Multiple Carboxylase Deficiency
Pyruvate Carboxylase Deficiency Disease
Holocarboxylase Synthetase Deficiency
Carbon-Nitrogen Ligases
Methylmalonyl-CoA Decarboxylase
Biotinidase Deficiency
Carbon-Carbon Ligases
Biotin
Biotinidase
Ligases
Pyruvate Carboxylase
Crotonates
Amino Acid Metabolism, Inborn Errors
Acetyl-CoA Carboxylase
Carboxy-Lyases
Ribulose-Bisphosphate Carboxylase
Phosphoenolpyruvate Carboxylase
Propionates
Bioavailability of biotin given orally to humans in pharmacologic doses. (1/8)
BACKGROUND: Patients with carboxylase deficiency are treated with pharmacologic doses of biotin. OBJECTIVE: We sought to determine the bioavailability of biotin at pharmacologic doses. DESIGN: Biotin was administered orally (2.1, 8.2, or 81.9 micromol) or intravenously (18.4 micromol) to 6 healthy adults in a crossover design with > or =2 wk between each biotin administration. Before and after each administration, timed 24-h urine samples were collected. Urinary biotin and biotin metabolites were analyzed by an HPLC avidin-binding assay. RESULTS: Urinary recoveries of biotin plus metabolites were similar (approximately 50%) after the 2 largest oral doses and the 1 intravenous dose, suggesting 100% bioavailability of the 2 largest oral doses. For unexplained reasons, the apparent recovery of the smallest oral dose was about twice that of the other doses. For all 4 doses, biotin accounted for >50% of the total of biotin and biotin metabolites in urine. Bisnorbiotin (13-23%), biotin-d,l-sulfoxide (5-13%), bisnorbiotin methyl ketone (3-9%), and biotin sulfone (1-3%) accounted for the remainder. The percentage excretion of biotin was greater when biotin was administered intravenously and for the largest oral dose than for the 2 smallest oral doses. CONCLUSION: Our data provide evidence that oral biotin is completely absorbed even when pharmacologic doses are administered. Biotin metabolites account for a substantial portion of total urinary excretion and must be considered in bioavailability studies. We speculate that renal losses of biotin (as a percentage of the dose administered) are moderately elevated when pharmacologic doses of biotin are administered. (+info)Effectiveness of nystatin in polysymptomatic patients. A randomized, double-blind trial with nystatin versus placebo in general practice. (2/8)
BACKGROUND: Antifungal therapy has been claimed to be effective in polysymptomatic patients with diffuse symptoms from multiple body systems and even well defined diseases, traditionally not related to fungi. Hypersensitivity to fungus proteins and mycotoxins has been proposed as the cause. METHODS: We conducted a 4-week randomized, double-blind, placebo-controlled study in 116 individuals selected by a 7-item questionnaire to determine whether the antifungal agent nystatin given orally was superior to placebo. At the onset of the study, the patients were free to select either their regular diet or a sugar- and yeast-free diet, which resulted in four different subgroups: nystatin + diet (ND); placebo + diet (PD); nystatin (N); and placebo (P). RESULTS: Nystatin was significantly better than placebo in reduction of the overall symptom score (P < 0.003). In six of the 45 individually recorded symptoms, the improvement was significant (P < 0.01). All three active treatment groups reduced their overall symptom scores significantly (P < 0.0001), while the placebo regimen had no effect (P = 0.83). The benefit of diet was significant within both the nystatin (ND > N) and the placebo groups (PD > P). CONCLUSIONS: Nystatin is superior to placebo in reducing localized and systemic symptoms in individuals with presumed fungus hypersensitivity as selected by a 7-item questionnaire. This superiority is probably enhanced even further by a sugar- and yeast-free diet. (+info)Reduced histone biotinylation in multiple carboxylase deficiency patients: a nuclear role for holocarboxylase synthetase. (3/8)
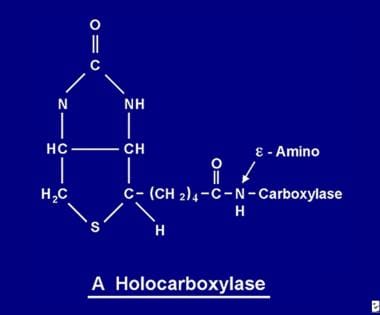
The attachment of biotin to apocarboxylases is catalyzed by holocarboxylase synthetase (HCS). An inherited deficiency of HCS results in the disorder 'multiple carboxylase deficiency', which is characterized by reduced activity of all biotin-dependent carboxylases. Here we show that the majority of HCS localizes to the nucleus rather than the cytoplasm based on immunofluorescence studies with antibodies to peptides and full length HCS and on the expression of recombinant HCS. Subnuclear fractionations indicate that HCS is associated with chromatin and the nuclear lamina, the latter in a discontinuous distribution in high salt-extracted nuclear membranes. During mitosis, HCS resolves into ring-like particles which co-localize with lamin B. Nuclear HCS retains its biotinylating activity and was shown to biotinylate purified histones in vitro. Significantly, fibroblasts from patients with HCS deficiency are severely deficient in histone biotinylation in addition to the deficiency of carboxylase activities. We propose that the role of HCS in histone modification may be linked to the participation of biotin in the regulation of gene expression or cell division and that affected patients may have additional disease beyond that due to the effect on carboxylases. (+info)Biotin responsive multiple carboxylase deficiency presenting as diabetic ketoacidosis. (4/8)
Multiple carboxylase deficiency (MCD) is a rare inherited metabolic disease of biotin dependency due to deficiency of holocarboxylase synthetase (HCS) or biotinidase deficiency. A 30-month-old female patient who presented with the initial features of diabetic ketoacidosis (severe metabolic acidosis, ketosis, and hyperglycemia), lactic acidemia, moderate hyperammonemia, and generalized organic aciduria is described. Associated symptoms and signs included erythematous skin rashes, alopecia and developmental delay. The patient responded dramatically to treatment with biotin (10 mg/day) showing normalization of clinical symptoms and most biochemical abnormalities. Based on the urine organic profile by gas chromatography/ mass spectrometry (GC/MS), the diagnosis of MCD was made. A plasma tandem mass study confirmed this diagnosis. The biotinase activity in serum was normal, indicating that this was a rare case of late-onset HCS deficiency. (+info)Paradoxical regulation of biotin utilization in brain and liver and implications for inherited multiple carboxylase deficiency. (5/8)
Holocarboxylase synthetase (HCS) catalyzes the biotinylation of five carboxylases in human cells, and mutations of HCS cause multiple carboxylase deficiency (MCD). Although HCS also participates in the regulation of its own mRNA levels, the relevance of this mechanism to normal metabolism or to the MCD phenotype is not known. In this study, we show that mRNA levels of enzymes involved in biotin utilization, including HCS, are down-regulated during biotin deficiency in liver while remaining constitutively expressed in brain. We propose that this mechanism of regulation is aimed at sparing the essential function of biotin in the brain at the expense of organs such as liver and kidney during biotin deprivation. In MCD, it is possible that some of the manifestations of the disease may be associated with down-regulation of biotin utilization in liver because of the impaired activity of HCS and that high dose biotin therapy may in part be important to overcoming the adverse regulatory impact in such organs. (+info)Effect of biotin deficiency and supplementation on lipid metabolism in rats: saturated fatty acids. (6/8)
The effects of biotin deficiency and supplementation upon saturated fatty acids in serum, liver, cerebrum, and cerebellum of rats were investigated. Serum total fatty acids were reduced in deficient animals to 29% of normal. The percentage composition of some odd-chain fatty acids was increased in the serum and liver of deficient rats. The relative composition of most saturated fatty acids with carbon-chain lengths greater than or equal to 22 were increased in serum and liver of deficient rats. In contrast, there were no differences in the brain saturated fatty acids of deficient animals compared to those of normal and supplemented animals. These results may indicate that alterations in saturated fatty acids do not play a major role in the neurologic abnormalities in human biotin or inherited multiple-carboxylase deficiencies. (+info)Clustering of mutations in the biotin-binding region of holocarboxylase synthetase in biotin-responsive multiple carboxylase deficiency. (7/8)
Holocarboxylase synthetase (HCS) catalyses the biotinylation of the four biotin-dependent carboxylases found in humans. A deficiency in HCS results in biotin-responsive multiple carboxylase deficiency (MCD). We have identified six different point mutations in the HCS gene in nine patients with MCD. Two of the mutations are frequent among the MCD patients analyzed. Four of the mutations cluster in the putative biotin-binding domain as deduced from the corresponding Escherichia coli enzyme and consistent with an explanation for biotin-responsiveness based on altered affinity for biotin. The two others may define an additional domain involved in biotin-binding or biotin-mediated stabilization of the protein. (+info)Profound biotinidase deficiency caused by a point mutation that creates a downstream cryptic 3' splice acceptor site within an exon of the human biotinidase gene. (8/8)
Biotinidase recycles the vitamin biotin from biocytin upon the degradation of the biotin-dependent carboxylases. We have identified a novel point mutation within the biotinidase gene that encodes the signal peptide in two unrelated individuals with profound biotinidase deficiency. Sequence analysis of genomic DNA from these individuals revealed a G to A transition (G100-->A) located 57 bases downstream of the authentic splice acceptor site in exon B. Although this mutation predicts a G34S substitution, it also generates a 3' splice acceptor site. Sequence of the PCR-amplified cDNA from the homozygous child revealed that all the product was shorter than that of normal individuals and was the result of aberrant splicing. The aberrantly spliced transcript lacked 57 bases, including a second in-frame ATG, that encode most of the putative signal peptide and results in an in-frame deletion of 19 amino acids. The mutation results in failure to secrete the aberrant protein into the blood. This is the first reported example in which a point mutation creates a cryptic 3' splice acceptor site motif that is used preferentially over the upstream authentic splice site. The preferential usage of the downstream splice site is not consistent with the 5'-3' scanning model, but is consistent with the exon definition model of RNA splicing. (+info)Multiple carboxylase deficiency (MCD) is a rare genetic disorder that affects the body's ability to metabolize certain amino acids, particularly those that contain sulfur. It is caused by mutations in the genes responsible for producing enzymes involved in the biotin-dependent carboxylation reactions, which are critical for various metabolic processes in the body.
There are two major types of MCD:
1. Profound multiple carboxylase deficiency (also known as Type II biotinidase deficiency): This form is more severe and is caused by a defect in the holocarboxylase synthetase enzyme, which is responsible for attaching biotin to several carboxylases.
2. Biotin-responsive multiple carboxylase deficiency (also known as Type I biotinidase deficiency): This form is milder and is caused by a defect in the biotinidase enzyme, which recycles biotin in the body. However, it can be treated with biotin supplementation.
Symptoms of MCD may include:
* Developmental delay
* Seizures
* Hypotonia (low muscle tone)
* Ataxia (lack of coordination)
* Rash
* Hair loss
* Acidosis (high levels of acid in the body)
* Coma and even death, if left untreated
Early diagnosis and treatment with biotin supplementation can significantly improve outcomes for individuals with MCD.
Pyruvate carboxylase deficiency disease is a rare inherited metabolic disorder that affects the body's ability to break down proteins, fats, and carbohydrates for energy. It is caused by mutations in the Pyruvate Carboxylase (PC) gene, which provides instructions for making an enzyme called pyruvate carboxylase. This enzyme plays a critical role in gluconeogenesis, a process that takes place in the liver and kidneys to produce glucose, a simple sugar that is a primary source of energy for the body.
In pyruvate carboxylase deficiency disease, the enzyme's activity is significantly reduced or absent, leading to an accumulation of toxic levels of certain metabolic intermediates, such as lactic acid and pyruvic acid, in the blood and other tissues. This accumulation can cause a range of symptoms, including developmental delay, seizures, poor muscle tone, difficulty breathing, and feeding problems.
The severity of pyruvate carboxylase deficiency disease varies widely, depending on the degree of enzyme activity that is affected. Some individuals may have mild symptoms, while others may experience severe, life-threatening complications. Treatment typically involves managing symptoms and providing supportive care to help prevent complications. In some cases, a low-protein diet or supplementation with certain vitamins and minerals may be recommended to help reduce the accumulation of toxic metabolites.
Holocarboxylase Synthetase Deficiency (HCD) is a rare genetic disorder of biotin metabolism, characterized by the body's inability to properly utilize the vitamin biotin. Biotin plays a crucial role in various essential functions, such as the breakdown of fats, proteins, and carbohydrates, as well as the regulation of gene expression.
Holocarboxylase synthetase is an enzyme responsible for attaching biotin to four different carboxylases, which are necessary for these vital processes. In Holocarboxylase Synthetase Deficiency, this enzyme is either partially or completely nonfunctional due to mutations in the HLCS gene.
The symptoms of HCD can vary widely but often include:
1. Feeding difficulties and poor growth in infancy
2. Severe metabolic acidosis
3. Ketoacidosis
4. Delayed development
5. Hypotonia (low muscle tone)
6. Skin rashes
7. Hair loss
8. Neurological symptoms, such as seizures and ataxia (loss of coordination and balance)
If left untreated, Holocarboxylase Synthetase Deficiency can lead to severe complications, including developmental delays, neurological damage, and even death. However, with early diagnosis and proper treatment involving biotin supplementation, many of these symptoms can be managed, and the progression of the disorder can be slowed or stopped.
Carbon-Nitrogen (C-N) ligases are a class of enzymes that catalyze the joining of a carbon atom from a donor molecule to a nitrogen atom in an acceptor molecule through a process called ligase reaction. This type of enzyme plays a crucial role in various biological processes, including the biosynthesis of amino acids, nucleotides, and other biomolecules that contain both carbon and nitrogen atoms.
C-N ligases typically require ATP or another energy source to drive the reaction forward, as well as cofactors such as metal ions or vitamins to facilitate the chemical bond formation between the carbon and nitrogen atoms. The specificity of C-N ligases varies depending on the enzyme, with some acting only on specific donor and acceptor molecules while others have broader substrate ranges.
Examples of C-N ligases include glutamine synthetase, which catalyzes the formation of glutamine from glutamate and ammonia, and asparagine synthetase, which catalyzes the formation of asparagine from aspartate and ammonia. Understanding the function and regulation of C-N ligases is important for understanding various biological processes and developing strategies to modulate them in disease states.
Methylmalonyl-CoA decarboxylase is a mitochondrial enzyme that plays a crucial role in the metabolism of certain amino acids and fatty acids. Specifically, it catalyzes the conversion of methylmalonyl-CoA to propionyl-CoA through the decarboxylation of the thioester bond.
The reaction is as follows:
Methylmalonyl-CoA → Propionyl-CoA + CO2
This enzyme requires biotin as a cofactor, and its activity is reduced in individuals with methylmalonic acidemia, a rare inherited metabolic disorder caused by mutations in the MMAB or MCEE genes that encode subunits of the methylmalonyl-CoA decarboxylase enzyme complex.
Deficiency of this enzyme leads to an accumulation of methylmalonic acid and methylmalonyl-CoA, which can cause metabolic acidosis, hyperammonemia, and other symptoms associated with the disorder.
Biotinidase deficiency is a genetic disorder that affects the body's ability to recycle and reuse biotin, a type of B vitamin. Biotinidase is an enzyme that helps release biotin from proteins in the food we eat and recycle it for use by the body.
In people with biotinidase deficiency, the biotinidase enzyme is either partially or completely missing, leading to a decrease in available biotin. This can result in a variety of symptoms, including seizures, developmental delays, hearing and vision loss, skin rashes, hair loss, and muscle weakness.
There are two main types of biotinidase deficiency: partial deficiency and profound deficiency. Partial deficiency means that some biotinidase activity is present, but not enough to prevent symptoms. Profound deficiency means that there is little or no biotinidase activity, resulting in more severe symptoms.
Biotinidase deficiency can be diagnosed through a blood test that measures the level of biotinidase enzyme activity. Treatment typically involves taking biotin supplements to replace the missing biotin and prevent symptoms from developing or worsening. With early diagnosis and treatment, people with biotinidase deficiency can often lead normal lives.
Carbon-carbon ligases are a type of enzyme that catalyze the formation of carbon-carbon bonds between two molecules. These enzymes play important roles in various biological processes, including the biosynthesis of natural products and the metabolism of carbohydrates and lipids.
Carbon-carbon ligases can be classified into several categories based on the type of reaction they catalyze. For example, aldolases catalyze the condensation of an aldehyde or ketone with another molecule to form a new carbon-carbon bond and a new carbonyl group. Other examples include the polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs), which are large multienzyme complexes that catalyze the sequential addition of activated carbon units to form complex natural products.
Carbon-carbon ligases are important targets for drug discovery and development, as they play critical roles in the biosynthesis of many disease-relevant molecules. Inhibitors of these enzymes have shown promise as potential therapeutic agents for a variety of diseases, including cancer, infectious diseases, and metabolic disorders.
Biotin is a water-soluble vitamin, also known as Vitamin B7 or Vitamin H. It is a cofactor for several enzymes involved in metabolism, particularly in the synthesis and breakdown of fatty acids, amino acids, and carbohydrates. Biotin plays a crucial role in maintaining healthy skin, hair, nails, nerves, and liver function. It is found in various foods such as nuts, seeds, whole grains, milk, and vegetables. Biotin deficiency is rare but can occur in people with malnutrition, alcoholism, pregnancy, or certain genetic disorders.
Biotinidase is an enzyme that is responsible for the release of biotin, a vital nutrient, from proteins in the body. Biotin is essential for various metabolic processes, including the synthesis of fatty acids and glucose. Biotinidase deficiency can lead to serious health problems, such as seizures, developmental delays, and hearing and vision loss. Therefore, biotinidase levels are often measured in newborn screening tests to identify babies who may be at risk for this rare but treatable condition.
Ligases are a group of enzymes that catalyze the formation of a covalent bond between two molecules, usually involving the joining of two nucleotides in a DNA or RNA strand. They play a crucial role in various biological processes such as DNA replication, repair, and recombination. In DNA ligases, the enzyme seals nicks or breaks in the phosphodiester backbone of the DNA molecule by catalyzing the formation of an ester bond between the 3'-hydroxyl group and the 5'-phosphate group of adjacent nucleotides. This process is essential for maintaining genomic integrity and stability.
Pyruvate carboxylase is a biotin-containing enzyme that plays a crucial role in gluconeogenesis, the process of generating new glucose molecules from non-carbohydrate sources. The enzyme catalyzes the conversion of pyruvate to oxaloacetate, an important intermediate in several metabolic pathways, particularly in the liver, kidneys, and brain.
The reaction catalyzed by pyruvate carboxylase is as follows:
Pyruvate + CO2 + ATP + H2O → Oxaloacetate + ADP + Pi + 2H+
In this reaction, pyruvate reacts with bicarbonate (HCO3-) to form oxaloacetate, consuming one molecule of ATP in the process. The generation of oxaloacetate provides a key entry point for non-carbohydrate precursors, such as lactate and certain amino acids, to enter the gluconeogenic pathway.
Pyruvate carboxylase deficiency is a rare but severe genetic disorder that can lead to neurological impairment and developmental delays due to the disruption of energy metabolism in the brain.
Crotonates are a group of organic compounds that contain a carboxylic acid functional group (-COOH) attached to a crotyl group, which is a type of alkyl group with the structure -CH=CH-CH\_{2}-. Crotyl groups are derived from crotonic acid or its derivatives.
Crotonates can be found in various natural and synthetic compounds, including some pharmaceuticals, agrochemicals, and other industrial chemicals. They can exist as salts, esters, or other derivatives of crotonic acid.
In medical contexts, crotonates may refer to certain medications or chemical compounds used for research purposes. For example, sodium crotylate is a salt of crotonic acid that has been studied for its potential anti-inflammatory and analgesic effects. However, it is not widely used in clinical practice.
It's worth noting that the term "crotonates" may not have a specific medical definition on its own, as it refers to a broad class of compounds with varying properties and uses.
Inborn errors of amino acid metabolism refer to genetic disorders that affect the body's ability to properly break down and process individual amino acids, which are the building blocks of proteins. These disorders can result in an accumulation of toxic levels of certain amino acids or their byproducts in the body, leading to a variety of symptoms and health complications.
There are many different types of inborn errors of amino acid metabolism, each affecting a specific amino acid or group of amino acids. Some examples include:
* Phenylketonuria (PKU): This disorder affects the breakdown of the amino acid phenylalanine, leading to its accumulation in the body and causing brain damage if left untreated.
* Maple syrup urine disease: This disorder affects the breakdown of the branched-chain amino acids leucine, isoleucine, and valine, leading to their accumulation in the body and causing neurological problems.
* Homocystinuria: This disorder affects the breakdown of the amino acid methionine, leading to its accumulation in the body and causing a range of symptoms including developmental delay, intellectual disability, and cardiovascular problems.
Treatment for inborn errors of amino acid metabolism typically involves dietary restrictions or supplementation to manage the levels of affected amino acids in the body. In some cases, medication or other therapies may also be necessary. Early diagnosis and treatment can help prevent or minimize the severity of symptoms and health complications associated with these disorders.
Acetyl-CoA carboxylase (ACCA) is a biotin-dependent enzyme that plays a crucial role in fatty acid synthesis. It catalyzes the conversion of acetyl-CoA to malonyl-CoA, which is the first and rate-limiting step in the synthesis of long-chain fatty acids. The reaction catalyzed by ACCA is as follows:
acetyl-CoA + HCO3- + ATP + 2H+ --> malonyl-CoA + CoA + ADP + Pi + 2H2O
ACCA exists in two isoforms, a cytosolic form (ACC1) and a mitochondrial form (ACC2). ACC1 is primarily involved in fatty acid synthesis, while ACC2 is responsible for the regulation of fatty acid oxidation. The activity of ACCA is regulated by several factors, including phosphorylation/dephosphorylation, allosteric regulation, and transcriptional regulation. Dysregulation of ACCA has been implicated in various metabolic disorders, such as obesity, insulin resistance, and non-alcoholic fatty liver disease.
Carboxy-lyases are a class of enzymes that catalyze the removal of a carboxyl group from a substrate, often releasing carbon dioxide in the process. These enzymes play important roles in various metabolic pathways, such as the biosynthesis and degradation of amino acids, sugars, and other organic compounds.
Carboxy-lyases are classified under EC number 4.2 in the Enzyme Commission (EC) system. They can be further divided into several subclasses based on their specific mechanisms and substrates. For example, some carboxy-lyases require a cofactor such as biotin or thiamine pyrophosphate to facilitate the decarboxylation reaction, while others do not.
Examples of carboxy-lyases include:
1. Pyruvate decarboxylase: This enzyme catalyzes the conversion of pyruvate to acetaldehyde and carbon dioxide during fermentation in yeast and other organisms.
2. Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO): This enzyme is essential for photosynthesis in plants and some bacteria, as it catalyzes the fixation of carbon dioxide into an organic molecule during the Calvin cycle.
3. Phosphoenolpyruvate carboxylase: Found in plants, algae, and some bacteria, this enzyme plays a role in anaplerotic reactions that replenish intermediates in the citric acid cycle. It catalyzes the conversion of phosphoenolpyruvate to oxaloacetate and inorganic phosphate.
4. Aspartate transcarbamylase: This enzyme is involved in the biosynthesis of pyrimidines, a class of nucleotides. It catalyzes the transfer of a carboxyl group from carbamoyl aspartate to carbamoyl phosphate, forming cytidine triphosphate (CTP) and fumarate.
5. Urocanase: Found in animals, this enzyme is involved in histidine catabolism. It catalyzes the conversion of urocanate to formiminoglutamate and ammonia.
Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) is a crucial enzyme in the Calvin cycle, which is a process that plants use to convert carbon dioxide into glucose during photosynthesis. RuBisCO catalyzes the reaction between ribulose-1,5-bisphosphate and carbon dioxide, resulting in the formation of two molecules of 3-phosphoglycerate, which can then be converted into glucose.
RuBisCO is considered to be the most abundant enzyme on Earth, making up as much as 50% of the soluble protein found in leaves. It is a large and complex enzyme, consisting of eight small subunits and eight large subunits that are arranged in a barrel-shaped structure. The active site of the enzyme, where the reaction between ribulose-1,5-bisphosphate and carbon dioxide takes place, is located at the interface between two large subunits.
RuBisCO also has a secondary function as an oxygenase, which can lead to the production of glycolate, a toxic compound for plants. This reaction occurs when the enzyme binds with oxygen instead of carbon dioxide and is more prevalent in environments with low carbon dioxide concentrations and high oxygen concentrations. The glycolate produced during this process needs to be recycled through a series of reactions known as photorespiration, which can result in significant energy loss for the plant.
Phosphoenolpyruvate carboxylase (PEP-carboxylase or PEPC) is a biotin-dependent enzyme that plays a crucial role in the carbon fixation process of photosynthesis, specifically in the C4 and CAM (Crassulacean Acid Metabolism) plant pathways. It is also found in some bacteria and archaea.
PEP-carboxylase catalyzes the irreversible reaction between phosphoenolpyruvate (PEP) and bicarbonate (HCO3-) to form oxaloacetate and inorganic phosphate (Pi). This reaction helps to initiate the carbon fixation process by incorporating atmospheric carbon dioxide into an organic molecule, which can then be used for various metabolic processes.
In C4 plants, PEP-carboxylase is primarily located in the mesophyll cells where it facilitates the initial fixation of CO2 onto PEP, forming oxaloacetate. This oxaloacetate is then reduced to malate, which is subsequently transported to bundle sheath cells for further metabolism and additional carbon fixation by another enzyme, ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO).
In CAM plants, PEP-carboxylase operates at night to fix CO2 into malate, which is stored in vacuoles. During the day, malate is decarboxylated, releasing CO2 for RuBisCO-mediated carbon fixation while conserving water through reduced stomatal opening.
PEP-carboxylase is also found in some non-photosynthetic bacteria and archaea, where it contributes to various metabolic pathways such as gluconeogenesis, anaplerotic reactions, and the glyoxylate cycle.
Propionates, in a medical context, most commonly refer to a group of medications that are used as topical creams or gels to treat fungal infections of the skin. Propionic acid and its salts, such as propionate, are the active ingredients in these medications. They work by inhibiting the growth of fungi, which causes the infection. Common examples of propionate-containing medications include creams used to treat athlete's foot, ringworm, and jock itch.
It is important to note that there are many different types of medications and compounds that contain the word "propionate" in their name, as it refers to a specific chemical structure. However, in a medical context, it most commonly refers to antifungal creams or gels.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
 Multiple carboxylase deficiency
Multiple carboxylase deficiency Prenatal Diagnosis and Fetal Therapy: Practice Essentials, Fetus as Patient, Options for Prenatal Diagnosis
Prenatal Diagnosis and Fetal Therapy: Practice Essentials, Fetus as Patient, Options for Prenatal Diagnosis Biotinidase deficiency: MedlinePlus Genetics
Biotinidase deficiency: MedlinePlus Genetics Resources for Newborn Screening in New Hampshire - Save Babies
Resources for Newborn Screening in New Hampshire - Save Babies Baby’s Pregnancy Calendar
Baby’s Pregnancy Calendar Organic Acid Disorders
Organic Acid Disorders Icd 10 Code For Anion Gap Metabolic Acidosis | DiabetesTalk.Net
Icd 10 Code For Anion Gap Metabolic Acidosis | DiabetesTalk.Net Biotinidase deficiency (Concept Id: C0220754)
- MedGen - NCBI
Biotinidase deficiency (Concept Id: C0220754)
- MedGen - NCBI WOLVIT Tablets - Kusum Local
WOLVIT Tablets - Kusum Local DDrare: Database of Drug Development for Rare Diseases
DDrare: Database of Drug Development for Rare Diseases Information of NSP Disorders - Delaware Health and Social Services - State of Delaware
Information of NSP Disorders - Delaware Health and Social Services - State of Delaware LOINC Part LP15301-2 3-Hydroxyisovalerate
LOINC Part LP15301-2 3-Hydroxyisovalerate Порушення метаболізму амінокислот з розгалуженим ланцюгом - Педіатрія - MSD Manual Professional Edition
Порушення метаболізму амінокислот з розгалуженим ланцюгом - Педіатрія - MSD Manual Professional Edition SMPDB
SMPDB Pesquisa | Portal Regional da BVS
Pesquisa | Portal Regional da BVS Antonyms for DEFICIENTLY - Thesaurus.net
Antonyms for DEFICIENTLY - Thesaurus.net Psoriasis and Acidic Blood: The Cause Is Deficiency of B vitamins | PsoriasisDietPlan.com
Psoriasis and Acidic Blood: The Cause Is Deficiency of B vitamins | PsoriasisDietPlan.com