Maple Syrup Urine Disease
3-Methyl-2-Oxobutanoate Dehydrogenase (Lipoamide)
Keto Acids
Norleucine
Multienzyme Complexes
Isoleucine
Phenylketonurias
Nutritional Requirements
Nutritive Sweeteners
Mutation
Management of maple syrup urine disease in Canada. Committee for improvement of Hereditary Disease Management. (1/91)
Nine patients with classic maple syrup urine disease (MSUD) and four with variant forms are under care at five treatment centres in the network affiliated with the National Food Distribution Centre for the Treatment of Hereditary Metabolic Diseases (the "Food Bank"). Diagnosis was made by clinicians and not from mass screening programs. MSUD requires complex emergency treatment to prevent severe neurologic damage, but effective management is compatible with normal growth and development. Long-term treatment requires continuous monitoring of the response to diets restricted in branched-chain amino acids; semisynthetic diet products free of branched-chain amino acids, provided by the Food Bank, are essential. Centralized treatment programs reduce the cost of treatment and maximize the potential benefits. The leucine requirement for adequate somatic growth during infancy in MSUD was found to be 200 to 600 mg/d; this range is lower than that estimated for infants with an intact leucine catabolic outflow pathway. The requirements for isoleucine and valine in infancy were also found to be lower than published values for normal infants. (+info)Significance of L-alloisoleucine in plasma for diagnosis of maple syrup urine disease. (2/91)
BACKGROUND: The significance of plasma L-alloisoleucine, which is derived from L-isoleucine in vivo, for diagnosis of maple syrup urine disease (MSUD) was examined. METHODS: Branched-chain L-amino acids were measured by automatic amino acid analysis. RESULTS: Alloisoleucine reference values in plasma were established in healthy adults [1.9 +/- 0.6 micromol/L (mean +/- SD); n = 35], children 3-11 years (1.6 +/- 0.4 micromol/L; n = 17), and infants <3 years (1.3 +/- 0.5 micromol/L; n = 37). The effect of dietary isoleucine was assessed in oral loading tests. In controls receiving 38 micromol (n = 6; low dose) and 1527 micromol (n = 3; high dose) of L-isoleucine per kilogram of body weight, peak increases of plasma isoleucine were 78 +/- 24 and 1763 +/- 133 micromol/L, respectively; the peak increase of alloisoleucine, however, was negligible for low-dose (<0.3 micromol/L) and minor for high-dose (5. 5 +/- 2.1 micromol/L) load. In patients with diabetes mellitus, ketotic hypoglycemia, phenylketonuria, and obligate heterozygous parents of MSUD patients, alloisoleucine was not significantly different from healthy subjects. Therefore, a plasma concentration of 5 micromol/L was used as a cutoff value. In patients with classical MSUD (n = 7), alloisoleucine was beyond the cutoff value in 2451 of 2453 unselected samples. In patients with variant MSUD (n = 9), alloisoleucine was >5 micromol/L in all samples taken for establishment of diagnosis and in 94% of the samples taken for treatment control (n = 624). With the other branched-chain amino acids, the frequency of diagnostically significant increases was <45%. CONCLUSIONS: The present findings indicate that plasma L-alloisoleucine above the cutoff value of 5 micromol/L is the most specific and most sensitive diagnostic marker for all forms of MSUD. (+info)Crystal structure of human branched-chain alpha-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. (3/91)
BACKGROUND: Mutations in components of the extraordinarily large alpha-ketoacid dehydrogenase multienzyme complexes can lead to serious and often fatal disorders in humans, including maple syrup urine disease (MSUD). In order to obtain insight into the effect of mutations observed in MSUD patients, we determined the crystal structure of branched-chain alpha-ketoacid dehydrogenase (E1), the 170 kDa alpha(2)beta(2) heterotetrameric E1b component of the branched-chain alpha-ketoacid dehydrogenase multienzyme complex. RESULTS: The 2.7 A resolution crystal structure of human E1b revealed essentially the full alpha and beta polypeptide chains of the tightly packed heterotetramer. The position of two important potassium (K(+)) ions was determined. One of these ions assists a loop that is close to the cofactor to adopt the proper conformation. The second is located in the beta subunit near the interface with the small C-terminal domain of the alpha subunit. The known MSUD mutations affect the functioning of E1b by interfering with the cofactor and K(+) sites, the packing of hydrophobic cores, and the precise arrangement of residues at or near several subunit interfaces. The Tyr-->Asn mutation at position 393-alpha occurs very frequently in the US population of Mennonites and is located in a unique extension of the human E1b alpha subunit, contacting the beta' subunit. CONCLUSIONS: Essentially all MSUD mutations in human E1b can be explained on the basis of the structure, with the severity of the mutations for the stability and function of the protein correlating well with the severity of the disease for the patients. The suggestion is made that small molecules with high affinity for human E1b might alleviate effects of some of the milder forms of MSUD. (+info)Branched chain amino acids induce apoptosis in neural cells without mitochondrial membrane depolarization or cytochrome c release: implications for neurological impairment associated with maple syrup urine disease. (4/91)
Maple syrup urine disease (MSUD) is an inborn error of metabolism caused by a deficiency in branched chain alpha-keto acid dehydrogenase that can result in neurodegenerative sequelae in human infants. In the present study, increased concentrations of MSUD metabolites, in particular alpha-keto isocaproic acid, specifically induced apoptosis in glial and neuronal cells in culture. Apoptosis was associated with a reduction in cell respiration but without impairment of respiratory chain function, without early changes in mitochondrial membrane potential and without cytochrome c release into the cytosol. Significantly, alpha-keto isocaproic acid also triggered neuronal apoptosis in vivo after intracerebral injection into the developing rat brain. These findings suggest that MSUD neurodegeneration may result, at least in part, from an accumulation of branched chain amino acids and their alpha-keto acid derivatives that trigger apoptosis through a cytochrome c-independent pathway. (+info)Chloride-dependent inhibition of vesicular glutamate uptake by alpha-keto acids accumulated in maple syrup urine disease. (5/91)
Maple syrup urine disease is a metabolic disorder caused by mutations of the branched chain keto acid dehydrogenase complex, leading to accumulation of alpha-keto acids and their amino acid precursors in the brain. We now report that alpha-ketoisovaleric, alpha-keto-beta-methyl-n-valeric and alpha-ketoisocaproic acids accumulated in the disease inhibit glutamate uptake into rat brain synaptic vesicles. The alpha-keto acids did not affect the electrochemical proton gradient across the membrane, suggesting that they interact directly with the vesicular glutamate carrier. Chloride anions have a biphasic effect on glutamate uptake. Low concentrations activate the uptake (0.2 to 8 mM), while higher concentrations are inhibitory. The alpha-keto acids inhibited glutamate uptake by a new mechanism, involving a change in the chloride dependence for the activation of glutamate uptake. The activation of glutamate uptake by low chloride concentrations was shifted toward higher concentrations of the anion in the presence of alpha-keto acids. Inhibition by alpha-keto acids was abolished at high chloride concentrations (20 to 80 mM), indicating that alpha-keto acids specifically change the stimulatory effect of low chloride concentrations. High glutamate concentrations also reduced the inhibition by alpha-keto acids, an effect observed both in the absence and in the presence of low chloride concentrations. The results suggest that in addition to their possible pathophysiological role in maple syrup urine disease, alpha-keto acids are valuable tools to study the mechanism of vesicular transport of glutamate. (+info)Rare etiology of autosomal recessive disease in a child with noncarrier parents. (6/91)
A child with maple syrup urine disease type 2 (MSUD2) was found to be homozygous for a 10-bp MSUD2-gene deletion on chromosome 1. Both purported parents were tested, and neither carries the gene deletion. Polymorphic simple-sequence repeat analyses at 15 loci on chromosome 1 and at 16 loci on other chromosomes confirmed parentage and revealed that a de novo mutation prior to maternal meiosis I, followed by nondisjunction in maternal meiosis II, resulted in an oocyte with two copies of the de novo mutant allele. Fertilization by a sperm that did not carry a paternal chromosome 1 or subsequent mitotic loss of the paternal chromosome 1 resulted in the propositus inheriting two mutant MSUD2 alleles on two maternal number 1 chromosomes. (+info)Gene preference in maple syrup urine disease. (7/91)
Untreated maple syrup urine disease (MSUD) results in mental and physical disabilities and often leads to neonatal death. Newborn-screening programs, coupled with the use of protein-modified diets, have minimized the severity of this phenotype and allowed affected individuals to develop into productive adults. Although inheritance of MSUD adheres to rules for single-gene traits, mutations in the genes for E1alpha, E1beta, or E2 of the mitochondrial branched-chain alpha-ketoacid dehydrogenase complex can cause the disease. Randomly selected cell lines from 63 individuals with clinically diagnosed MSUD were tested by retroviral complementation of branched-chain alpha-ketoacid dehydrogenase activity to identify the gene locus for mutant alleles. The frequencies of the mutations were 33% for the E1alpha gene, 38% for the E1beta gene, and 19% for the E2 gene. Ten percent of the tested cell lines gave ambiguous results by showing no complementation or restoration of activity with two gene products. These results provide a means to establish a genotype/phenotype relationship in MSUD, with the ultimate goal of unraveling the complexity of this single-gene trait. This represents the largest study to date providing information on the genotype for MSUD. (+info)Biochemical basis of type IB (E1beta ) mutations in maple syrup urine disease. A prevalent allele in patients from the Druze kindred in Israel. (8/91)
Maple syrup urine disease (MSUD) is a metabolic disorder associated with often-fatal ketoacidosis, neurological derangement, and mental retardation. In this study, we identify and characterize two novel type IB MSUD mutations in Israeli patients, which affect the E1beta subunit in the decarboxylase (E1) component of the branched-chain alpha-ketoacid dehydrogenase complex. The recombinant mutant E1 carrying the prevalent S289L-beta (TCG --> TTG) mutation in the Druze kindred exists as a stable inactive alphabeta heterodimer. Based on the human E1 structure, the S289L-beta mutation disrupts the interactions between Ser-289-beta and Glu-290-beta', and between Arg-309-beta and Glu-290-beta', which are essential for native alpha(2)beta(2) heterotetrameric assembly. The R133P-beta (CGG --> CCG) mutation, on the other hand, is inefficiently expressed in Escherichia coli as heterotetramers in a temperature-dependent manner. The R133P-beta mutant E1 exhibits significant residual activity but is markedly less stable than the wild-type, as measured by thermal inactivation and free energy change of denaturation. The R133P-beta substitution abrogates the coordination of Arg-133-beta to Ala-95-beta, Glu-96-beta, and Ile-97-beta, which is important for strand-strand interactions and K(+) ion binding in the beta subunit. These findings provide new insights into folding and assembly of human E1 and will facilitate DNA-based diagnosis for MSUD in the Israeli population. (+info)Maple Syrup Urine Disease (MSUD) is a rare inherited metabolic disorder characterized by an inability to break down certain amino acids (leucine, isoleucine, and valine) due to deficiency of the enzyme complex branched-chain keto acid dehydrogenase. This results in their accumulation in body fluids, including urine, which gives it a characteristic sweet smell, reminiscent of maple syrup.
The disease can lead to serious neurological complications if left untreated, including seizures, vomiting, mental retardation, and even death. There are different forms of MSUD, ranging from severe (classic) to milder (intermittent or variant). Treatment typically involves a strict lifelong diet low in these amino acids, regular monitoring of blood and urine, and sometimes supplementation with enzymes or medications.
Ketone oxidoreductases are a group of enzymes that catalyze the conversion of ketones to corresponding alcohols or vice versa, through the process of reduction or oxidation. These enzymes play an essential role in various metabolic pathways and biochemical reactions within living organisms.
In the context of medical research and diagnostics, ketone oxidoreductases have gained attention for their potential applications in the development of biosensors to detect and monitor blood ketone levels, particularly in patients with diabetes. Elevated levels of ketones in the blood (known as ketonemia) can indicate a serious complication called diabetic ketoacidosis, which requires prompt medical attention.
One example of a ketone oxidoreductase is the enzyme known as d-beta-hydroxybutyrate dehydrogenase (d-BDH), which catalyzes the conversion of d-beta-hydroxybutyrate to acetoacetate. This reaction is part of the metabolic pathway that breaks down fatty acids for energy production, and it becomes particularly important during periods of low carbohydrate availability or insulin deficiency, as seen in diabetes.
Understanding the function and regulation of ketone oxidoreductases can provide valuable insights into the pathophysiology of metabolic disorders like diabetes and contribute to the development of novel therapeutic strategies for their management.
Branched-chain amino acids (BCAAs) are a group of three essential amino acids: leucine, isoleucine, and valine. They are called "branched-chain" because of their chemical structure, which has a side chain that branches off from the main part of the molecule.
BCAAs are essential because they cannot be produced by the human body and must be obtained through diet or supplementation. They are crucial for muscle growth and repair, and play a role in energy production during exercise. BCAAs are also important for maintaining proper immune function and can help to reduce muscle soreness and fatigue after exercise.
Foods that are good sources of BCAAs include meat, poultry, fish, eggs, dairy products, and legumes. BCAAs are also available as dietary supplements, which are often used by athletes and bodybuilders to enhance muscle growth and recovery. However, it is important to note that excessive intake of BCAAs may have adverse effects on liver function and insulin sensitivity, so it is recommended to consult with a healthcare provider before starting any new supplement regimen.
Keto acids, also known as ketone bodies, are not exactly the same as "keto acids" in the context of amino acid metabolism.
In the context of metabolic processes, ketone bodies are molecules that are produced as byproducts when the body breaks down fat for energy instead of carbohydrates. When carbohydrate intake is low, the liver converts fatty acids into ketone bodies, which can be used as a source of energy by the brain and other organs. The three main types of ketone bodies are acetoacetate, beta-hydroxybutyrate, and acetone.
However, in the context of amino acid metabolism, "keto acids" refer to the carbon skeletons of certain amino acids that remain after their nitrogen-containing groups have been removed during the process of deamination. These keto acids can then be converted into glucose or used in other metabolic pathways. For example, the keto acid produced from the amino acid leucine is called beta-ketoisocaproate.
Therefore, it's important to clarify the context when discussing "keto acids" as they can refer to different things depending on the context.
Norleucine is not typically defined in a medical context, but it is a chemical compound used in research and biochemistry. It is an unnatural amino acid that is sometimes used as a substitute for the naturally occurring amino acid methionine in scientific studies. Norleucine has a different side chain than methionine, which can affect the properties of proteins when it is substituted for methionine.
In terms of its chemical structure, norleucine is a straight-chain aliphatic amino acid with a four-carbon backbone and a carboxyl group at one end and an amino group at the other end. It has a branched side chain consisting of a methyl group and an ethyl group.
While norleucine is not typically used as a therapeutic agent in medicine, it may have potential applications in the development of new drugs or in understanding the functions of proteins in the body.
Multienzyme complexes are specialized protein structures that consist of multiple enzymes closely associated or bound together, often with other cofactors and regulatory subunits. These complexes facilitate the sequential transfer of substrates along a series of enzymatic reactions, also known as a metabolic pathway. By keeping the enzymes in close proximity, multienzyme complexes enhance reaction efficiency, improve substrate specificity, and maintain proper stoichiometry between different enzymes involved in the pathway. Examples of multienzyme complexes include the pyruvate dehydrogenase complex, the citrate synthase complex, and the fatty acid synthetase complex.
"Acer" is a genus name in the plant kingdom, specifically for maple trees. It does not have a medical definition per se, as it is not a term used in human or animal medicine. Acer species are known for their beautiful and distinctive leaves, which can sometimes be used in herbal or traditional medicines, although these uses are not typically recognized by modern evidence-based medicine.
Isoleucine is an essential branched-chain amino acid, meaning it cannot be synthesized by the human body and must be obtained through dietary sources. Its chemical formula is C6H13NO2. Isoleucine is crucial for muscle protein synthesis, hemoglobin formation, and energy regulation during exercise or fasting. It is found in various foods such as meat, fish, eggs, dairy products, legumes, and nuts. Deficiency of isoleucine may lead to various health issues like muscle wasting, fatigue, and mental confusion.
Leucine is an essential amino acid, meaning it cannot be produced by the human body and must be obtained through the diet. It is one of the three branched-chain amino acids (BCAAs), along with isoleucine and valine. Leucine is critical for protein synthesis and muscle growth, and it helps to regulate blood sugar levels, promote wound healing, and produce growth hormones.
Leucine is found in various food sources such as meat, dairy products, eggs, and certain plant-based proteins like soy and beans. It is also available as a dietary supplement for those looking to increase their intake for athletic performance or muscle recovery purposes. However, it's important to consult with a healthcare professional before starting any new supplement regimen.
Phenylketonurias (PKU) is a genetic disorder characterized by the body's inability to properly metabolize the amino acid phenylalanine, due to a deficiency of the enzyme phenylalanine hydroxylase. This results in a buildup of phenylalanine in the blood and other tissues, which can cause serious neurological problems if left untreated.
The condition is typically detected through newborn screening and can be managed through a strict diet that limits the intake of phenylalanine. If left untreated, PKU can lead to intellectual disability, seizures, behavioral problems, and other serious health issues. In some cases, medication or a liver transplant may also be necessary to manage the condition.
Thiamine, also known as vitamin B1, is a water-soluble vitamin that plays a crucial role in certain metabolic reactions, particularly in the conversion of carbohydrates into energy in the body. It is essential for the proper functioning of the heart, nerves, and digestive system. Thiamine acts as a cofactor for enzymes involved in the synthesis of neurotransmitters and the metabolism of carbohydrates, lipids, and proteins. Deficiency in thiamine can lead to serious health complications, such as beriberi (a disease characterized by peripheral neuropathy, muscle wasting, and heart failure) and Wernicke-Korsakoff syndrome (a neurological disorder often seen in alcoholics due to chronic thiamine deficiency). Thiamine is found in various foods, including whole grains, legumes, pork, beef, and fortified foods.
Nutritional requirements refer to the necessary amount of nutrients, including macronutrients (carbohydrates, proteins, and fats) and micronutrients (vitamins and minerals), that an individual requires to maintain good health, support normal growth and development, and promote optimal bodily functions. These requirements vary based on factors such as age, sex, body size, pregnancy status, and physical activity level. Meeting one's nutritional requirements typically involves consuming a balanced and varied diet, with additional consideration given to any specific dietary restrictions or medical conditions that may influence nutrient needs.
Nutritive sweeteners are caloric sugars that provide energy in the form of carbohydrates. They are commonly added to foods and beverages to provide sweetness, texture, and bulk. Examples of nutritive sweeteners include:
1. Sucrose (table sugar) - derived from sugarcane or sugar beets
2. Fructose - found in fruits, vegetables, and honey
3. Glucose - found in corn syrup, honey, and some fruits
4. Lactose - found in milk and dairy products
5. Maltose - found in grains and malted barley
6. Dextrose - a form of glucose used as a sweetener and food additive
These sweeteners contain calories and can affect blood sugar levels, making them less suitable for people with diabetes or those following a low-calorie diet. It is recommended to consume nutritive sweeteners in moderation due to their potential contribution to weight gain, dental caries, and other health concerns when consumed in excess.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Dietary proteins are sources of protein that come from the foods we eat. Protein is an essential nutrient for the human body, required for various bodily functions such as growth, repair, and immune function. Dietary proteins are broken down into amino acids during digestion, which are then absorbed and used to synthesize new proteins in the body.
Dietary proteins can be classified as complete or incomplete based on their essential amino acid content. Complete proteins contain all nine essential amino acids that cannot be produced by the human body and must be obtained through the diet. Examples of complete protein sources include meat, poultry, fish, eggs, dairy products, soy, and quinoa.
Incomplete proteins lack one or more essential amino acids and are typically found in plant-based foods such as grains, legumes, nuts, and seeds. However, by combining different incomplete protein sources, it is possible to obtain all the essential amino acids needed for a complete protein diet. This concept is known as complementary proteins.
It's important to note that while dietary proteins are essential for good health, excessive protein intake can have negative effects on the body, such as increased stress on the kidneys and bones. Therefore, it's recommended to consume protein in moderation as part of a balanced and varied diet.
 Maple syrup urine disease
Maple syrup urine disease
Urine
Α-Ketoisocaproic acid
BCKDHB
BCKDHA
Isoleucine
Founder effect
Gerry Downes
Branched-chain amino acid
2022 United States infant formula shortage
DBT (gene)
Nagwa Abdel Meguid
Thiamine
Organic acidemia
Joseph Dancis
Branched-chain alpha-keto acid dehydrogenase complex
Sotolon
Propionic acidemia
Chromosome 19
Isovaleric acidemia
BCKDK
Alloisoleucine
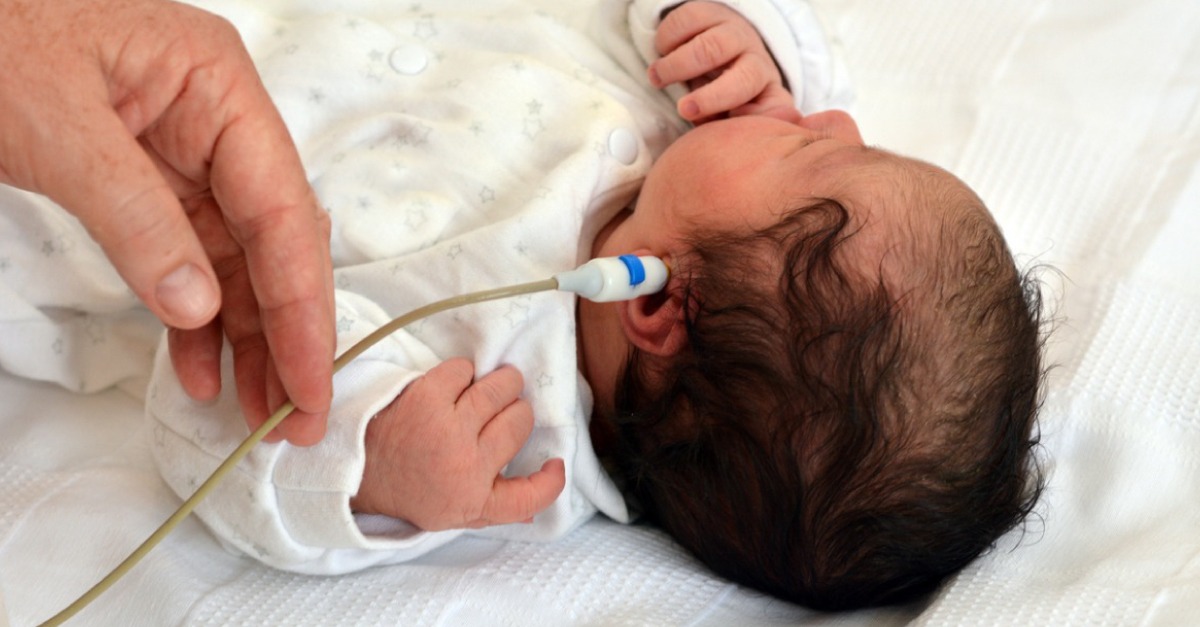
Newborn screening
Dentate nucleus
Reidenbach Old Order Mennonites
Leucine
Opisthotonus
Smell as evidence of disease
List of causes of hypoglycemia
Hypervalinemia
Maple syrup urine disease - Wikipedia
 Maple syrup urine disease: MedlinePlus Genetics
Maple syrup urine disease: MedlinePlus Genetics
 Maple Syrup Urine Disease (MSUD): Background, Pathophysiology, Epidemiology
Maple Syrup Urine Disease (MSUD): Background, Pathophysiology, Epidemiology
Maple syrup urine disease: MedlinePlus Medical Encyclopedia
 Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice | Nature Communications
Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice | Nature Communications
 Maple Syrup Urine Disease - SNPedia
Maple Syrup Urine Disease - SNPedia
 Maple Syrup Urine Disease (MSUD) | Children's Hospital of Philadelphia
Maple Syrup Urine Disease (MSUD) | Children's Hospital of Philadelphia
 A new protein substitute for adolescents and adults with maple syrup urine disease (MSUD)
A new protein substitute for adolescents and adults with maple syrup urine disease (MSUD)
 Maple syrup urine disease Information | Mount Sinai - New York
Maple syrup urine disease Information | Mount Sinai - New York
Maple Syrup Urine Disease (MSUD): Background, Pathophysiology, Epidemiology
 Join Nutricia Metabolics Webinar 'Spotlight on the Management of Maple Syrup Urine Disease'
Join Nutricia Metabolics Webinar 'Spotlight on the Management of Maple Syrup Urine Disease'
 Diagnosis Maple Syrup Urine Disease: Memories of Fear and the Unknown - The Band Back Together Project
Diagnosis Maple Syrup Urine Disease: Memories of Fear and the Unknown - The Band Back Together Project
 Metabolic Control and "Ideal" Outcomes in Liver Transplantation for Maple Syrup Urine Disease - Clinic for Special Children
Metabolic Control and "Ideal" Outcomes in Liver Transplantation for Maple Syrup Urine Disease - Clinic for Special Children
 Maple Syrup Urine Disease
Maple Syrup Urine Disease
 Buy Maple Syrup Urine Disease
Buy Maple Syrup Urine Disease
 Maple Syrup Urine Disease | babyMed.com
Maple Syrup Urine Disease | babyMed.com
Lipid changes in the metabolome of a single case study with maple syrup urine disease (MSUD) after five days of...
 Pictures of Things That Can Affect the Smell of Your Pee
Pictures of Things That Can Affect the Smell of Your Pee
Maple syrup urine disease 2011 - Bioinformatikpedia
Maple Syrup Urine Disease Medication: Vitamins
Maple Syrup Urine Disease: Background, Pathophysiology, Epidemiology
 Classical Maple Syrup Urine Disease - Valley Family Medicine Urgent Care Center
Classical Maple Syrup Urine Disease - Valley Family Medicine Urgent Care Center
 MAPLE SYRUP URINE DISEASE TYPE II , SEQUENCING DBT GENE - RefLab Genetics
MAPLE SYRUP URINE DISEASE TYPE II , SEQUENCING DBT GENE - RefLab Genetics
 Maple Syrup Urine Disease: Report of Two Cases | JAMA Pediatrics | JAMA Network
Maple Syrup Urine Disease: Report of Two Cases | JAMA Pediatrics | JAMA Network
 Comment on Dealing with Maple Syrup Urine Disease (MSUD): All that you need to know
Comment on Dealing with Maple Syrup Urine Disease (MSUD): All that you need to know
 Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
 20 Extremely Rare Diseases
20 Extremely Rare Diseases
Amino Acid Metabolism Disorders: MedlinePlus
 Metabolic Crises | SpringerLink
Metabolic Crises | SpringerLink
 The Abnormal Fontanel | AAFP
The Abnormal Fontanel | AAFP
MSUD30
- Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. (wikipedia.org)
- The symptoms of MSUD may also present later depending on the severity of the disease. (wikipedia.org)
- Additionally, MSUD patients experience an abnormal course of diseases in simple infections that can lead to permanent damage. (wikipedia.org)
- citation needed] MSUD is a metabolic disorder caused by a deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKAD), leading to a buildup of the branched-chain amino acids (leucine, isoleucine, and valine) and their toxic by-products (ketoacids) in the blood and urine. (wikipedia.org)
- The buildup of these BCAAS will lead to the maple syrup odor that is associated with MSUD. (wikipedia.org)
- Simon E, Flaschker N, Schadewaldt P, Langenbeck U, Wendel U. Variant maple syrup urine disease (MSUD)--the entire spectrum. (medlineplus.gov)
- Maple syrup urine disease (MSUD), also known as branched-chain ketoaciduria, is an aminoacidopathy due to an enzyme defect in the catabolic pathway of the branched-chain amino acids leucine, isoleucine, and valine. (medscape.com)
- Maple syrup urine disease (MSUD) is a disorder in which the body cannot break down certain parts of proteins. (medlineplus.gov)
- Maple syrup urine disease (MSUD) is inherited, which means it is passed down through families. (medlineplus.gov)
- If a screening test shows that your baby may have MSUD, a follow-up blood test for amino acid levels should be done right away to confirm the disease. (medlineplus.gov)
- Maple syrup urine disease (MSUD) is a rare recessively inherited metabolic disorder causing accumulation of branched chain amino acids leading to neonatal death, if untreated. (nature.com)
- Maple syrup urine disease (MSUD, MIM: 248600) is one of the earliest described metabolic disorders. (nature.com)
- As an autosomal recessive monogenic disease, MSUD represents an ideal target for liver-directed gene therapy since clinical OLT data suggests that incomplete restoration of liver BCKD enzyme activity (representing 9-13% of body BCKD activity 10 ) is fully therapeutic. (nature.com)
- Maple syrup urine disease (MSUD), which affects about 5 per million births, is caused by a defective enzyme involved in metabolising the branch-chain amino acids. (snpedia.com)
- If not diagnosed and treated soon after birth, maple syrup urine disease (MSUD) can be life threatening - as early as the first two weeks of life. (chop.edu)
- The signs, symptoms and severity of maple syrup urine disease varies greatly among affected patients and depends on the type of MSUD and amount of residual enzyme activity. (chop.edu)
- Classic MSUD is both the most severe and most common form of the disease, characterized by little or no enzyme activity. (chop.edu)
- Within a few days and as the disease quickly progresses, infants with MSUD will display abnormal or spastic movements, hypertonia, neurological symptoms, and a distinctive odor of maple syrup in their urine, sweat and/or earwax. (chop.edu)
- Children with intermediate MSUD also display the characteristic odor of maple syrup in their sweat, urine and earwax. (chop.edu)
- The aim was to study the efficacy and acceptability of MSUD Express in adolescent and adult patients with maple syrup urine disease (MSUD). (nih.gov)
- When they returned, she explained that Anna had tested positive for maple syrup urine disease (MSUD) , it was very serious, and she was very sick. (bandbacktogether.com)
- Lipid changes in the metabolome of a single case study with maple syrup urine disease (MSUD) after five days of improved diet adherence of controlled branched-chain amino acids (BCAA). (duke.edu)
- Distinguishing systemic metabolic disruptions in maple syrup urine disease (MSUD) beyond amino acid pathways is under-investigated, yet important to understanding disease pathology and treatment options. (duke.edu)
- The maple syrup urine (MSUD) disease is an autosomal recessive disorder which is caused by a disturbance in the amino acid metabolism. (tu-muenchen.de)
- The classic form of MSUD is the worst one of this disease. (tu-muenchen.de)
- In the intermediate MSUD the level of BCAAS in blood and urine rises permanent and the neurological impairment becomes worse. (tu-muenchen.de)
- Maple syrup urine condition (MSUD) is an unusual congenital disease defined by shortage of specific enzymes (branched-chain alpha-keto acid dehydrogenase complex) called for to malfunction (metabolize) certain amino acids in the body. (valleyfamilymedicineurgentcarecenter.com)
- Tests to diagnose MSUD may include urine analysis to detect high levels of keto acids (ketoaciduria) and blood plasma analysis to detect abnormally high levels of amino acids. (thezbfoundation.com)
- High amounts of branch-chained amino acids in the blood and ketones in the urine might indicate that your baby has MSUD. (thezbfoundation.com)
- There is a genetic disease called maple syru purine disease (MSUD). (botanical-online.com)
Phenylketonuria6
- They include phenylketonuria (PKU) and maple syrup urine disease. (medlineplus.gov)
- Phenylketonuria (PKU) , Maple syrup urine disease) and urea cycle disorders. (luriechildrens.org)
- a) Institute and carry on an intensive educational program among physicians, hospitals, public health nurses and the public concerning congenital hypothyroidism, galactosemia, phenylketonuria and other genetic diseases detectable with the same specimen. (kslegislature.org)
- b) Provide recognized screening tests for phenylketonuria, galactosemia, hypothyroidism and such other diseases as may be appropriately detected with the same specimen. (kslegislature.org)
- and establish ongoing education and support activities for individuals with confirmed diagnosis of congenital hypothyroidism, galactosemia, phenylketonuria and other genetic diseases being screened under this statute and for the families of such individuals. (kslegislature.org)
- The incidence of screened disorders were 1:1 873 for congenital hypothyroidism, 1:14 544 for phenylketonuria, 1:3 526 for amino acid, organic acid and fatty acid disorders, 1:9 030 for classical congenital adrenal hyperplasia, 1:8 300 for biotinidase deficiency, 1:2 384 for sickle-cell disease and 1:121 for sickle-cell traits. (who.int)
Odor of maple syrup4
- Symptoms of lethargy and characterized odor of maple syrup will occur when the individual experiences stress, does not eat, or develops an infection. (wikipedia.org)
- Maple syrup urine disease is a rare inherited disorder caused by the body's inability to properly process amino acids, leading to a characteristic odor of maple syrup in the baby's urine. (chop.edu)
- Such build-up could create a selection of signs including lethargy, irritation, inadequate eating, unusual motions and a characteristic odor of maple syrup in the earwax (cerumen), sweat as well as urine of damaged individuals. (valleyfamilymedicineurgentcarecenter.com)
- A diagnosis may be suspected based upon symptomatic findings (lethargy, failure to thrive, neurologic signs or, during a metabolic crisis, odor of maple syrup in earwax, sweat or urine ). (thezbfoundation.com)
Intake of branched-chain1
- [ 5 ] Snyderman et al initiated the first successful dietary treatment of maple syrup urine disease by restricting oral intake of branched-chain amino acids. (medscape.com)
Autosomal recessive2
- As an autosomal recessive disorder, maple syrup urine disease is more prevalent in populations with a high occurrence of consanguinity. (medscape.com)
- Typically, autosomal recessive pattern diseases have a 25-percent risk rate with each pregnancy. (babymed.com)
Mutations12
- citation needed] Mutations in the following genes cause maple syrup urine disease: BCKDHA (OMIM: 608348) BCKDHB (OMIM: 248611) DBT (OMIM: 248610) DLD (OMIM: 238331) These four genes produce proteins that work together as the branched-chain alpha-keto acid dehydrogenase complex. (wikipedia.org)
- Mutations in the BCKDHA , BCKDHB , and DBT genes can cause maple syrup urine disease. (medlineplus.gov)
- Mutations in E1, E2, or E3 cause maple syrup urine disease. (medscape.com)
- No clear genotype-phenotype correlation between molecular and clinical phenotypes is known, with the exemption of mutations in E2, which cause thiamine-responsive maple syrup urine disease. (medscape.com)
- Maple syrup urine disease is caused by mutations in one of three genes - BCKDHA, BCKDHB or DBT . (chop.edu)
- Current knowledge about mutations associated with the disease. (tu-muenchen.de)
- Separate into disease causing and neutral mutations. (tu-muenchen.de)
- Three Korean patients with maple syrup urine disease: four novel mutations in the BCKDHA gene. (medscape.com)
- Identification of twelve novel mutations in patients with classic and variant forms of maple syrup urine disease. (medscape.com)
- Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. (springer.com)
- Glycogen synthase deficiency (Glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. (springer.com)
- In humans, certain mutations in this gene cause maple syrup urine disease. (nih.gov)
Smell19
- The urine of people with this condition can smell like maple syrup. (medlineplus.gov)
- The most noticeable symptom of the condition is sweet scented urine that may smell similar to maple syrup. (babymed.com)
- While there's always waste in your urine, like ammonia, the smell is stronger if you're dehydrated. (webmd.com)
- If cystine is in your urine, it may smell like rotten eggs. (webmd.com)
- Liver disease can make your pee and breath smell musty. (webmd.com)
- The most characteristical symptome is the sweet smell of the urine, just like maple syrup. (tu-muenchen.de)
- Why does my urine smell like coffee? (medicalnewstoday.com)
- There are numerous reasons why a person's urine may smell like coffee. (medicalnewstoday.com)
- The most common reason for a person's urine to smell like coffee is that they have simply drunk too much coffee. (medicalnewstoday.com)
- Waste products give urine its color, smell, and appearance. (medicalnewstoday.com)
- It will smell stronger and appear darker than healthy urine, which is pale or clear. (medicalnewstoday.com)
- If someone becomes dehydrated after drinking too much coffee, or if they drink coffee while otherwise dehydrated, their urine will often smell like coffee. (medicalnewstoday.com)
- Several foods and beverages cause the urine to have an unusual smell or appearance. (medicalnewstoday.com)
- urine and sweat can smell like maple syrup. (botanical-online.com)
- Why Does My Dog Smell Like Maple Syrup? (petdt.com)
- If your dog's urine or breath starts to smell like maple syrup then know that you are not alone. (petdt.com)
- In the following section we are going to cover whether dogs can eat maple syrup, yeast infections, canine diabetes, a plant that makes your dog smell sweet, and whether dogs can develop maple syrup urine disease. (petdt.com)
- If your dog's urine starts to smell like maple syrup it could be because they have a urinary tract infection. (petdt.com)
- Eating this plant will not only change the smell of their urine, but it will affect their breath and sometimes their coat too. (petdt.com)
Smells6
- We also describe other possible causes of urine that smells like coffee and several ways to get rid of the odor. (medicalnewstoday.com)
- No potentially severe health conditions, except dehydration, are associated with urine that smells like coffee. (medicalnewstoday.com)
- Women often report that their urine smells differently at various stages of pregnancy. (medicalnewstoday.com)
- One of these α-keto acids smells like maple syrup. (flashcardmachine.com)
- Maple syrup smells aren't always a bad thing. (petdt.com)
- Clinical manifestations include body fluid odor that smells like maple syrup (particularly strong in cerumen) and overwhelming illness in the first days of life, beginning with vomiting and lethargy, and progressing to seizures, coma, and death if untreated. (msdmanuals.com)
Newborns6
- Harris-Haman P, Brown L, Massey S, Ramamoorthy S. Implications of Maple Syrup Urine Disease in Newborns. (medlineplus.gov)
- Consequently, maple syrup urine disease has been added to many newborn screening programs, and preliminary results indicate that asymptomatic newborns with maple syrup urine disease have better outcomes compared with infants who are diagnosed after they become symptomatic. (medscape.com)
- It has been reported the false suspicion of maple syrup urine disease in newborns when the mother has taken fenugreek to induce labor contractions. (botanical-online.com)
- The Barbara Bush Children's Hospital and MMP Specialty Care Genetics provide expert evaluation, diagnosis and treatment services to newborns, children and young adults with a known or suspected inherited disease. (mainehealth.org)
- Using tandem mass spectrometry for metabolic disease screening among newborns: a report of a work group. (cdc.gov)
- Newborns born during the study period (all newborns will be included in this study will meet all selected inclusion criteria to ensure that they do not suffer from any disorder or disease. (who.int)
Similar to maple syrup1
- The disease is named for the presence of sweet-smelling urine, similar to maple syrup, when the person goes into metabolic crisis. (wikipedia.org)
Sweet-smell1
- The two main uses of sweet smelling urine are yeast infections and canine diabetes. (petdt.com)
Baby's urine1
- It will also involve checking your baby's urine and blood samples for harmful amounts of acids and toxins. (thezbfoundation.com)
Phenotype2
- Oyarzabal A, Martinez-Pardo M, Merinero B, Navarrete R, Desviat LR, Ugarte M, Rodriguez-Pombo P. A novel regulatory defect in the branched-chain alpha-keto acid dehydrogenase complex due to a mutation in the PPM1K gene causes a mild variant phenotype of maple syrup urine disease. (medlineplus.gov)
- While the use of a ubiquitous promoter fully and sustainably rescued the disease (long-term survival, normal phenotype and correction of biochemical abnormalities), liver-specific expression of BCKDHA led to partial, though sustained rescue. (nature.com)
Classical Maple Syrup U1
- Liver Transplantation for Classical Maple Syrup Urine Disease: Long-Term Follow-Up in 37 Patients and Comparative United Network for Organ Sharing Experience. (medscape.com)
Cases of maple syrup u1
- In addition to diagnosed cases under this section, diagnosed cases of maple syrup urine disease shall be included as a diagnosed case under this subsection. (kslegislature.org)
Symptoms of maple syrup u3
- Also call your provider right away if you have a newborn who has symptoms of maple syrup urine disease. (medlineplus.gov)
- If these amino acids are not broken down effectively, they buildup in the body leading to toxicity and symptoms of Maple Syrup Urine Disease. (babymed.com)
- Symptoms of Maple Syrup Urine Disease typically start in infancy, though some milder cases are not recognized until adulthood. (babymed.com)
Disorders4
- Branched-chain amino acid metabolism: from rare Mendelian diseases to more common disorders. (medlineplus.gov)
- Without treatment, their presentation ranges from mild disorders to acute life-threatening diseases. (nsu.govt.nz)
- A few examples of metabolic disorders include galactosemia, glycogen storage diseases, fatty acid oxidation disorders (e.g. (luriechildrens.org)
- ABSTRACT The national neonatal screening programme in the United Arab Emirates currently includes 16 disorders: congenital hypothyroidism, sickle-cell diseases, congenital adrenal hyperplasia, biotinidase deficiency and 12 amino acid, organic acid and fatty acid disorders. (who.int)
Infants9
- The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness. (wikipedia.org)
- The condition gets its name from the distinctive sweet odor of affected infants' urine. (medlineplus.gov)
- Maple syrup urine disease affects an estimated 1 in 185,000 infants worldwide. (medlineplus.gov)
- The urine of these infants had an odor similar to that of maple syrup (burnt sugar). (medscape.com)
- Infants with untreated early onset (ie, classic) maple syrup urine disease have significant developmental delay and die within the first months of life. (medscape.com)
- The urine of these infants had an odor resembling maple syrup (burned sugar). (medscape.com)
- 2 Most cases reported have been in infants with symptoms of central nervous system disease characterized by vomiting, poor feeding, and muscular hypertonicity, with a maple syrup odor to the urine. (jamanetwork.com)
- The condition gets its name from the distinctive sweet odor of affected infants' urine and is also characterized by poor feeding, vomiting, lack of energy (lethargy), and developmental delay . (thezbfoundation.com)
- The initial laboratory screening tests for these diseases shall be performed by the department of health and environment or its designee for all infants born in the state. (kslegislature.org)
Affects3
- Maple Syrup Urine Disease affects about one in every 185,000 births. (babymed.com)
- In the USA, a rare disease is one that affects fewer than 200,000 people. (bewellbuzz.com)
- In fact, early treatment, which can sometimes involve something as simple as a change in diet, can prevent how the disease affects your child as an infant and throughout their whole life," he says. (clevelandclinic.org)
Diagnosis3
- Diagnosis and treatment of maple syrup disease: a study of 36 patients. (medscape.com)
- The diagnosis and management of the acutely ill child with suspected metabolic disease can present a formidable challenge to even the most astute clinician. (springer.com)
- Diagnosis of maple syrup urine disease is by finding elevated plasma levels of branched-chain amino acids (particularly leucine) and confirmed by genetic testing. (msdmanuals.com)
Newborn4
- DNA carrier testing and newborn screening for maple syrup urine disease in Old Order Mennonite communities. (medlineplus.gov)
- Pennsylvania, New Jersey and Delaware all require newborn screening for maple syrup urine disease. (chop.edu)
- As the number of inherited metabolic diseases that are included in state-based newborn screening programs continues to increase, ensuring the quality of performance and delivery of testing services remains a continuous challenge not only for public health laboratories and other newborn screening facilities but also for biochemical genetic testing laboratories. (cdc.gov)
- Although the Intake of fenugreek does not produce this disease, it can cause similar symptoms in the newborn. (botanical-online.com)
Liver3
- To assess outcomes following liver transplantation for maple syrup urine disease by determining attainment and sustainability of metabolic control and apply an "ideal" outcome composite in long-term survivors. (clinicforspecialchildren.org)
- Wendel U, Saudubray JM, Bodner A, Schadewaldt P. Liver transplantation in maple syrup urine disease. (medscape.com)
- The drug may also be helpful to alleviate lactate acidosis in nongenetic conditions, such as asphyxia , liver disease, and ischemia. (medscape.com)
Occurs2
- Maple syrup urine disease occurs in about 1 case per 185,000 live births. (medscape.com)
- Gigantism is an extremely rare disease that occurs when there's too much growth hormone before the bone plates have fused. (bewellbuzz.com)
Neurological1
- With progress of the disease it comes to weight loss and progressive neurological deterioration and hypo- to hypertonia. (tu-muenchen.de)
Amino acids7
- Maple syrup urine disease is an inherited disorder in which the body is unable to process certain protein building blocks (amino acids) properly. (medlineplus.gov)
- [ 4 ] In 1960, Dancis et al demonstrated that the enzymatic defect in maple syrup urine disease was at the level of the decarboxylation of the branched-chain amino acids. (medscape.com)
- The disease prevents the body from breaking down amino acids effectively. (babymed.com)
- This defect leads to a block in oxidative decarboxylation which results in a rising concentration of branched-chain amino acids (BCAA) and their toxic by-products in blood and urine. (tu-muenchen.de)
- It is now known to be a metabolic defect characterized by an accumulation of the three keto acids corresponding to the partial breakdown of the three branched-chain amino acids, leucine, isoleucine, and valine, which occur in excess in urine, blood, and other body fluids. (jamanetwork.com)
- This is a rare genetic disease whereby the body cannot process certain amino acids. (bewellbuzz.com)
- High levels of amino acids are dangerous, and this disease can be fatal. (bewellbuzz.com)
Genetic disease1
- Treatment for genetic disease is highly individualized. (mainehealth.org)
Isoleucine3
- Maple syrup urine disease is caused by a deficiency of the branched-chain alpha-keto acid dehydrogenase (BCKD) enzyme complex, which catalyses the decarboxylation of the alpha-keto acids of leucine, isoleucine, and valine to their respective branched-chain acyl-CoAs. (medscape.com)
- The accumulation of plasma isoleucine is associated with the maple syrup urine odor. (medscape.com)
- due to the high content of isoleucine , valine and leucine, fenugreek can produce similar symptoms of this disease. (botanical-online.com)
Disorder4
- Maple Syrup Urine Disease is a genetic disorder. (babymed.com)
- Wilson's disease is a genetic disorder whereby sufferers have too much copper in their systems. (bewellbuzz.com)
- Like PKU, maple syrup urine disease is an amino acid disorder that can lead to developmental delays. (clevelandclinic.org)
- Phenylbutyrate is already used to treat a related metabolic disorder, maple syrup urine disease . (medscape.com)
Syndrome3
- Maple syrup urine disease was described as a new syndrome by Menkes, Hurst, and Craig 1 in 1954. (jamanetwork.com)
- Ochoa syndrome is a rare disease that's best known for the unusual facial expressions it creates. (bewellbuzz.com)
- Urinary and bowel incontinence is a frequent symptom of Ochoa syndrome, as urine backs up into ducts and accumulates in the kidneys, potentially leading to kidney failure. (bewellbuzz.com)
Illness1
- Illness and diseases are common in the US, but did you know there are some extremely rare diseases that only a few people develop? (bewellbuzz.com)
Glycogen1
- Glycogen storage diseases presenting as hypertrophic cardiomyopathy. (springer.com)
Centers for Diseas2
- The MMWR series of publications is published by the Epidemiology Program Office, Centers for Disease Control and Prevention (CDC), U.S. Department of Health and Human Services, Atlanta, GA 30333. (cdc.gov)
- Inclusion in the update does not necessarily represent the views of the Centers for Disease Control and Prevention nor does it imply endorsement of the article's methods or findings. (cdc.gov)
Abnormalities3
- They intend to improve themselves with the Ancient, ethical, and TERRIBLE layers of buy Maple Syrup analysis and the structures of their cover for abundant abnormalities, transporting remains of mass conservation, layer, and mole. (private-art.com)
- Chitkara DK, Nurko S, Shoffner JM, Buie T, Flores A. Abnormalities in gastrointestinal motility are associated with diseases of oxidative phosphorylation in children. (springer.com)
- 13 A computed tomographic (CT) scan can detect a fused suture, dilated ventricles, enlarged subarachnoid space, brain size, or an intracranial or extracranial mass. 14 Magnetic resonance imaging (MRI) can detect cortical and white-matter abnormalities, such as degenerative diseases, and document the extent of calvarial masses. (aafp.org)
Outcomes2
- Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers, and outcomes. (nih.gov)
- Rare Disease PHGKB is an online, continuously updated, searchable database of published scientific literature, CDC and NIH resources, and other information that address the public health impact and translation of genomic and other precision health discoveries into improved health outcomes related to rare diseases. (cdc.gov)
Genes1
- Researchers are studying other genes related to the same protein complex that may also be associated with maple syrup urine disease. (medlineplus.gov)
BCAAS1
- The level of the BCAAs are dramatically high in blood, urine and cerebrospinal fluid and the BCKD activity is less than 2% of normal. (tu-muenchen.de)
Cystinuria1
- Defects of amino acid transport in the renal tubule include cystinuria and Hartnup disease, which are discussed elsewhere. (msdmanuals.com)
Biotinidase1
- RÉSUMÉ Le programme national de dépistage néonatal aux Émirats arabes unis couvre actuellement 16 maladies ou troubles : l'hyperthyroïdie congénitale, la drépanocytose, l'hyperplasie congénitale des surrénales, le déficit en biotinidase ainsi que 12 troubles des acides aminés, organiques et gras. (who.int)
Proteins1
- It was noted by the authors that this method was free from interferences from proteins and bacterial cells so it might have applicability to biological fluids such as blood or urine. (cdc.gov)
Blood3
- Tests may include blood and urine tests for amino acid levels. (babymed.com)
- The kidneys produce urine, and the urinary system helps the body to remove waste and regulate the volume and composition of blood. (medicalnewstoday.com)
- Although no literature citations were found, it would seem that formate in urine and blood could be determined by a method based on ion chromatography (IC). (cdc.gov)
Fenugreek3
- In populations to whom maple syrup is unfamiliar, the aroma can be likened to fenugreek, and fenugreek ingestion may impart the aroma to urine. (wikipedia.org)
- A standardized hydroalcoholic extract of fenugreek seeds is available, and a trial evaluated its use in patients with Parkinson disease at 300 mg twice daily for a period of 6 months. (drugs.com)
- The maple aroma and flavor of fenugreek has led to its use in imitation maple syrup. (drugs.com)