hepatic and plasma triacylglycerol (TG) concentrations and plasma NEFA fluxes were not different between chylomicron-deficient mice and controls. The contribution from DNL to the hepatic TG pool was only modestly higher in chylomicron-deficient mice [12+/-2.1% (n=7) compared with 3.7+/-1.0% (n=9); means+/-S.E.M.], whereas cholesterogenesis was markedly elevated. The fractional contribution from plasma NEFA to hepatic TG was greatly elevated in the chylomicron-deficient animals (62% compared with 23%). Accordingly, 73% of hepatic TG was neither from DNL nor from plasma NEFA in controls, presumably reflecting prior contribution from chylomicron remnants, compared with only 26% in the chylomicron-deficient group. The long-term contribution from DNL to adipose fat stores reached approximately the same steady-state values (approximately 30%) in the two groups. Body fat accumulation was much lower in chylomicron-deficient animals; thus, whole-body absolute DNL was significantly lower. We conclude that plasma and hepatic TG pools and hepatic secretion of apoB-containing particles are maintained at normal levels in chylomicron-deficient mice, not by de novo fatty acid synthesis, but by more avid re-esterification of plasma NEFA, replacing the normally predominant contribution from chylomicrons, and that some dietary fat can be absorbed by apoB-independent mechanisms. (+info)Anderson's disease: exclusion of apolipoprotein and intracellular lipid transport genes. (6/482)
Anderson's disease is a rare, hereditary hypocholesterolemic syndrome characterized by chronic diarrhea, steatorrhea, and failure to thrive associated with the absence of apo B48-containing lipoproteins. To further define the molecular basis of the disease, we studied 8 affected subjects in 7 unrelated families of North African origin after treatment with a low-fat diet. Lipid loading of intestinal biopsies persisted, but the pattern and extent of loading was variable among the patients. Electron microscopy showed lipoprotein-like particles in membrane-bound compartments, the densities (0.65 to 7.5 particles/mu(2)) and the mean diameters (169 to 580 nm) of which were, in general, significantly larger than in a normal fed subject (0.66 particles/mu(2), 209 nm mean diameter). There were also large lipid particles having diameters up to 7043 nm (average diameters from 368 to 2127 nm) that were not surrounded by a membrane. Rarely, lipoprotein-like particles 50 to 150 nm in diameter were observed in the intercellular spaces. Intestinal organ culture showed that apo B and apo AIV were synthesized with apparently normal molecular weights and that small amounts were secreted in lipid-bound forms (density <1.006 g/mL). Normal microsomal triglyceride transfer protein (MTP) and activity were also detected in intestinal biopsies. Segregation analyses of 4 families excluded, as a cause of the disease, significant regions of the genome surrounding the genes for apo AI, AIV, B, CI, CII, CIII, and E, as were the genes encoding 3 proteins involved in intracellular lipid transport, MTP, and fatty acid binding proteins 1 and 2. The results suggest that a factor other than apoproteins and MTP are important for human intestinal chylomicron assembly and secretion. (+info)Hypokalaemic paralysis. (7/482)
Hypokalaemic paralysis is a relatively uncommon but potentially life-threatening clinical syndrome. If recognised and treated appropriately, patients recover without any clinical sequellae. The syndrome of hypokalaemic paralysis represents a heterogeneous group of disorders characterised clinically by hypokalaemia and acute systemic weakness. Most cases are due to familial or primary hypokalaemic periodic paralysis; sporadic cases are associated with numerous other conditions including barium poisoning, hyperthyroidism, renal disorders, certain endocrinopathies and gastrointestinal potassium losses. The age of onset, race, family history, medications, and underlying disease states can help in identifying the cause of hypokalaemic paralysis. Initial therapy of the patient with hypokalaemic paralysis includes potassium replacement and search for underlying aetiology. Further management depends on the aetiology of hypokalaemia, severity of symptoms, and duration of disease. This review presents the differential diagnosis for hypokalaemic paralysis and discusses management of the syndrome. (+info)Intestinal failure defined by measurements of intestinal energy and wet weight absorption. (8/482)
BACKGROUND AND AIMS: Intestinal failure defined by the minimal energy and wet weight absorption required to avoid home parenteral nutrition (HPN) is not well described. Thus the aim of this study was to identify the minimal level of gut function necessary to avoid parenteral support using objective measurements of intestinal function. METHODS: Energy (bomb calorimetry) and wet weight absorption were measured during 48 hour balance studies in 45 HPN patients with intestinal failure and in 44 non-HPN borderline patients with a short bowel or malabsorption exceeding 2 MJ/day. RESULTS: In the non-HPN patients, the lower 5% confidence interval of the absorption of energy was 84% of the basal metabolic rate (BMR, the Harris-Benedict equations), equivalent to 4.9 MJ/day. Wet weight absorption was 1.4 kg/day. The HPN patients absorbed less of either or both. The non-HPN patients absorbed 24-86% (range) of the energy and 23-95% of the wet weight. Absorption in the HPN patients ranged from below 0% (net secretion) in patients with very short bowels to 100% absorption of an insufficient oral intake in patients with pseudo-obstruction. Non-HPN patients who absorbed less than half of their intake avoided HPN by hyperphagia (200-400% of BMR equivalent to 10-24 MJ/day, and 3-7 kg/day of wet weight). CONCLUSION: Intestinal failure was accurately measured as absorption below 1.4 kg/day of wet weight and 84% of the calculated BMR (depending on weight, sex and age), which is equal to 4.9 MJ/day. Intestinal absorption, expressed as a percentage of intake, did not discriminate between patients with and without intestinal failure, except for patients who absorbed less than 25% of their intake. (+info)Malabsorption syndromes refer to a group of disorders in which the small intestine is unable to properly absorb nutrients from food, leading to various gastrointestinal and systemic symptoms. This can result from a variety of underlying conditions, including:
1. Mucosal damage: Conditions such as celiac disease, inflammatory bowel disease (IBD), or bacterial overgrowth that cause damage to the lining of the small intestine, impairing nutrient absorption.
2. Pancreatic insufficiency: A lack of digestive enzymes produced by the pancreas can lead to poor breakdown and absorption of fats, proteins, and carbohydrates. Examples include chronic pancreatitis or cystic fibrosis.
3. Bile acid deficiency: Insufficient bile acids, which are necessary for fat emulsification and absorption, can result in steatorrhea (fatty stools) and malabsorption. This may occur due to liver dysfunction, gallbladder removal, or ileal resection.
4. Motility disorders: Abnormalities in small intestine motility can affect nutrient absorption, as seen in conditions like gastroparesis, intestinal pseudo-obstruction, or scleroderma.
5. Structural abnormalities: Congenital or acquired structural defects of the small intestine, such as short bowel syndrome, may lead to malabsorption.
6. Infections: Certain bacterial, viral, or parasitic infections can cause transient malabsorption by damaging the intestinal mucosa or altering gut flora.
Symptoms of malabsorption syndromes may include diarrhea, steatorrhea, bloating, abdominal cramps, weight loss, and nutrient deficiencies. Diagnosis typically involves a combination of clinical evaluation, laboratory tests, radiologic imaging, and sometimes endoscopic procedures to identify the underlying cause. Treatment is focused on addressing the specific etiology and providing supportive care to manage symptoms and prevent complications.
Tropical sprue is a malabsorption disorder that is most commonly found in tropical or subtropical regions. It is characterized by symptoms such as chronic diarrhea, weight loss, and fatigue, which are caused by the impaired absorption of nutrients in the small intestine.
The exact cause of tropical sprue is not known, but it is thought to be related to an infection or other environmental factor that damages the lining of the small intestine. This damage can interfere with the absorption of nutrients, particularly fat-soluble vitamins and minerals such as vitamin B12, iron, and folate.
Tropical sprue is typically treated with a combination of antibiotics to eliminate any potential infectious causes, as well as a diet that is high in nutrients and low in fat. In severe cases, supplementation with fat-soluble vitamins and other nutrients may be necessary. With appropriate treatment, most people with tropical sprue are able to recover and manage their symptoms.
Celiac disease is a genetic autoimmune disorder in which the consumption of gluten, a protein found in wheat, barley, and rye, leads to damage in the small intestine. In people with celiac disease, their immune system reacts to gluten by attacking the lining of the small intestine, leading to inflammation and destruction of the villi - finger-like projections that help absorb nutrients from food.
This damage can result in various symptoms such as diarrhea, bloating, fatigue, anemia, and malnutrition. Over time, if left untreated, celiac disease can lead to serious health complications, including osteoporosis, infertility, neurological disorders, and even certain types of cancer.
The only treatment for celiac disease is a strict gluten-free diet, which involves avoiding all foods, beverages, and products that contain gluten. With proper management, individuals with celiac disease can lead healthy lives and prevent further intestinal damage and related health complications.
The jejunum is the middle section of the small intestine, located between the duodenum and the ileum. It is responsible for the majority of nutrient absorption that occurs in the small intestine, particularly carbohydrates, proteins, and some fats. The jejunum is characterized by its smooth muscle structure, which allows it to contract and mix food with digestive enzymes and absorb nutrients through its extensive network of finger-like projections called villi.
The jejunum is also lined with microvilli, which further increase the surface area available for absorption. Additionally, the jejunum contains numerous lymphatic vessels called lacteals, which help to absorb fats and fat-soluble vitamins into the bloodstream. Overall, the jejunum plays a critical role in the digestion and absorption of nutrients from food.
Intestinal absorption refers to the process by which the small intestine absorbs water, nutrients, and electrolytes from food into the bloodstream. This is a critical part of the digestive process, allowing the body to utilize the nutrients it needs and eliminate waste products. The inner wall of the small intestine contains tiny finger-like projections called villi, which increase the surface area for absorption. Nutrients are absorbed into the bloodstream through the walls of the capillaries in these villi, and then transported to other parts of the body for use or storage.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
Lactose intolerance is a digestive condition in which the body has difficulty digesting lactose, a sugar found in milk and dairy products. This occurs due to a deficiency or insufficiency of lactase, an enzyme produced by the small intestine that breaks down lactose into simpler sugars (glucose and galactose) for absorption. When there is not enough lactase to digest the consumed lactose, it passes undigested into the large intestine, where it is fermented by bacteria, leading to various gastrointestinal symptoms.
The symptoms of lactose intolerance may include bloating, cramps, diarrhea, nausea, and gas, usually occurring within 30 minutes to two hours after consuming dairy products. The severity of these symptoms can vary depending on the amount of lactose consumed and an individual's level of lactase deficiency or insufficiency.
Lactose intolerance is not life-threatening but can cause discomfort and may affect a person's quality of life. It is essential to manage the condition through dietary modifications, such as consuming smaller amounts of dairy products, choosing lactose-free or reduced-lactose options, or using lactase enzyme supplements before eating dairy products. In some cases, a healthcare professional may recommend additional management strategies based on an individual's specific needs and medical history.
 Pediatric Malabsorption Syndromes: Background, Pathophysiology, Epidemiology
Pediatric Malabsorption Syndromes: Background, Pathophysiology, Epidemiology Malabsorption (Syndrome): Symptoms, Causes & Treatment
Malabsorption (Syndrome): Symptoms, Causes & Treatment Malabsorption Syndromes - MSD Manual Professional Edition
Malabsorption Syndromes - MSD Manual Professional Edition Malabsorption Syndromes
Malabsorption Syndromes Malabsorption Syndrome, Runting/Stunting - El Sitio Avicola
Malabsorption Syndrome, Runting/Stunting - El Sitio Avicola Malabsorption - Wikipedia
Malabsorption - Wikipedia Upper GI and small bowel series: MedlinePlus Medical Encyclopedia
Upper GI and small bowel series: MedlinePlus Medical Encyclopedia The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study
The low FODMAP diet improves gastrointestinal symptoms in patients with irritable bowel syndrome: a prospective study Vitamin B1 (Thiamine) Deficiency - StatPearls - NCBI Bookshelf
Vitamin B1 (Thiamine) Deficiency - StatPearls - NCBI Bookshelf Treatment of Tuberculosis and Tuberculosis Infection in Adults and Children
Treatment of Tuberculosis and Tuberculosis Infection in Adults and Children Bone Mineral Density Testing | Osteoporosis Canada
Bone Mineral Density Testing | Osteoporosis Canada Pathological Anatomy (LZ-H) 2023/2024 - University of Bologna
Pathological Anatomy (LZ-H) 2023/2024 - University of Bologna Beagle | Breeds A to Z | The Kennel Club
Beagle | Breeds A to Z | The Kennel Club urofacial syndrome - Ontology Browser - Rat Genome Database
urofacial syndrome - Ontology Browser - Rat Genome Database Advanced Search Results - Public Health Image Library(PHIL)
Advanced Search Results - Public Health Image Library(PHIL) Birthing Center | Columbus Regional Health
Birthing Center | Columbus Regional Health Grierson-Gopalan Syndrome - Physiopedia
Grierson-Gopalan Syndrome - Physiopedia Weight Reduction Programs and Devices - Medical Clinical Policy Bulletins | Aetna
Weight Reduction Programs and Devices - Medical Clinical Policy Bulletins | Aetna