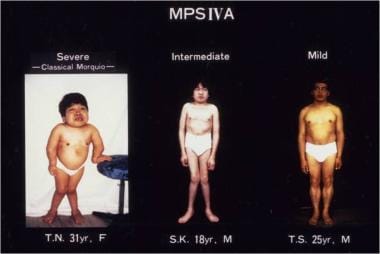
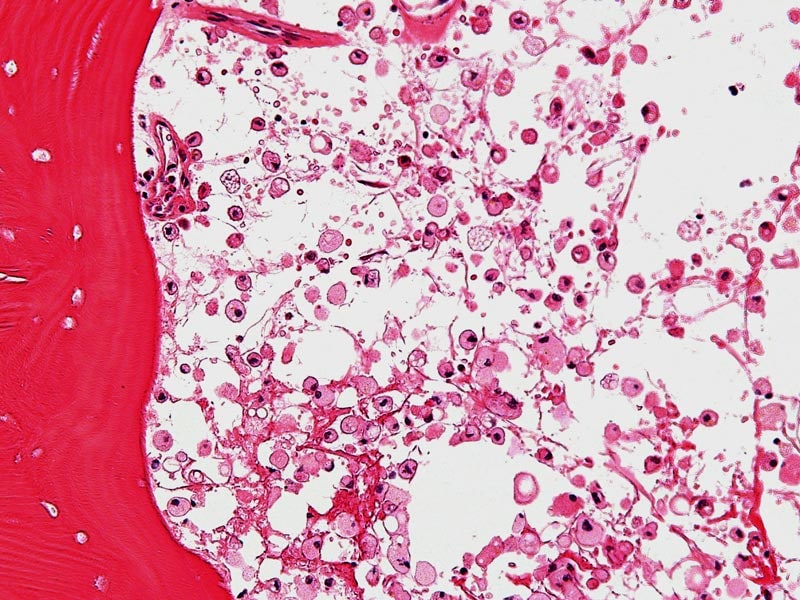
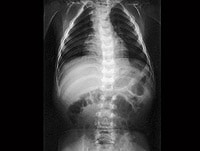
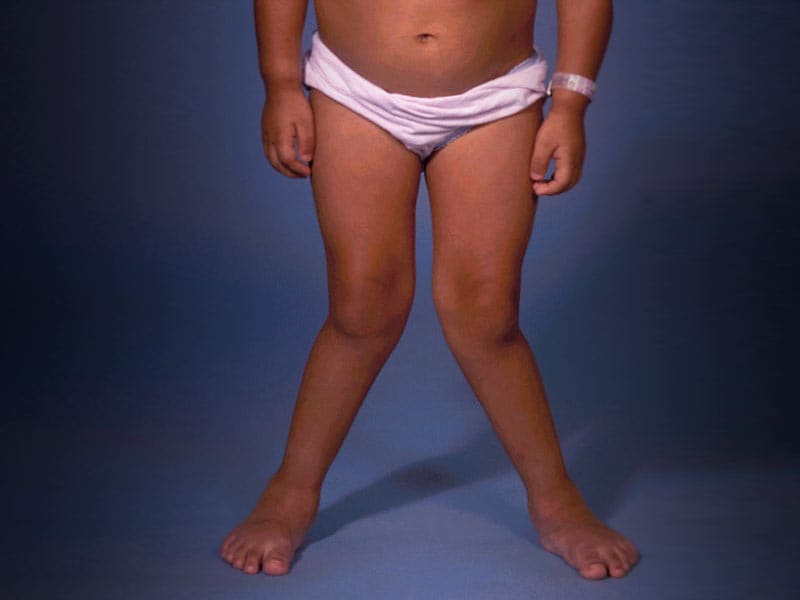
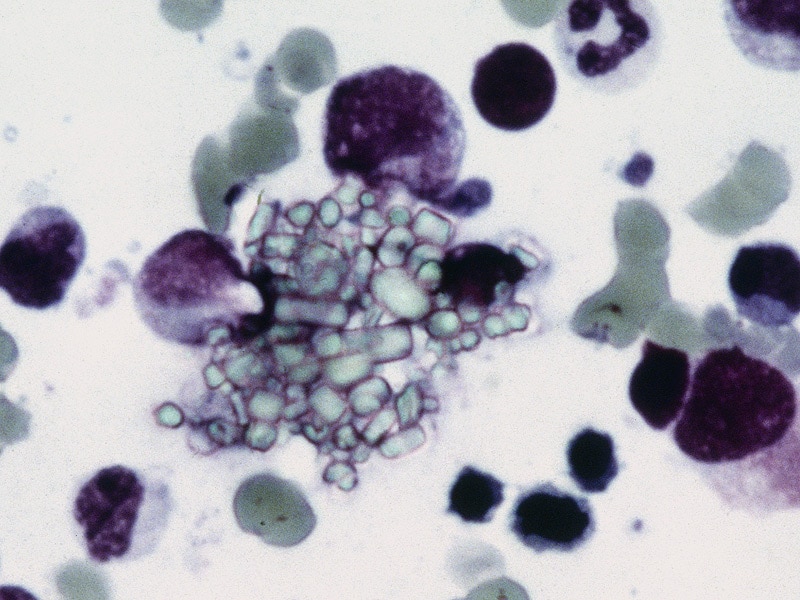
Mucolipidoses
Mucolipidosis type IV: the origin of the disease in the Ashkenazi Jewish population. (1/185)
Mucolipidosis type IV (MLIV) is a neurodegenerative lysosomal storage disease in which most of the patients diagnosed hitherto are Ashkenazi Jews. The basic metabolic defect causing this disease is still unknown and the relevant gene has not yet been mapped or cloned. Seventeen Israel Ashkenazi families with MLIV patients had been interviewed to study their family origin. Although the families immigrated to Israel from various European countries they all could trace their roots three to four generations back to northern Poland or the immediate neighbouring country, Lithuania. Furthermore, there are only one or two ultraorthodox families among the 70-80 Ashkenazi families with MLIV patients worldwide, a marked under-representation of this group which constitutes at least 10% of the Ashkenazi population. This data indicate that MLIV mutation occurred only around the 18th and 19th centuries, after the major expansion of this population, in a founder in this defined European region belonging to a more modern, secular family. (+info)Mucolipidosis IV consists of one complementation group. (2/185)
Mucolipidosis IV (MLIV) is an autosomal recessive disorder of unknown etiology characterized by severe visual impairment and psychomotor retardation. Recently, there has been considerable interest in positional cloning of the MLIV gene. It is unknown whether MLIV is a genetically homogenous disorder. In this paper, we present experiments that determined whether the MLIV phenotype in fibroblasts could be corrected by fusing normal cells to MLIV cells and fusing fibroblasts from pairs of patients. All of our MLIV patients fulfilled several diagnostic criteria that we developed. In addition, we found high sensitivity to chloroquine in cultured fibroblasts from MLIV patients. We found that normal cells corrected the MLIV phenotype. Fusion products of normal and MLIV fibroblasts, but not MLIV fibroblasts themselves, were relatively protected against chloroquine selection. In addition, 74% of the normal-to-patient fusion products had reduced levels or total loss of MLIV characteristic autofluorescence. However, there was no complementation of the phenotype in fibroblast cultures from any of our MLIV patients, including those of non-Jewish ancestry. In fusion products of MLIV cultures from 24 patients, 90-100% of the cells remained autofluorescent. These results indicate that all of our known MLIV patients, regardless of ancestry or severity of the developmental defect, have a single mutated gene. (+info)Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. (3/185)
Mucolipidosis type IV (MLIV) is a lysosomal storage disorder characterized by severe neurologic and ophthalmologic abnormalities. It is a rare autosomal recessive disease, and the majority of patients diagnosed, to date, are of Ashkenazi Jewish descent. We have mapped the MLIV gene to chromosome 19p13.2-13.3 by linkage analysis with 15 markers in 13 families. A maximum LOD score of 5.51 with no recombinants was observed with marker D19S873. Several markers in the linked interval also displayed significant linkage disequilibrium with the disorder. We constructed haplotypes in 26 Ashkenazi Jewish families and demonstrate the existence of two founder chromosomes in this population. The localization of MLIV to chromosome 19 will permit genetic prenatal diagnosis in affected families and will aid in the isolation of the disease gene. (+info)Orthopaedic management in four cases of mucolipidosis type III. (4/185)
Four patients with mucolipidosis type III, three of them brothers, were seen initially in the first two decades of life. Their main symptoms were carpal tunnel syndrome, trigger fingers and generalized joint stiffness. Radiographs showed spinal deformities and hip dysplasia, but these were not causing pain. Carpal tunnel syndrome was treated surgically but joint stiffness and hip and knee contractures were managed by physiotherapy. Up to the age of 24 none of these patients has had pelvic osteotomy for hip dysplasia; this operation, not yet reported in mucolipidosis type III, may eventually be necessary. (+info)Molecular basis of GM1 gangliosidosis and Morquio disease, type B. Structure-function studies of lysosomal beta-galactosidase and the non-lysosomal beta-galactosidase-like protein. (5/185)
GM1 gangliosidosis and Morquio B disease are distinct disorders both clinically and biochemically yet they arise from the same beta-galactosidase enzyme deficiency. On the other hand, galactosialidosis and sialidosis share common clinical and biochemical features, yet they arise from two separate enzyme deficiencies, namely, protective protein/cathepsin A and neuraminidase, respectively. However distinct, in practice these disorders overlap both clinically and biochemically so that easy discrimination between them is sometimes difficult. The principle reason for this may be found in the fact that these three enzymes form a unique complex in lysosomes that is required for their stability and posttranslational processing. In this review, I focus mainly on the primary and secondary beta-galactosidase deficiency states and offer some hypotheses to account for differences between GM1 gangliosidosis and Morquio B disease. (+info)Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC) (6/185)
Mucolipidosis IIIC, or variant pseudo-Hurler polydystrophy, is an autosomal recessive disease of lysosomal hydrolase trafficking. Unlike the related diseases, mucolipidosis II and IIIA, the enzyme affected in mucolipidosis IIIC (N-Acetylglucosamine-1-phosphotransferase [GlcNAc-phosphotransferase]) retains full transferase activity on synthetic substrates but lacks activity on lysosomal hydrolases. Bovine GlcNAc-phosphotransferase has recently been isolated as a multisubunit enzyme with the subunit structure alpha(2)beta(2)gamma(2). We cloned the cDNA for the human gamma-subunit and localized its gene to chromosome 16p. We also showed, in a large multiplex Druze family that exhibits this disorder, that MLIIIC also maps to this chromosomal region. Sequence analysis of the gamma-subunit cDNA in patients from 3 families identified a frameshift mutation, in codon 167 of the gamma subunit, that segregated with the disease, indicating MLIIIC results from mutations in the phosphotransferase gamma-subunit gene. This is to our knowledge the first description of the molecular basis for a human mucolipidosis and suggests that the gamma subunit functions in lysosomal hydrolase recognition. (+info)Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. (7/185)
Sialidosis is an autosomal recessive disease caused by the genetic deficiency of lysosomal sialidase, which catalyzes the hydrolysis of sialoglycoconjugates. The disease is associated with progressive impaired vision, macular cherry-red spots and myoclonus (sialidosis type I) or with skeletal dysplasia, Hurler-like phenotype, dysostosis multiplex, mental retardation and hepatosplenomegaly (sialidosis type II). We have analyzed the genomic DNA from nine sialidosis patients of multiple ethnic origin in order to find mutations responsible for the enzyme deficiency. The activity of the identified variants was studied by transgenic expression. One patient had a frameshift mutation (G623delG deletion), which introduced a stop codon, truncating 113 amino acids. All others had missense mutations: G679G-->A (Gly227Arg), C893C-->T (Ala298Val), G203G-->T (Gly68Val), A544A-->G (Ser182Gly) C808C-->T (Leu270Phe) and G982G-->A (Gly328Ser). We have modeled the three-dimensional structure of sialidase based on the atomic coordinates of the homologous bacterial sialidases, located the positions of mutations and estimated their potential effect. This analysis showed that five mutations are clustered in one region on the surface of the sialidase molecule. These mutations dramatically reduce the enzyme activity and cause a rapid intralysosomal degradation of the expressed protein. We hypothesize that this region may be involved in the interface of sialidase binding with lysosomal cathepsin A and/or beta-galactosidase in their high-molecular-weight complex required for the expression of sialidase activity in the lysosome. (+info)Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. (8/185)
Mucolipidosis type IV (MLIV) is an autosomal recessive lysosomal storage disorder characterized by severe psychomotor retardation and ophthalmologic abnormalities, including corneal opacity, retinal degeneration, and strabismus. Unlike the situation in other lysosomal disorders, the accumulation of heterogeneous storage material observed in MLIV does not result from a block in the catabolic pathways but is due to an ill-defined transport defect in the late steps of endocytosis. With the aim of cloning the MLIV gene, we searched in the 19p13.2-13.3 region, where the locus previously had been assigned by linkage mapping. In this region, we have identified a novel gene that is mutated in all patients with MLIV who were enrolled in our study. One patient was homozygous for the splice-acceptor mutation, and another was homozygous for a deletion removing the first six exons of the gene. In addition, four compound heterozygotes for these two mutations were identified. Haplotype analysis indicates that we have identified the two major founder mutations, which account for >95% of MLIV chromosomes in Ashkenazi Jewish patients. The gene, ML4, encodes a protein named "mucolipidin, " which localizes on the plasma membrane and, in the carboxy-terminal region, shows homologies to polycystin-2, the product of the polycystic kidney disease 2 gene (PKD2) and to the family of transient receptor potential Ca(2+) channels. Mucolipidin is likely to play an important role in endocytosis. (+info)Mucolipidoses are a group of inherited metabolic disorders characterized by the accumulation of complex carbohydrates (muco-) and fatty substances (lipids) in various tissues and cells (-oses). This is due to deficiency in enzymes that help break down these substances within lysosomes, which are organelles responsible for recycling and breaking down waste materials inside the cell.
There are four main types of mucolipidoses (I, II, III, and IV), each resulting from specific genetic mutations affecting different enzymes or proteins involved in the lysosomal degradation pathway. The symptoms, severity, and age of onset can vary widely among these types, ranging from mild to severe and including developmental delays, bone abnormalities, vision and hearing loss, heart problems, and coarse facial features.
Mucolipidoses are typically inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition. However, mucolipidosis II is caused by X-linked inheritance, where a single copy of the mutated gene on the X chromosome is enough to cause the disorder.
Early diagnosis and management of mucolipidoses can help improve quality of life and slow disease progression. Treatment options include physical therapy, occupational therapy, speech therapy, medications for symptom management, and in some cases, enzyme replacement therapy or bone marrow transplantation.
 Mucolipidosis
Mucolipidosis
Mucolipidosis type IV
I-cell
Medical genetics of Jews
List of OMIM disorder codes
Sialidosis
Gastrin
List of conditions with craniosynostosis
Naomi Amir
Achlorhydria
MCOLN1
Mucopolysaccharidosis
Pacman dysplasia
Pseudo-Hurler polydystrophy
MCOLN3
Transient receptor potential channel
I-cell disease
Nicotinic acid adenine dinucleotide phosphate
Cherry-red spot
Ion channel
List of MeSH codes (C05)
List of MeSH codes (C10)
Lysosomal storage disease
List of MeSH codes (C18)
List of MeSH codes (C16)
Megalocornea
Nuclear mitochondrial DNA segment
N-acetylglucosamine-1-phosphate transferase
List of diseases (M)
History of eugenics
Mucolipidosis - Wikipedia
 Mucolipidosis III alpha/beta: MedlinePlus Genetics
Mucolipidosis III alpha/beta: MedlinePlus Genetics
 Cure Mucolipidosis - National Organization for Rare Disorders
Cure Mucolipidosis - National Organization for Rare Disorders
 Mucolipidosis I | Boston Children's Hospital
Mucolipidosis I | Boston Children's Hospital
 Inclusion-Cell (I-Cell) Disease (Mucolipidosis Type II): Background, Pathophysiology, Epidemiology
Inclusion-Cell (I-Cell) Disease (Mucolipidosis Type II): Background, Pathophysiology, Epidemiology
Sialidosis (Mucolipidosis I): Background, Pathophysiology, Epidemiology
 Mucolipidosis II (I cell Disorder) Market Size, Demand & Analysis By 2030
Mucolipidosis II (I cell Disorder) Market Size, Demand & Analysis By 2030
 Mucolipidosis Type IV | Related Diseases (MPS Society)
Mucolipidosis Type IV | Related Diseases (MPS Society)
 Mucolipidosis type IV
Mucolipidosis type IV
Sialidosis (Mucolipidosis I) Medication
 Mucolipidosis type II - Global Genes
Mucolipidosis type II - Global Genes
Sialidosis (Mucolipidosis I): Background, Pathophysiology, Epidemiology
 Cherry red spot in sialidosis (mucolipidosis type I) | Hereditary Ocular Diseases
Cherry red spot in sialidosis (mucolipidosis type I) | Hereditary Ocular Diseases
 Impaired myelination and reduced brain ferric iron in the mouse model of mucolipidosis IV | Disease Models & Mechanisms | The...
Impaired myelination and reduced brain ferric iron in the mouse model of mucolipidosis IV | Disease Models & Mechanisms | The...
 Chapter 160. Mucopolysaccharidosis, Glycoproteinosis, and Mucolipidosis | Rudolph's Pediatrics, 22e | AccessPediatrics | McGraw...
Chapter 160. Mucopolysaccharidosis, Glycoproteinosis, and Mucolipidosis | Rudolph's Pediatrics, 22e | AccessPediatrics | McGraw...
Metabolic Problems: MedlinePlus
 General Data Protection Regulation Compliance - 23andMe Europe
General Data Protection Regulation Compliance - 23andMe Europe
Median Neuropathy Clinical Presentation: History, Physical, Causes
Macular cherry-red spot and corneal haze in sialidosis (mucolipidosis type 1) [1]<...
 Greenwood Genetic Center
Greenwood Genetic Center
Evidence Mounting for Eliglustat in Gaucher's Disease
Characterization and expression analysis of mcoln1.1 and mcoln1.2, the putative zebrafish co-orthologs of the gene responsible...
 Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ<...
Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ<...
 Glycobiology of cell death: when glycans and lectins govern cell fate | Cell Death & Differentiation
Glycobiology of cell death: when glycans and lectins govern cell fate | Cell Death & Differentiation
 ICD-10 Code for Niemann-Pick disease- E75.24- Codify by AAPC
ICD-10 Code for Niemann-Pick disease- E75.24- Codify by AAPC
 Snapshot of Science for May 2018
Snapshot of Science for May 2018
Genetic Brain Disorders | MedlinePlus
 Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - USSialidosis6
- However, type I (sialidosis) is now classified as a glycoproteinosis, and type IV (Mucolipidosis type IV) is now classified as a gangliosidosis. (wikipedia.org)
- Mucolipidosis I is also known as sialidosis. (childrenshospital.org)
- Sialidosis, also known as mucolipidosis type I (ML I), is a rare inherited lysosomal storage disease that has clinical and histologic findings similar to the mucopolysaccharidoses and the sphingolipidoses. (medscape.com)
- Sphranger J, Gehler J, Cantz M. Mucolipidosis I--a sialidosis. (medscape.com)
- Heroman JW, Rychwalski P, Barr CC. Cherry red spot in sialidosis (mucolipidosis type I) . Arch Ophthalmol. (arizona.edu)
- Goldberg, MF 2008, ' Macular cherry-red spot and corneal haze in sialidosis (mucolipidosis type 1) [1] ', Archives of ophthalmology , vol. 126, no. 12. (johnshopkins.edu)
Cure Mucolipidosis2
- Cure Mucolipidosis is a global organization that is committed to the identification and treatment of Mucolipidosis through education, advocacy and research. (globalgenes.org)
- Cure Mucolipidosis will form partnerships with Science, Medicine and industry and will work towards finding a cure for people affected by Mucolipidosis globally. (globalgenes.org)
Ashkenazi Jews1
- Medical genetics of Ashkenazi Jews mucolipidoses at NINDS RESERVED, INSERM US14 -- ALL RIGHTS. (wikipedia.org)
Lysosomes4
- Conditions that cause molecules to build up inside lysosomes, including mucolipidosis III alpha/beta, are called lysosomal storage disorders. (medlineplus.gov)
- The signs and symptoms of mucolipidosis III alpha/beta are most likely due to the shortage of digestive enzymes inside lysosomes and the effects these enzymes have outside the cell. (medlineplus.gov)
- Mucolipidosis I is caused by a mutation on the NEU1 gene, resulting in a deficiency in an enzyme known as neuraminidase 1, which lysosomes require to properly break down large sugar molecules inside the body's cells. (childrenshospital.org)
- In individuals with mucolipidosis II, lysosomes do not perform their normal role effectively, leading to the accumulation of certain substances within the cell. (databridgemarketresearch.com)
Autosomal recessive2
- The mucolipidoses are inherited in an autosomal recessive manner, that is, they occur only when a child inherits two copies of the defective gene, one from each parent. (wikipedia.org)
- Mucolipidosis type IV (MLIV) is an autosomal recessive lysosomal storage disorder caused by mutations in the MCOLN1 gene coding for mucolipin-1 (TRPML1). (ox.ac.uk)
Gaucher1
- The Ashkenazi Jewish Panel includes the following diseases: Bloom syndrome, Canavan disease, Fanconi anemia type C, familial dysautonomia, Gaucher disease, glycogen storage disease type 1a, Mucolipidosis IV, Neimann-Pick disease, and Tay-Sachs disease. (cdc.gov)
Pseudo-Hurler Polydy2
- Mucolipidosis II (ML II), also known as I-Cell disease, and Mucolipidosis IIIA (ML IIIA), also known as Pseudo-Hurler Polydystrophy, are lysosomal storage disorders caused by a deficiency of N-acetylglucosamine-1-phosphotransferase (NAPT). (ggc.org)
- a deficiency or defect in this enzyme results in two forms of mucolipidoses, I-cell disease, and pseudo-Hurler polydystrophy. (theodora.com)
Disease10
- Mucolipidosis III alpha/beta and mucolipidosis II alpha/beta represent two ends of a spectrum of disease severity. (medlineplus.gov)
- Children who show signs and symptoms of Mucolipidosis I at birth typically have a more severe form of the disease. (childrenshospital.org)
- Spranger and Wiedermann subsequently classified this disease as mucolipidosis type II (ML II) because it had clinical characteristics that included mucopolysaccharidoses and sphingolipidoses. (medscape.com)
- Initially classified as a lipomucopolysaccharidosis, this disease was later classified into the group of similar diseases now known as the mucolipidoses. (medscape.com)
- I-cell disease (mucolipidosis II) (I cell disorder) refers to a rare inherited metabolic disorder that is generally characterized by coarse facial features such as mental retardation and skeletal abnormalities. (databridgemarketresearch.com)
- The increase in the incidences of I-cell disease (mucolipidosis II) (I cell Disorder) among population across the globe acts as one of the major factors driving the growth of global mucolipidosis II (I cell disorder) market. (databridgemarketresearch.com)
- Mucolipidosis II, also known as I-cell disease, is a rare inherited genetic disorder that falls under the broader category of lysosomal storage diseases. (databridgemarketresearch.com)
- Mucolipidosis type IV (ML IV) also known as ganglioside sialidase deficiency and sialolipidosis, is an inherited lysosomal storage disease, belonging to the group of oligosaccharidosis that affects many organs and tissues, including the nervous system. (mpssociety.org.uk)
- 3) Inherited pathogenic alleles of LYSET can cause a severe inherited disease which resembles GlcNAc-1-phosphotransferase deficiency (i.e., mucolipidosis type II). (stanford.edu)
- Mucolipidosis II/I-Cell disease) - Alzheimer disease - Hermansky Pudlak Syndrome (pigmentation bleeding disorder) - Cancer - virus and bacterial infections - ARC syndrome -Microvillar inclusion Disease. (umcutrecht.nl)
Disorder10
- Mucolipidosis III alpha/beta is a disorder that affects many parts of the body. (medlineplus.gov)
- Mucolipidosis III alpha/beta is a rare disorder, although its exact prevalence is unknown. (medlineplus.gov)
- Mutations in the GNPTAB gene can also cause a similar but more severe disorder called mucolipidosis II alpha/beta . (medlineplus.gov)
- Mucolipidosis I (ML I) is a rare, inherited disorder. (childrenshospital.org)
- Data Bridge Market Research analyses that the global mucolipidosis II (I cell disorder) market which was USD 12.80 billion in 2022, is expected to reach USD 15.80 billion by 2030, and is expected to undergo a CAGR of 3.9% during the forecast period 2023-2030. (databridgemarketresearch.com)
- Abnormal Curvature of the Spine" dominates the symptoms segment of the global mucolipidosis II (I cell disorder) market owing to the rising incidences of genetic diseases and other related issues. (databridgemarketresearch.com)
- Mucolipidosis type IV ( ML IV ), like other types of mucolipidosis is an inherited neurodegenerative lysosomal storage disorder. (chemeurope.com)
- Mutations in the α/β subunit precursor gene cause the severe lysosomal storage disorder mucolipidosis II (ML II) or the more moderate mucolipidosis III alpha/beta (ML III α/β), while mutations in the γ subunit gene cause the mildest disorder, mucolipidosis III gamma (ML III γ). (wustl.edu)
- GlcNAc-1-phosphotransferase deficiency leads to the severe lysosomal storage disorder mucolipidosis II (MLII). (stanford.edu)
- Their son Christopher was born with Mucolipidosis II, a very rare, terminal genetic disorder which caused both physical and developmental disabilities. (ncpd.org)
MLIV1
- Mucolipidosis IV (MLIV) is caused by mutations in the gene MCOLN1. (escholarship.org)
Mucopolysaccharidoses1
- When originally named, the mucolipidoses derived their name from the similarity in presentation to both mucopolysaccharidoses and sphingolipidoses. (wikipedia.org)
Metabolic disorders1
- Mucolipidosis is a group of inherited metabolic disorders that affect the body's ability to carry out the normal turnover of various materials within cells. (wikipedia.org)
Patients1
- Myelination deficits and severely dysmorphic corpus callosum were present early and resembled white matter pathology in mucolipidosis IV patients. (escholarship.org)
Mutations2
- Mutations in the GNPTAB gene cause mucolipidosis III alpha/beta. (medlineplus.gov)
- Mutations in the GNPTAB gene that cause mucolipidosis III alpha/beta result in reduced activity of GlcNAc-1-phosphotransferase. (medlineplus.gov)
Neurodegenerative1
- These results indicate the early involvement of glia, and challenge the traditional view of mucolipidosis IV as an overtly neurodegenerative condition. (escholarship.org)
Abnormalities2
- People with mucolipidosis III alpha/beta often have heart valve abnormalities and mild clouding of the clear covering of the eye ( cornea ). (medlineplus.gov)
- A rare severe form of mucolipidosis characterized by growth retardation skeletal abnormalities (dysostosis multiplex craniosynostosis contractures of the joints and osteopenia) facial dysmorphism stiff skin obstructive airway cardiomegaly and severe global developmental delay. (globalgenes.org)
Genetic1
- Spranger JW, Wiedemann HR. The genetic mucolipidoses. (medscape.com)
Deficiency2
- Mucolipidosis types II and III (ML II and ML III) result from a deficiency of the enzyme N-acetylglucosamine-1-phosphotransferase, which phosphorylates target carbohydrate residues on N-linked glycoproteins. (wikipedia.org)
- Mucolipidosis I (acid neuraminidase deficiency). (medscape.com)
Mutation1
- This type of mucolipidosis is caused by mutation of a non-selective cation channel, TRPML1 . (chemeurope.com)
Collaboration2
- There are currently no approved therapies which reverse the effects of Mucolipidosis I. Current approaches involve managing specific symptoms through targeted therapies and collaboration between specialists. (childrenshospital.org)
- To promote and support a global multi-stakeholder collaboration for Mucolipidosis. (globalgenes.org)
Mouse2
- Early evidence of delayed oligodendrocyte maturation in the mouse model of mucolipidosis type IV. (harvard.edu)
- Using an accurate mouse model of mucolipidosis IV, we observed early behavioral deficits which were accompanied by activation of microglia and astrocytes. (escholarship.org)
Type IV1
- Characterization and expression analysis of mcoln1.1 and mcoln1.2, the putative zebrafish co-orthologs of the gene responsible for human mucolipidosis type IV. (ox.ac.uk)
Result1
- Poor mental capacities, and difficulty reaching physical developmental milestones may also be result of mucolipidosis. (wikipedia.org)
People1
- Osteoporosis and progressive joint problems also cause bone pain that becomes more severe over time in people with mucolipidosis III alpha/beta. (medlineplus.gov)
Article1
- See the equivalent section in the main mucolipidosis article . (chemeurope.com)
Individuals2
- Individuals with mucolipidosis III alpha/beta grow slowly and have short stature. (medlineplus.gov)
- Individuals with mucolipidosis III alpha/beta generally survive into adulthood, but they may have a shortened lifespan. (medlineplus.gov)
Treatment1
- Our Vision To serve as a resource for stakeholders in the work, identification, treatment, and continual developments towards a cure for Mucolipidosis. (globalgenes.org)
Work1
- We work with the broad array of world-class specialists at Boston Children's to optimize the care we provide your child with Mucolipidosis I. (childrenshospital.org)