Multiple Endocrine Neoplasia Type 1
Multiple Endocrine Neoplasia Type 2a
Multiple Endocrine Neoplasia Type 2b
Multiple Endocrine Neoplasia
Proto-Oncogene Proteins c-ret
Carcinoma, Medullary
Gastrinoma
Pheochromocytoma
Hyperparathyroidism, Primary
Zollinger-Ellison Syndrome
Germ-Line Mutation
Hyperparathyroidism
Proto-Oncogene Proteins
Pituitary Neoplasms
Insulinoma
Receptor Protein-Tyrosine Kinases
Angiofibroma
Pancreatic Neoplasms
Pedigree
Chromosomes, Human, Pair 11
Bone Demineralization, Pathologic
Neuroendocrine Tumors
Adenoma, Islet Cell
Calcitonin
Metanephrine
Carcinoid Tumor
von Hippel-Lindau Disease
Hirschsprung Disease
Glucagonoma
Drosophila Proteins
Genetic Testing
Parathyroid Glands
Ganglioneuroma
Prolactinoma
Paraganglioma
Mutation
Proto-Oncogenes
Loss of Heterozygosity
Cyclin-Dependent Kinase Inhibitor p18
Vipoma
Chromosomes, Human, Pair 10
Normetanephrine
Neoplastic Syndromes, Hereditary
Mutation, Missense
Lipoma
Acromegaly
Parathyroid Diseases
Polymorphism, Single-Stranded Conformational
Neoplasm Proteins
Point Mutation
Gastrins
Heterozygote
Neoplasms, Multiple Primary
Hyperplasia
Phenotype
Genes, Tumor Suppressor
Growth Hormone-Secreting Pituitary Adenoma
Exons
Pancreaticoduodenectomy
Polymerase Chain Reaction
Codon
Parathyroid Hormone
Thyroid Gland
Endocrine System Diseases
Octreotide
Endocrine Glands
Endocrine System
Cyclin-Dependent Kinase Inhibitor p27
Frameshift Mutation
Genetic Markers
Adrenal Glands
Chromosome Mapping
Gene Deletion
Carcinoma
Tomography, X-Ray Computed
Base Sequence
Japan
Alleles
Family Health
Somatostatin
Genetic Linkage
3T3 Cells
Cell Transformation, Neoplastic
Follow-Up Studies
Sequence Analysis, DNA
Molecular Sequence Data
Pancreas
Polymorphism, Restriction Fragment Length
Genotype
PC12 Cells
Genetic Association Studies
Immunohistochemistry
Magnetic Resonance Imaging
Paraganglioma, Extra-Adrenal
Genetic Predisposition to Disease
Asian Continental Ancestry Group
Endocrine Disruptors
Gene Expression Regulation, Neoplastic
Carcinoma, Neuroendocrine
Cervical Intraepithelial Neoplasia
Polymorphism, Genetic
Intestinal ganglioneuromatosis and multiple endocrine neoplasia type 2B: implications for treatment. (1/71)
Three infants, who presented with intestinal obstruction due to diffuse transmural intestinal ganglioneuromatosis, are described. Mutation analysis of exon 16 of the RET proto-oncogene revealed germline M918T and thus, a molecular diagnosis of multiple endocrine neoplasia type 2B (MEN 2B). Two infants developed medullary carcinoma of the thyroid. The third had a prophylactic thyroidectomy despite no obvious thyroid masses and normal calcitonin concentrations, but microscopic multifocal medullary carcinoma was found on histological examination. Early recognition of intestinal ganglioneuromatosis with germline RET M918T mutation in pseudo-Hirschsprung's disease is an indication for prophylactic thyroidectomy. (+info)Biological and biochemical properties of Ret with kinase domain mutations identified in multiple endocrine neoplasia type 2B and familial medullary thyroid carcinoma. (2/71)
Several mutations were identified in the kinase domain of the RET proto-oncogene in patients with multiple endocrine neoplasia (MEN) 2B, familial medullary thyroid carcinoma (FMTC) or sporadic medullary thyroid carcinoma. We introduced seven mutations (glutamic acid 768-->aspartic acid (E768D), valine 804-->leucine (V804L), alanine 883-->phenylalanine (A883F), serine 891-->alanine (S891A), methionine 918-->threonine (M918T), alanine 919-->proline (A919P) and E768D/A919P) into the short and long isoforms of RET cDNA and transfected the mutant cDNAs into NIH3T3 cells. The transforming activity of the long isoform of Ret with each mutation was much higher that that of its short isoform. Based on the levels of the transforming activity, these mutant RET genes were classified into two groups; a group with high transforming activity (A883F, M918T and E768D/A919P) and a group with low transforming activity (E768D, V804L, S891A and A919P) (designated high group and low group). Interestingly, the level of transforming activity correlated with clinical phenotypes; high group Ret with the A883F or M918T mutation and low group Ret with the E768D, V804L or S891A mutation were associated with the development of MEN 2B and FMTC, respectively. In addition, we found that substitution of phenylalanine for tyrosine 905 present in the kinase domain abolished both transforming and autophosphorylation activities of low group Ret whereas it did not affect the activities of high group Ret. (+info)Sympathoadrenal hyperplasia causes renal malformations in Ret(MEN2B)-transgenic mice. (3/71)
The tyrosine kinase receptor Ret is expressed in the ureteric bud and is required for normal renal development. Constitutive loss of Ret, its co-receptor gfralpha-1, or the ligand glial cell line-derived neurotrophic factor results in renal agenesis. Transgenic embryos that express a constitutively active form of Ret (Ret(MEN2B)) under the control of the dopamine-beta-hydroxylase (DbetaH) promoter develop profound neuroglial hyperplasia of their sympathetic ganglia and adrenal medullae. Embryos from two independent DbetaH-Ret(MEN2B)-transgenic lines exhibit renal malformations. In contrast with ret-/- embryos, renal maldevelopment in DbetaH-Ret(MEN2B)-transgenic embryos results from primary changes in sympathoadrenal organs extrinsic to the kidney. The ureteric bud invades the metanephric mesenchyme normally, but subsequent bud branching and nephrogenesis are retarded, resulting in severe renal hypoplasia. Ablation of sympathoadrenal precursors restores normal renal growth in vivo and in vitro. We postulate that disruption of renal development results because Ret(MEN2B) derived from the hyperplastic nervous tissue competes with endogenous renal Ret for gfralpha-1 or other signaling components. This hypothesis is supported by the observation that renal malformations, which do not normally occur in a transgenic line with low levels of DbetaH-Ret(MEN2B) expression, arise in a gdnf+/- background. However, renal maldevelopment was not recapitulated in kidneys that were co-cultured with explanted transgenic ganglia in vitro. Our observations illustrate a novel pathogenic mechanism for renal dysgenesis that may explain how putative activating mutations of the RET gene can produce a phenotype usually associated with RET deficiency. (+info)Medullary thyroid carcinoma in Northern Ireland, 1967-1997. (4/71)
BACKGROUND: The experience of managing medullary thyroid carcinoma (MTC) in a specialist endocrine surgery unit was reviewed. METHODS: The case records of 38 patients (19 male, 19 female) treated over a 30 year period were studied. RESULTS: There were 23 (60.5%) patients with sporadic MTC while the remainder had familial MTC--12 multiple endocrine neoplasia (MEN) type 2A, two MEN type 2B, one non-MEN familial medullary thyroid carcinoma (FMTC). Sporadic MTC patients were significantly older at presentation (median 56 years, interquartile range 41.5-61.3 years) compared to MEN 2A patients (median 26 years interquartile range 17.5-34 years) and had more advanced stage of disease. Survival of MTC patients was significantly worse in sporadic disease than in those with MEN 2A (P < 0.0001). All familial cases had bilateral multifocal tumour whereas in sporadic patients only unilateral disease was seen. The availability of genetic testing now allows early identification of affected members of familial MTC kindreds. This has led to total thyroidectomy being performed on the basis of positive genetic screening alone in three patients (two MEN 2A, one FMTC), in all of whom widespread C-cell hyperplasia and microscopic multifocal invasive MTC were identified histologically. CONCLUSIONS: The management of MTC has changed during the study period with total thyroidectomy recommended as the primary procedure of choice for all patients. In the familial setting, positive genetic testing now allows thyroidectomy to be performed at an early pre-clinical stage, with the hope of permanent cure. (+info)Multiple endocrine neoplasia type 2B mutation in human RET oncogene induces medullary thyroid carcinoma in transgenic mice. (5/71)
Multiple endocrine neoplasia type 2B (MEN 2B) is a familial cancer syndrome, in which the cardinal feature is medullary thyroid carcinoma (MTC), a malignant tumor arising from the calcitonin producing thyroid C-cells. MEN 2B is associated with a germline point mutation in the RET proto-oncogene, leading to a Met-->Thr substitution at codon 918 in the kinase domain, which alters the substrate specificity of the protein. We used the human calcitonin gene (CALC-I) promoter to generate transgenic mice expressing either the human RET oncogene with the MEN2B-specific 918 Met-->Thr mutation (CALC-MEN2B-RET) or the human non-mutated RET proto-oncogene (CALC-WT-RET) in the C-cells. At 20 - 22 months of age three out of eight CALC-MEN2B-RET transgenic founders presented with macroscopic bilateral MTC. In two founders nodular C-cell hyperplasia (CCH) was observed. Thyroid abnormalities were never observed in CALC-WT-RET transgenic mice or control non-transgenic mice analysed at this age. In some mice from established CALC-MEN2B-RET transgenic lines nodular CCH was observed from 8 months on whereas MTC was detected in 13% of mice from one CALC-MEN2B-RET line, from the age of 11 months on. These results show for the first time that the MEN2B mutation in the RET oncogene predisposes mice for MTC. (+info)Tissue-specific carcinogenesis in transgenic mice expressing the RET proto-oncogene with a multiple endocrine neoplasia type 2A mutation. (6/71)
Germ line mutations of the RET proto-oncogene are responsible for the development of multiple endocrine neoplasia type 2A (MEN 2A), an inherited cancer syndrome characterized by medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia. To study the mechanism of tissue-specific tumor development by RET with a MEN2A (cysteine 634-->arginine) mutation, we generated transgenic mice by introducing the RET-MEN2A gene fused to Moloney murine leukemia virus long terminal repeat. Expression of the transgene and its product was detected at variable levels in a variety of tissues including thyroid, heart, liver, colon, parotid gland, and brain. All of 29 mice analyzed developed thyroid C-cell hyperplasia or medullary carcinoma, accompanying high levels of serum calcitonin. In addition, development of mammary or parotid gland adenocarcinoma was observed in one-half of the transgenic mice. RET dimerization and its complex formation with Shc and Grb2 adaptor proteins were detected in tumor tissues. Unexpectedly, no tumor formation was found in other tissues despite RET-MEN2A expression where RET dimerization was undetectable. Because these tissues but not tumors expressed glial cell line-derived neurotrophic factor family receptor alpha (GFR alpha) at high levels, this suggested that GFR alpha expression may interfere in the dimerization of the RET-MEN2A mutant proteins, leading to tissue-specific tumor development in vivo. (+info)The Ron oncogenic activity induced by the MEN2B-like substitution overcomes the requirement for the multifunctional docking site. (7/71)
Oncogenic activation of the Ron tyrosine kinase (Macrophage Stimulating Protein receptor) relies on substitutions of two highly conserved residues in the catalytic domain (D1232V and M1254T), which result in ligand-independent activation of the receptor, in vivo tumorigenesis and metastasis. We show here that the Y/F conversion of the Y1317 residue in the kinase domain impairs tumorigenic and metastatic properties of Ron activated by the MEN2B-like mutation (RonM1254T), but not by other two oncogenic substitutions. Furthermore, RonM1254T lacking the multifunctional docking site retains transforming and metastatic activity. These data reveal that the transforming activity of RonM1254T mutant is dependent on Y1317 phosphorylation, suggesting a shift in intramolecular substrate specificity. Consistently, a shift of RonM1254T kinase substrate specificity was observed by in vitro peptide phosphorylation assays and in vivo receptor auto-phosphorylation. The Y1317 phosphorylation elicits by itself activation of PI-3K/Akt and MAPK signalling pathways. Our data indicate that the accomplishment of the full oncogenic phenotype of RonM1254T requires the phosphorylation both of the canonical C-terminal docking site and of the unique Y1317 residue in the tyrosine kinase domain. (+info)Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. (8/71)
OBJECTIVE: The aim of this prospective study is to update our knowledge of the chronology of pheochromocytoma occurrence in multiple endocrine neoplasia type 2 (MEN 2), and to better manage MEN 2 patients after the genetic diagnosis. DESIGN: Eighty-seven non-index gene carrier MEN 2 patients were included in this prospective study: 84 patients with MEN 2A (from 52 families) and 3 with MEN 2B (from 3 families). METHODS: Medullary thyroid carcinoma (MTC) was diagnosed by measuring plasma calcitonin in basal conditions or after pentagastrin stimulation. The search for pheochromocytoma consisted of clinical evaluation, 24 h determination of urinary catecholamines and adrenal imaging. The mean age at genetic diagnosis of MEN 2 was 14.0+/-7.0 years, the mean duration for the follow-up was 7.6+/-2.8 years. RESULTS: All 87 patients had a MTC detected at the same time as the genetic diagnosis was made. Urinary catecholamine measurements led to the diagnosis of pheochromocytoma and a combination of imaging techniques enabled the correct localization of both unilateral or bilateral adrenal involvement. Pheochromocytoma was detected simultaneously with MTC in only seven patients, and seven others were detected throughout the follow-up. Of the 14 patients with pheochromocytoma, 11 had bilateral involvement: nine were initially bilateral and two became so during follow-up. CONCLUSION: This study demonstrates that in MEN 2, MTC is the lesion which appears earliest. Pheochromocytoma develops later during the evolution of the disease, and necessitates regular clinical and biological monitoring throughout follow-up. Determination of urinary and/or plasma catecholamines and metanephrines should be performed to detect pheochromocytoma. Imaging techniques lead to the detection of both unilateral and bilateral pheochromocytoma, thus making video-assisted laparoscopic adrenalectomy possible. (+info)Multiple Endocrine Neoplasia Type 1 (MEN1) is a rare inherited disorder characterized by the development of tumors in various endocrine glands. These tumors can be benign or malignant and may lead to overproduction of hormones, causing a variety of symptoms. The three main endocrine glands affected in MEN1 are:
1. Parathyroid glands: Over 90% of individuals with MEN1 develop multiple parathyroid tumors (parathyroid hyperplasia), leading to primary hyperparathyroidism, which results in high levels of calcium in the blood.
2. Pancreas: Up to 80% of individuals with MEN1 develop pancreatic neuroendocrine tumors (PNETs). These tumors can produce and release various hormones, such as gastrin, insulin, glucagon, and vasoactive intestinal peptide (VIP), leading to specific clinical syndromes like Zollinger-Ellison syndrome, hypoglycemia, or watery diarrhea.
3. Pituitary gland: Approximately 30-40% of individuals with MEN1 develop pituitary tumors, most commonly prolactinomas, which can cause menstrual irregularities, galactorrhea (milk production), and visual field defects.
MEN1 is caused by mutations in the MEN1 gene, located on chromosome 11, and it is inherited in an autosomal dominant manner. This means that a person has a 50% chance of inheriting the disease-causing mutation from an affected parent. The diagnosis of MEN1 typically requires meeting specific clinical criteria or having a positive genetic test for a pathogenic MEN1 gene variant. Regular monitoring and early intervention are crucial in managing this condition to prevent complications and improve outcomes.
Multiple Endocrine Neoplasia Type 2a (MEN 2A) is a rare genetic disorder characterized by the development of tumors in various endocrine glands. It is caused by a mutation in the RET gene. The condition typically involves the following three endocrine glands:
1. Medullary Thyroid Carcinoma (MTC): Almost all patients with MEN 2A develop this type of thyroid cancer, which arises from the parafollicular cells (also known as C cells) of the thyroid gland.
2. Pheochromocytomas: These are tumors that develop in the adrenal glands, usually in the chromaffin cells. They can cause the release of excessive amounts of catecholamines, leading to hypertension and other symptoms. Approximately 50% of MEN 2A patients will develop pheochromocytomas.
3. Primary Parathyroid Hyperplasia or Adenomas: Overactivity of the parathyroid glands can lead to hyperparathyroidism, which results in increased calcium levels in the blood (hypercalcemia). This occurs in about 20% of MEN 2A patients.
MEN 2A is an autosomal dominant disorder, meaning that if one parent has the condition, there is a 50% chance their offspring will inherit the mutated gene and develop the disease. Early detection and treatment of the associated tumors can significantly improve patient outcomes.
Multiple Endocrine Neoplasia Type 2b (MEN 2b) is a rare genetic disorder characterized by the development of tumors in various endocrine glands. It is caused by a mutation in the RET gene. The condition is typically diagnosed in childhood or early adulthood and is often marked by the presence of medullary thyroid carcinoma (MTC), pheochromocytomas, and multiple mucosal neuromas.
MTC is a cancer of the parafollicular cells of the thyroid gland, which can cause overproduction of calcitonin. Pheochromocytomas are tumors that develop in the adrenal glands and can lead to excessive production of catecholamines, resulting in hypertension and other symptoms. Mucosal neuromas are benign growths that occur on the mucous membranes, such as those lining the mouth, tongue, and eyelids.
Individuals with MEN 2b may also develop other features, such as Marfanoid habitus (tall and thin build, long limbs, and flexible joints), gastrointestinal autonomic dysfunction, and megacolon. The condition is inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the mutated gene from an affected parent.
Multiple Endocrine Neoplasia (MEN) is a group of inherited disorders characterized by the development of tumors in various endocrine glands, which can lead to overproduction of hormones. There are two main types: MEN type 1 and MEN type 2.
MEN type 1, also known as Wermer's syndrome, is caused by mutations in the MEN1 gene. It typically involves tumors in the parathyroid glands (leading to hyperparathyroidism), pancreas (often gastrinomas or insulinomas), and pituitary gland. Some individuals may also develop tumors in other organs, such as the adrenal glands, lungs, or thyroid gland.
MEN type 2, which includes MEN type 2A and MEN type 2B, is caused by mutations in the RET gene. MEN type 2A involves medullary thyroid carcinoma (MTC), pheochromocytomas (tumors of the adrenal glands), and parathyroid tumors. MEN type 2B includes MTC, pheochromocytomas, neuromas (nerve tissue tumors), and distinctive physical features such as a marfanoid habitus and mucosal neuromas.
Early detection and management of these tumors are crucial to prevent complications from hormone excess or tumor invasion. Regular screening and monitoring are recommended for individuals with MEN, even if they do not have symptoms. Treatment typically involves surgical removal of the affected glands or tumors, along with medications to manage hormonal imbalances.
Proto-oncogene proteins c-RET are a group of gene products that play crucial roles in the development and functioning of the nervous system, as well as in other tissues. The c-RET proto-oncogene encodes a receptor tyrosine kinase, which is a type of enzyme that helps transmit signals from the outside to the inside of cells. This receptor is activated by binding to its ligands, leading to the activation of various signaling pathways that regulate cell growth, differentiation, and survival.
Mutations in the c-RET proto-oncogene can lead to its overactivation, resulting in the conversion of this gene into an oncogene. Oncogenes are genes that have the potential to cause cancer when they are mutated or abnormally expressed. Activating mutations in c-RET have been implicated in several types of human cancers, including multiple endocrine neoplasia type 2 (MEN2), papillary thyroid carcinoma, and certain types of lung and kidney cancers. These mutations can lead to the constitutive activation of c-RET, resulting in uncontrolled cell growth and tumor formation.
Medullary carcinoma is a type of cancer that develops in the neuroendocrine cells of the thyroid gland. These cells produce hormones that help regulate various bodily functions. Medullary carcinoma is a relatively rare form of thyroid cancer, accounting for about 5-10% of all cases.
Medullary carcinoma is characterized by the presence of certain genetic mutations that cause the overproduction of calcitonin, a hormone produced by the neuroendocrine cells. This overproduction can lead to the formation of tumors in the thyroid gland.
Medullary carcinoma can be hereditary or sporadic. Hereditary forms of the disease are caused by mutations in the RET gene and are often associated with multiple endocrine neoplasia type 2 (MEN 2), a genetic disorder that affects the thyroid gland, adrenal glands, and parathyroid glands. Sporadic forms of medullary carcinoma, on the other hand, are not inherited and occur randomly in people with no family history of the disease.
Medullary carcinoma is typically more aggressive than other types of thyroid cancer and tends to spread (metastasize) to other parts of the body, such as the lymph nodes, lungs, and liver. Symptoms may include a lump or nodule in the neck, difficulty swallowing, hoarseness, and coughing. Treatment options may include surgery, radiation therapy, and chemotherapy. Regular monitoring of calcitonin levels is also recommended to monitor the effectiveness of treatment and detect any recurrence of the disease.
A gastrinoma is a rare type of tumor that originates from the delta cells of the endocrine system, which are typically found in the pancreas and duodenum (the first part of the small intestine). These tumors produce excessive amounts of the hormone gastrin, leading to a condition known as Zollinger-Ellison syndrome.
Zollinger-Ellison syndrome is characterized by severe gastric acid hypersecretion, multiple and/or large peptic ulcers, diarrhea, and gastroesophageal reflux disease (GERD). The excessive gastrin secreted by the gastrinoma stimulates the stomach to produce more acid, which can cause painful ulcers and other digestive issues.
Gastrinomas are often malignant (cancerous) and have a tendency to spread (metastasize) to other parts of the body, such as the liver and lymph nodes. Treatment typically involves surgical removal of the tumor, along with medications to manage acid production and prevent ulcers.
Pheochromocytoma is a rare type of tumor that develops in the adrenal glands, which are triangular-shaped glands located on top of each kidney. These tumors produce excessive amounts of hormones called catecholamines, including adrenaline and noradrenaline. This can lead to a variety of symptoms such as high blood pressure, sweating, headaches, rapid heartbeat, and anxiety.
Pheochromocytomas are typically slow-growing and can be benign or malignant (cancerous). While the exact cause of these tumors is not always known, some genetic factors have been identified that may increase a person's risk. Treatment usually involves surgical removal of the tumor, along with medications to manage symptoms and control blood pressure before and after surgery.
Adrenal gland neoplasms refer to abnormal growths or tumors in the adrenal glands. These glands are located on top of each kidney and are responsible for producing hormones that regulate various bodily functions such as metabolism, blood pressure, and stress response. Adrenal gland neoplasms can be benign (non-cancerous) or malignant (cancerous).
Benign adrenal tumors are called adenomas and are usually small and asymptomatic. However, some adenomas may produce excessive amounts of hormones, leading to symptoms such as high blood pressure, weight gain, and mood changes.
Malignant adrenal tumors are called adrenocortical carcinomas and are rare but aggressive cancers that can spread to other parts of the body. Symptoms of adrenocortical carcinoma may include abdominal pain, weight loss, and hormonal imbalances.
It is important to diagnose and treat adrenal gland neoplasms early to prevent complications and improve outcomes. Diagnostic tests may include imaging studies such as CT scans or MRIs, as well as hormone level testing and biopsy. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Primary hyperparathyroidism is a medical condition characterized by excessive secretion of parathyroid hormone (PTH) from one or more of the parathyroid glands in the neck. These glands are normally responsible for regulating calcium levels in the body by releasing PTH, which helps to maintain an appropriate balance of calcium and phosphate in the bloodstream.
In primary hyperparathyroidism, the parathyroid gland(s) become overactive and produce too much PTH, leading to elevated calcium levels (hypercalcemia) in the blood. This can result in a variety of symptoms, such as fatigue, weakness, bone pain, kidney stones, and cognitive impairment, although some individuals may not experience any symptoms at all.
The most common cause of primary hyperparathyroidism is a benign tumor called an adenoma that develops in one or more of the parathyroid glands. In rare cases, primary hyperparathyroidism can be caused by cancer of the parathyroid gland(s) or by enlargement of all four glands (four-gland hyperplasia). Treatment typically involves surgical removal of the affected parathyroid gland(s), which is usually curative.
Parathyroid neoplasms refer to abnormal growths in the parathyroid glands, which are small endocrine glands located in the neck, near or within the thyroid gland. These neoplasms can be benign (non-cancerous) or malignant (cancerous).
Benign parathyroid neoplasms are typically called parathyroid adenomas and are the most common type of parathyroid disorder. They result in overproduction of parathyroid hormone (PTH), leading to a condition known as primary hyperparathyroidism. Symptoms may include kidney stones, osteoporosis, fatigue, depression, and abdominal pain.
Malignant parathyroid neoplasms are called parathyroid carcinomas. They are rare but more aggressive than adenomas, with a higher risk of recurrence and metastasis. Symptoms are similar to those of benign neoplasms but may also include hoarseness, difficulty swallowing, and enlarged lymph nodes in the neck.
It is important to note that parathyroid neoplasms can only be definitively diagnosed through biopsy or surgical removal and subsequent histopathological examination.
Zollinger-Ellison Syndrome (ZES) is a rare digestive disorder that is characterized by the development of one or more gastrin-secreting tumors, also known as gastrinomas. These tumors are usually found in the pancreas and duodenum (the first part of the small intestine). Gastrinomas produce excessive amounts of the hormone gastrin, which leads to the overproduction of stomach acid.
The increased stomach acid can cause severe peptic ulcers, often multiple or refractory to treatment, in the duodenum and jejunum (the second part of the small intestine). ZES may also result in diarrhea due to the excess acid irritating the intestines. In some cases, gastrinomas can be malignant and metastasize to other organs such as the liver and lymph nodes.
The diagnosis of Zollinger-Ellison Syndrome typically involves measuring serum gastrin levels and performing a secretin stimulation test. Imaging tests like CT scans, MRI, or endoscopic ultrasounds may be used to locate the tumors. Treatment usually includes medications to reduce stomach acid production (such as proton pump inhibitors) and surgery to remove the gastrinomas when possible.
A germ-line mutation is a genetic change that occurs in the egg or sperm cells (gametes), and thus can be passed down from parents to their offspring. These mutations are present throughout the entire body of the offspring, as they are incorporated into the DNA of every cell during embryonic development.
Germ-line mutations differ from somatic mutations, which occur in other cells of the body that are not involved in reproduction. While somatic mutations can contribute to the development of cancer and other diseases within an individual, they are not passed down to future generations.
It's important to note that germ-line mutations can have significant implications for medical genetics and inherited diseases. For example, if a parent has a germ-line mutation in a gene associated with a particular disease, their offspring may have an increased risk of developing that disease as well.
Thyroid neoplasms refer to abnormal growths or tumors in the thyroid gland, which can be benign (non-cancerous) or malignant (cancerous). These growths can vary in size and may cause a noticeable lump or nodule in the neck. Thyroid neoplasms can also affect the function of the thyroid gland, leading to hormonal imbalances and related symptoms. The exact causes of thyroid neoplasms are not fully understood, but risk factors include radiation exposure, family history, and certain genetic conditions. It is important to note that most thyroid nodules are benign, but a proper medical evaluation is necessary to determine the nature of the growth and develop an appropriate treatment plan.
Hyperparathyroidism is a condition in which the parathyroid glands produce excessive amounts of parathyroid hormone (PTH). There are four small parathyroid glands located in the neck, near or within the thyroid gland. They release PTH into the bloodstream to help regulate the levels of calcium and phosphorus in the body.
In hyperparathyroidism, overproduction of PTH can lead to an imbalance in these minerals, causing high blood calcium levels (hypercalcemia) and low phosphate levels (hypophosphatemia). This can result in various symptoms such as fatigue, weakness, bone pain, kidney stones, and cognitive issues.
There are two types of hyperparathyroidism: primary and secondary. Primary hyperparathyroidism occurs when there is a problem with one or more of the parathyroid glands, causing them to become overactive and produce too much PTH. Secondary hyperparathyroidism develops as a response to low calcium levels in the body due to conditions like vitamin D deficiency, chronic kidney disease, or malabsorption syndromes.
Treatment for hyperparathyroidism depends on the underlying cause and severity of symptoms. In primary hyperparathyroidism, surgery to remove the overactive parathyroid gland(s) is often recommended. For secondary hyperparathyroidism, treating the underlying condition and managing calcium levels with medications or dietary changes may be sufficient.
Proto-oncogene proteins are normal cellular proteins that play crucial roles in various cellular processes, such as signal transduction, cell cycle regulation, and apoptosis (programmed cell death). They are involved in the regulation of cell growth, differentiation, and survival under physiological conditions.
When proto-oncogene proteins undergo mutations or aberrations in their expression levels, they can transform into oncogenic forms, leading to uncontrolled cell growth and division. These altered proteins are then referred to as oncogene products or oncoproteins. Oncogenic mutations can occur due to various factors, including genetic predisposition, environmental exposures, and aging.
Examples of proto-oncogene proteins include:
1. Ras proteins: Involved in signal transduction pathways that regulate cell growth and differentiation. Activating mutations in Ras genes are found in various human cancers.
2. Myc proteins: Regulate gene expression related to cell cycle progression, apoptosis, and metabolism. Overexpression of Myc proteins is associated with several types of cancer.
3. EGFR (Epidermal Growth Factor Receptor): A transmembrane receptor tyrosine kinase that regulates cell proliferation, survival, and differentiation. Mutations or overexpression of EGFR are linked to various malignancies, such as lung cancer and glioblastoma.
4. Src family kinases: Intracellular tyrosine kinases that regulate signal transduction pathways involved in cell proliferation, survival, and migration. Dysregulation of Src family kinases is implicated in several types of cancer.
5. Abl kinases: Cytoplasmic tyrosine kinases that regulate various cellular processes, including cell growth, differentiation, and stress responses. Aberrant activation of Abl kinases, as seen in chronic myelogenous leukemia (CML), leads to uncontrolled cell proliferation.
Understanding the roles of proto-oncogene proteins and their dysregulation in cancer development is essential for developing targeted cancer therapies that aim to inhibit or modulate these aberrant signaling pathways.
Pituitary neoplasms refer to abnormal growths or tumors in the pituitary gland, a small endocrine gland located at the base of the brain. These neoplasms can be benign (non-cancerous) or malignant (cancerous), with most being benign. They can vary in size and may cause various symptoms depending on their location, size, and hormonal activity.
Pituitary neoplasms can produce and secrete excess hormones, leading to a variety of endocrine disorders such as Cushing's disease (caused by excessive ACTH production), acromegaly (caused by excessive GH production), or prolactinoma (caused by excessive PRL production). They can also cause local compression symptoms due to their size, leading to headaches, vision problems, and cranial nerve palsies.
The exact causes of pituitary neoplasms are not fully understood, but genetic factors, radiation exposure, and certain inherited conditions may increase the risk of developing these tumors. Treatment options for pituitary neoplasms include surgical removal, radiation therapy, and medical management with drugs that can help control hormonal imbalances.
Insulinoma is a rare type of neuroendocrine tumor that originates from the beta cells of the pancreatic islets (islets of Langerhans). These tumors produce and secrete excessive amounts of insulin, leading to hypoglycemia (low blood sugar levels) even when the person hasn't eaten for a while. Insulinomas are typically slow-growing and benign (noncancerous), but about 10% of them can be malignant (cancerous) and may spread to other parts of the body. Common symptoms include sweating, confusion, dizziness, and weakness due to low blood sugar levels. The diagnosis is often confirmed through imaging tests like CT scans or MRI, and measuring insulin and C-peptide levels in the blood during a fasting test. Treatment usually involves surgical removal of the tumor.
A neuroma is not a specific type of tumor, but rather refers to a benign (non-cancerous) growth or swelling of nerve tissue. The most common type of neuroma is called a Morton's neuroma, which typically occurs between the third and fourth toes in the foot. It develops as a result of chronic irritation, compression, or trauma to the nerves leading to the toes, causing them to thicken and enlarge.
Morton's neuroma can cause symptoms such as pain, numbness, tingling, or burning sensations in the affected area. Treatment options for Morton's neuroma may include rest, ice, orthotics, physical therapy, medication, or in some cases, surgery. It is essential to consult a healthcare professional if you suspect you have a neuroma or are experiencing related symptoms.
Receptor Protein-Tyrosine Kinases (RTKs) are a type of transmembrane receptors found on the cell surface that play a crucial role in signal transduction and regulation of various cellular processes, including cell growth, differentiation, metabolism, and survival. They are called "tyrosine kinases" because they possess an intrinsic enzymatic activity that catalyzes the transfer of a phosphate group from ATP to tyrosine residues on target proteins, thereby modulating their function.
RTKs are composed of three main domains: an extracellular domain that binds to specific ligands (growth factors, hormones, or cytokines), a transmembrane domain that spans the cell membrane, and an intracellular domain with tyrosine kinase activity. Upon ligand binding, RTKs undergo conformational changes that lead to their dimerization or oligomerization, which in turn activates their tyrosine kinase activity. Activated RTKs then phosphorylate specific tyrosine residues on downstream signaling proteins, initiating a cascade of intracellular signaling events that ultimately result in the appropriate cellular response.
Dysregulation of RTK signaling has been implicated in various human diseases, including cancer, diabetes, and developmental disorders. As such, RTKs are important targets for therapeutic intervention in these conditions.
Angiofibroma is a benign tumor that most commonly occurs in the nasopharynx (the upper part of the throat behind the nose) in adolescents and young adults, particularly males. It is composed of blood vessels and fibrous tissue. Angiofibromas are also known as juvenile nasopharyngeal angiofibromas because they often occur in young people and originate in the nasopharynx.
These tumors can cause symptoms such as nosebleeds, nasal congestion, and difficulty breathing through the nose. In some cases, they may also cause hearing problems or double vision. Angiofibromas are typically treated with surgery to remove the tumor. Radiation therapy may also be used in some cases.
It is important to note that angiofibroma is a specific type of tumor that has distinct characteristics and is treated differently from other types of tumors. If you have any concerns about this condition or if you are experiencing symptoms that you think may be related to an angiofibroma, it is important to consult with a healthcare professional for proper diagnosis and treatment.
Pancreatic neoplasms refer to abnormal growths in the pancreas that can be benign or malignant. The pancreas is a gland located behind the stomach that produces hormones and digestive enzymes. Pancreatic neoplasms can interfere with the normal functioning of the pancreas, leading to various health complications.
Benign pancreatic neoplasms are non-cancerous growths that do not spread to other parts of the body. They are usually removed through surgery to prevent any potential complications, such as blocking the bile duct or causing pain.
Malignant pancreatic neoplasms, also known as pancreatic cancer, are cancerous growths that can invade and destroy surrounding tissues and organs. They can also spread (metastasize) to other parts of the body, such as the liver, lungs, or bones. Pancreatic cancer is often aggressive and difficult to treat, with a poor prognosis.
There are several types of pancreatic neoplasms, including adenocarcinomas, neuroendocrine tumors, solid pseudopapillary neoplasms, and cystic neoplasms. The specific type of neoplasm is determined through various diagnostic tests, such as imaging studies, biopsies, and blood tests. Treatment options depend on the type, stage, and location of the neoplasm, as well as the patient's overall health and preferences.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Human chromosome pair 11 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each member of the pair is a single chromosome, and together they contain the genetic material that is inherited from both parents. They are located on the eleventh position in the standard karyotype, which is a visual representation of the 23 pairs of human chromosomes.
Chromosome 11 is one of the largest human chromosomes and contains an estimated 135 million base pairs. It contains approximately 1,400 genes that provide instructions for making proteins, as well as many non-coding RNA molecules that play a role in regulating gene expression.
Chromosome 11 is known to contain several important genes and genetic regions associated with various human diseases and conditions. For example, it contains the Wilms' tumor 1 (WT1) gene, which is associated with kidney cancer in children, and the neurofibromatosis type 1 (NF1) gene, which is associated with a genetic disorder that causes benign tumors to grow on nerves throughout the body. Additionally, chromosome 11 contains the region where the ABO blood group genes are located, which determine a person's blood type.
It's worth noting that human chromosomes come in pairs because they contain two copies of each gene, one inherited from the mother and one from the father. This redundancy allows for genetic diversity and provides a backup copy of essential genes, ensuring their proper function and maintaining the stability of the genome.
Pathologic bone demineralization is a condition characterized by the loss of minerals, such as calcium and phosphate, from the bones. This process makes the bones more porous, weaker, and more susceptible to fractures. It can occur due to various medical conditions, including osteoporosis, hyperparathyroidism, Paget's disease of bone, and cancer that has spread to the bones (metastatic cancer).
In a healthy individual, the body constantly remodels the bones by removing old bone tissue (resorption) and replacing it with new tissue. This process is regulated by two types of cells: osteoclasts, which are responsible for bone resorption, and osteoblasts, which produce new bone tissue. In pathologic bone demineralization, there is an imbalance between the activity of these two cell types, with excessive resorption and inadequate formation of new bone tissue.
Pathologic bone demineralization can lead to a range of symptoms, including bone pain, fractures, loss of height, and a decreased ability to perform daily activities. Treatment for this condition depends on the underlying cause but may include medications that slow down bone resorption or promote bone formation, as well as lifestyle changes such as exercise and dietary modifications.
Thyroidectomy is a surgical procedure where all or part of the thyroid gland is removed. The thyroid gland is a butterfly-shaped endocrine gland located in the neck, responsible for producing hormones that regulate metabolism, growth, and development.
There are different types of thyroidectomy procedures, including:
1. Total thyroidectomy: Removal of the entire thyroid gland.
2. Partial (or subtotal) thyroidectomy: Removal of a portion of the thyroid gland.
3. Hemithyroidectomy: Removal of one lobe of the thyroid gland, often performed to treat benign solitary nodules or differentiated thyroid cancer.
Thyroidectomy may be recommended for various reasons, such as treating thyroid nodules, goiter, hyperthyroidism (overactive thyroid), or thyroid cancer. Potential risks and complications of the procedure include bleeding, infection, damage to nearby structures like the parathyroid glands and recurrent laryngeal nerve, and hypoparathyroidism or hypothyroidism due to removal of or damage to the parathyroid glands or thyroid gland, respectively. Close postoperative monitoring and management are essential to minimize these risks and ensure optimal patient outcomes.
Neuroendocrine tumors (NETs) are a diverse group of neoplasms that arise from cells of the neuroendocrine system, which is composed of dispersed neuroendocrine cells throughout the body, often in close association with nerves and blood vessels. These cells have the ability to produce and secrete hormones or hormone-like substances in response to various stimuli. NETs can occur in a variety of organs, including the lungs, pancreas, small intestine, colon, rectum, stomach, and thyroid gland, as well as in some less common sites such as the thymus, adrenal glands, and nervous system.
NETs can be functional or nonfunctional, depending on whether they produce and secrete hormones or hormone-like substances that cause specific symptoms related to hormonal excess. Functional NETs may give rise to a variety of clinical syndromes, such as carcinoid syndrome, Zollinger-Ellison syndrome, pancreatic neuroendocrine tumor syndrome (also known as Verner-Morrison or WDHA syndrome), and others. Nonfunctional NETs are more likely to present with symptoms related to the size and location of the tumor, such as abdominal pain, intestinal obstruction, or bleeding.
The diagnosis of NETs typically involves a combination of imaging studies, biochemical tests (e.g., measurement of serum hormone levels), and histopathological examination of tissue samples obtained through biopsy or surgical resection. Treatment options depend on the type, location, stage, and grade of the tumor, as well as the presence or absence of functional symptoms. They may include surgery, radiation therapy, chemotherapy, targeted therapy, and/or peptide receptor radionuclide therapy (PRRT).
An adenoma is a benign (noncancerous) tumor that develops from glandular epithelial cells. These types of cells are responsible for producing and releasing fluids, such as hormones or digestive enzymes, into the surrounding tissues. Adenomas can occur in various organs and glands throughout the body, including the thyroid, pituitary, adrenal, and digestive systems.
Depending on their location, adenomas may cause different symptoms or remain asymptomatic. Some common examples of adenomas include:
1. Colorectal adenoma (also known as a polyp): These growths occur in the lining of the colon or rectum and can develop into colorectal cancer if left untreated. Regular screenings, such as colonoscopies, are essential for early detection and removal of these polyps.
2. Thyroid adenoma: This type of adenoma affects the thyroid gland and may result in an overproduction or underproduction of hormones, leading to conditions like hyperthyroidism (overactive thyroid) or hypothyroidism (underactive thyroid).
3. Pituitary adenoma: These growths occur in the pituitary gland, which is located at the base of the brain and controls various hormonal functions. Depending on their size and location, pituitary adenomas can cause vision problems, headaches, or hormonal imbalances that affect growth, reproduction, and metabolism.
4. Liver adenoma: These rare benign tumors develop in the liver and may not cause any symptoms unless they become large enough to press on surrounding organs or structures. In some cases, liver adenomas can rupture and cause internal bleeding.
5. Adrenal adenoma: These growths occur in the adrenal glands, which are located above the kidneys and produce hormones that regulate stress responses, metabolism, and blood pressure. Most adrenal adenomas are nonfunctioning, meaning they do not secrete excess hormones. However, functioning adrenal adenomas can lead to conditions like Cushing's syndrome or Conn's syndrome, depending on the type of hormone being overproduced.
It is essential to monitor and manage benign tumors like adenomas to prevent potential complications, such as rupture, bleeding, or hormonal imbalances. Treatment options may include surveillance with imaging studies, medication to manage hormonal issues, or surgical removal of the tumor in certain cases.
Parathyroidectomy is a surgical procedure for the removal of one or more of the parathyroid glands. These glands are located in the neck and are responsible for producing parathyroid hormone (PTH), which helps regulate the levels of calcium and phosphorus in the body.
Parathyroidectomy is typically performed to treat conditions such as hyperparathyroidism, where one or more of the parathyroid glands become overactive and produce too much PTH. This can lead to high levels of calcium in the blood, which can cause symptoms such as weakness, fatigue, bone pain, kidney stones, and mental confusion.
There are different types of parathyroidectomy procedures, including:
* Partial parathyroidectomy: removal of one or more, but not all, of the parathyroid glands.
* Total parathyroidectomy: removal of all four parathyroid glands.
* Subtotal parathyroidectomy: removal of three and a half of the four parathyroid glands, leaving a small portion of one gland to prevent hypoparathyroidism (a condition where the body produces too little PTH).
The choice of procedure depends on the underlying condition and its severity. After the surgery, patients may need to have their calcium levels monitored and may require calcium and vitamin D supplements to maintain normal calcium levels in the blood.
An islet cell adenoma is a rare, typically benign tumor that develops in the islets of Langerhans, which are clusters of hormone-producing cells in the pancreas. The islets of Langerhans contain several types of cells, including beta cells that produce insulin, alpha cells that produce glucagon, and delta cells that produce somatostatin.
Islet cell adenomas can cause various endocrine disorders depending on the type of hormone-producing cells involved. For example, if the tumor consists mainly of beta cells, it may secrete excessive amounts of insulin, leading to hypoglycemia (low blood sugar). Conversely, if the tumor is composed primarily of alpha cells, it may produce too much glucagon, resulting in hyperglycemia (high blood sugar) and a condition known as glucagonoma.
Islet cell adenomas are usually slow-growing and small but can become quite large in some cases. They are typically diagnosed through imaging tests such as CT scans or MRI, and hormone levels may be measured to determine the type of cells involved. Treatment options include surgical removal of the tumor, medication to manage hormonal imbalances, and, in rare cases, radiofrequency ablation or embolization.
Calcitonin is a hormone that is produced and released by the parafollicular cells (also known as C cells) of the thyroid gland. It plays a crucial role in regulating calcium homeostasis in the body. Specifically, it helps to lower elevated levels of calcium in the blood by inhibiting the activity of osteoclasts, which are bone cells that break down bone tissue and release calcium into the bloodstream. Calcitonin also promotes the uptake of calcium in the bones and increases the excretion of calcium in the urine.
Calcitonin is typically released in response to high levels of calcium in the blood, and its effects help to bring calcium levels back into balance. In addition to its role in calcium regulation, calcitonin may also have other functions in the body, such as modulating immune function and reducing inflammation.
Clinically, synthetic forms of calcitonin are sometimes used as a medication to treat conditions related to abnormal calcium levels, such as hypercalcemia (high blood calcium) or osteoporosis. Calcitonin can be administered as an injection, nasal spray, or oral tablet, depending on the specific formulation and intended use.
Endocrine gland neoplasms refer to abnormal growths (tumors) that develop in the endocrine glands. These glands are responsible for producing hormones, which are chemical messengers that regulate various functions and processes in the body. Neoplasms can be benign or malignant (cancerous). Benign neoplasms tend to grow slowly and do not spread to other parts of the body. Malignant neoplasms, on the other hand, can invade nearby tissues and organs and may also metastasize (spread) to distant sites.
Endocrine gland neoplasms can occur in any of the endocrine glands, including:
1. Pituitary gland: located at the base of the brain, it produces several hormones that regulate growth and development, as well as other bodily functions.
2. Thyroid gland: located in the neck, it produces thyroid hormones that regulate metabolism and calcium balance.
3. Parathyroid glands: located near the thyroid gland, they produce parathyroid hormone that regulates calcium levels in the blood.
4. Adrenal glands: located on top of each kidney, they produce hormones such as adrenaline, cortisol, and aldosterone that regulate stress response, metabolism, and blood pressure.
5. Pancreas: located behind the stomach, it produces insulin and glucagon, which regulate blood sugar levels, and digestive enzymes that help break down food.
6. Pineal gland: located in the brain, it produces melatonin, a hormone that regulates sleep-wake cycles.
7. Gonads (ovaries and testicles): located in the pelvis (ovaries) and scrotum (testicles), they produce sex hormones such as estrogen, progesterone, and testosterone that regulate reproductive function and secondary sexual characteristics.
Endocrine gland neoplasms can cause various symptoms depending on the type and location of the tumor. For example, a pituitary gland neoplasm may cause headaches, vision problems, or hormonal imbalances, while an adrenal gland neoplasm may cause high blood pressure, weight gain, or mood changes.
Diagnosis of endocrine gland neoplasms typically involves a combination of medical history, physical examination, imaging studies such as CT or MRI scans, and laboratory tests to measure hormone levels. Treatment options may include surgery, radiation therapy, chemotherapy, or hormonal therapy, depending on the type and stage of the tumor.
Metanephrine is a catecholamine metabolite, specifically a derivative of epinephrine (adrenaline). It is formed in the body through the metabolic breakdown of epinephrine by the enzyme catechol-O-methyltransferase (COMT). Metanephrines, including metanephrine and normetanephrine, are primarily produced in the adrenal glands but can also be found in other tissues in smaller amounts.
Elevated levels of metanephrines in the blood or urine may indicate a pheochromocytoma, a rare tumor originating from the chromaffin cells of the adrenal medulla, or a paraganglioma, a similar type of tumor located outside the adrenal glands. These tumors can cause excessive production of catecholamines, including epinephrine and norepinephrine, leading to increased metanephrine levels.
It is essential to differentiate between metanephrine and normetanephrine as they have distinct clinical implications. Normetanephrine is a derivative of norepinephrine (noradrenaline), while metanephrine originates from epinephrine. The measurement of both free metanephrines and normetanephrines in plasma or urine is often used to diagnose and monitor pheochromocytomas and paragangliomas.
Duodenal neoplasms refer to abnormal growths in the duodenum, which is the first part of the small intestine that receives digestive secretions from the pancreas and bile duct. These growths can be benign or malignant (cancerous).
Benign neoplasms include adenomas, leiomyomas, lipomas, and hamartomas. They are usually slow-growing and do not spread to other parts of the body. However, they may cause symptoms such as abdominal pain, bleeding, or obstruction of the intestine.
Malignant neoplasms include adenocarcinomas, neuroendocrine tumors (carcinoids), lymphomas, and sarcomas. They are more aggressive and can invade surrounding tissues and spread to other parts of the body. Symptoms may include abdominal pain, weight loss, jaundice, anemia, or bowel obstruction.
The diagnosis of duodenal neoplasms is usually made through imaging tests such as CT scans, MRI, or endoscopy with biopsy. Treatment depends on the type and stage of the tumor and may include surgery, chemotherapy, radiation therapy, or a combination of these modalities.
A carcinoid tumor is a type of slow-growing neuroendocrine tumor that usually originates in the digestive tract, particularly in the small intestine. These tumors can also arise in other areas such as the lungs, appendix, and rarely in other organs. Carcinoid tumors develop from cells of the diffuse endocrine system (also known as the neuroendocrine system) that are capable of producing hormones or biologically active amines.
Carcinoid tumors can produce and release various hormones and bioactive substances, such as serotonin, histamine, bradykinins, prostaglandins, and tachykinins, which can lead to a variety of symptoms. The most common syndrome associated with carcinoid tumors is the carcinoid syndrome, characterized by flushing, diarrhea, abdominal cramping, and wheezing or difficulty breathing.
Carcinoid tumors are typically classified as functional or nonfunctional based on whether they produce and secrete hormones that cause symptoms. Functional carcinoid tumors account for approximately 30% of cases and can lead to the development of carcinoid syndrome, while nonfunctional tumors do not produce significant amounts of hormones and are often asymptomatic until they grow large enough to cause local or distant complications.
Treatment options for carcinoid tumors depend on the location, size, and extent of the tumor, as well as whether it is functional or nonfunctional. Treatment may include surgery, medications (such as somatostatin analogs, chemotherapy, or targeted therapies), and radiation therapy. Regular follow-up with imaging studies and biochemical tests is essential to monitor for recurrence and assess treatment response.
Von Hippel-Lindau (VHL) disease is a rare genetic disorder characterized by the development of tumors and cysts in various parts of the body. It is caused by mutations in the VHL gene, which leads to the abnormal growth of blood vessels, resulting in the formation of these tumors.
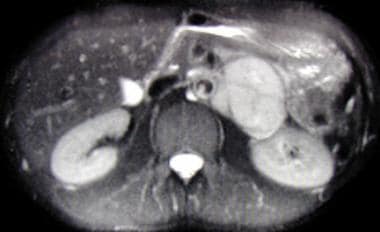
The tumors associated with VHL disease can develop in several organs, including the eyes (in the form of retinal hemangioblastomas), the brain and spinal cord (in the form of cerebellar hemangioblastomas and spinal cord hemangioblastomas), the adrenal glands (in the form of pheochromocytomas or paragangliomas), the kidneys (in the form of clear cell renal cell carcinomas), and the pancreas (in the form of serous cystadenomas or neuroendocrine tumors).
Individuals with VHL disease are at risk for developing multiple tumors over their lifetime, and the severity of the disease can vary widely from person to person. The diagnosis of VHL disease is typically made through genetic testing, family history, and imaging studies to detect the presence of tumors. Treatment may involve surgical removal of the tumors, radiation therapy, or other interventions depending on the location and size of the tumors. Regular monitoring and follow-up are essential for individuals with VHL disease to manage their condition effectively.
Hirschsprung disease is a gastrointestinal disorder that affects the large intestine, specifically the section known as the colon. This condition is congenital, meaning it is present at birth. It occurs due to the absence of ganglion cells (nerve cells) in the bowel's muscular wall, which are responsible for coordinating muscle contractions that move food through the digestive tract.
The affected segment of the colon cannot relax and propel the contents within it, leading to various symptoms such as constipation, intestinal obstruction, or even bowel perforation in severe cases. Common diagnostic methods include rectal suction biopsy, anorectal manometry, and contrast enema studies. Treatment typically involves surgical removal of the aganglionic segment and reattachment of the normal colon to the anus (known as a pull-through procedure).
A glucagonoma is a rare type of neuroendocrine tumor that originates from the alpha cells of the pancreas, where the hormone glucagon is produced. This tumor can lead to an overproduction of glucagon, resulting in a characteristic syndrome known as the "glucagonoma syndrome."
The symptoms of glucagonoma syndrome may include:
1. A distinctive rash called necrolytic migratory erythema, which is characterized by red, swollen, and painful skin lesions that can affect various parts of the body.
2. Weight loss
3. Diabetes or high blood sugar levels (hyperglycemia)
4. Anemia
5. Deep vein thrombosis (blood clots in the deep veins)
6. Depression and confusion
7. A decreased appetite
8. Fatigue and weakness
9. Diarrhea or steatorrhea (fatty stools)
10. High levels of amino acids, fatty acids, and zinc in the blood.
Glucagonomas are typically slow-growing tumors, but they can metastasize (spread) to other organs such as the liver, lymph nodes, and bones. Treatment options for glucagonoma may include surgery to remove the tumor, chemotherapy, targeted therapy, or radiation therapy. Regular follow-up care is essential to monitor the tumor's progression and manage any associated symptoms.
'Drosophila proteins' refer to the proteins that are expressed in the fruit fly, Drosophila melanogaster. This organism is a widely used model system in genetics, developmental biology, and molecular biology research. The study of Drosophila proteins has contributed significantly to our understanding of various biological processes, including gene regulation, cell signaling, development, and aging.
Some examples of well-studied Drosophila proteins include:
1. HSP70 (Heat Shock Protein 70): A chaperone protein involved in protein folding and protection from stress conditions.
2. TUBULIN: A structural protein that forms microtubules, important for cell division and intracellular transport.
3. ACTIN: A cytoskeletal protein involved in muscle contraction, cell motility, and maintenance of cell shape.
4. BETA-GALACTOSIDASE (LACZ): A reporter protein often used to monitor gene expression patterns in transgenic flies.
5. ENDOGLIN: A protein involved in the development of blood vessels during embryogenesis.
6. P53: A tumor suppressor protein that plays a crucial role in preventing cancer by regulating cell growth and division.
7. JUN-KINASE (JNK): A signaling protein involved in stress response, apoptosis, and developmental processes.
8. DECAPENTAPLEGIC (DPP): A member of the TGF-β (Transforming Growth Factor Beta) superfamily, playing essential roles in embryonic development and tissue homeostasis.
These proteins are often studied using various techniques such as biochemistry, genetics, molecular biology, and structural biology to understand their functions, interactions, and regulation within the cell.
Genetic testing is a type of medical test that identifies changes in chromosomes, genes, or proteins. The results of a genetic test can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Genetic tests are performed on a sample of blood, hair, skin, amniotic fluid (the fluid that surrounds a fetus during pregnancy), or other tissue. For example, a physician may recommend genetic testing to help diagnose a genetic condition, confirm the presence of a gene mutation known to increase the risk of developing certain cancers, or determine the chance for a couple to have a child with a genetic disorder.
There are several types of genetic tests, including:
* Diagnostic testing: This type of test is used to identify or confirm a suspected genetic condition in an individual. It may be performed before birth (prenatal testing) or at any time during a person's life.
* Predictive testing: This type of test is used to determine the likelihood that a person will develop a genetic disorder. It is typically offered to individuals who have a family history of a genetic condition but do not show any symptoms themselves.
* Carrier testing: This type of test is used to determine whether a person carries a gene mutation for a genetic disorder. It is often offered to couples who are planning to have children and have a family history of a genetic condition or belong to a population that has an increased risk of certain genetic disorders.
* Preimplantation genetic testing: This type of test is used in conjunction with in vitro fertilization (IVF) to identify genetic changes in embryos before they are implanted in the uterus. It can help couples who have a family history of a genetic disorder or who are at risk of having a child with a genetic condition to conceive a child who is free of the genetic change in question.
* Pharmacogenetic testing: This type of test is used to determine how an individual's genes may affect their response to certain medications. It can help healthcare providers choose the most effective medication and dosage for a patient, reducing the risk of adverse drug reactions.
It is important to note that genetic testing should be performed under the guidance of a qualified healthcare professional who can interpret the results and provide appropriate counseling and support.
The parathyroid glands are four small endocrine glands located in the neck, usually near or behind the thyroid gland. They secrete parathyroid hormone (PTH), which plays a critical role in regulating calcium and phosphate levels in the blood and bones. PTH helps maintain the balance of these minerals by increasing the absorption of calcium from food in the intestines, promoting reabsorption of calcium in the kidneys, and stimulating the release of calcium from bones when needed. Additionally, PTH decreases the excretion of calcium through urine and reduces phosphate reabsorption in the kidneys, leading to increased phosphate excretion. Disorders of the parathyroid glands can result in conditions such as hyperparathyroidism (overactive glands) or hypoparathyroidism (underactive glands), which can have significant impacts on calcium and phosphate homeostasis and overall health.
A ganglioneuroma is a type of benign (noncancerous) tumor that arises from the nerve cells called ganglia in the autonomic nervous system. These tumors typically develop in the abdomen or chest and are most commonly found in children and adolescents, although they can occur at any age.
Ganglioneuromas are composed of mature nerve cells (ganglion cells) and supporting tissue called stroma. They tend to grow slowly and usually do not cause any symptoms unless they become very large or press on nearby organs. In some cases, ganglioneuromas may produce hormones that can cause symptoms such as diarrhea, flushing, or heart palpitations.
While ganglioneuromas are generally benign, there is a small risk that they may become malignant (cancerous) and develop into a type of tumor called a ganglioneuroblastoma or neuroblastoma. For this reason, it is important to monitor these tumors closely and remove them if they grow too large or cause symptoms.
Treatment for ganglioneuromas typically involves surgical removal of the tumor. In some cases, radiation therapy or chemotherapy may also be recommended, particularly if there is a risk of malignant transformation.
A prolactinoma is a type of pituitary tumor that produces an excess amount of the hormone prolactin, leading to various symptoms. The pituitary gland, located at the base of the brain, is responsible for producing and releasing several hormones that regulate different bodily functions. Prolactin is one such hormone, primarily known for its role in stimulating milk production in women during lactation (breastfeeding).
Prolactinoma tumors can be classified into two types: microprolactinomas and macroprolactinomas. Microprolactinomas are smaller tumors, typically less than 10 millimeters in size, while macroprolactinomas are larger tumors, generally greater than 10 millimeters in size.
The overproduction of prolactin caused by these tumors can lead to several clinical manifestations, including:
1. Galactorrhea: Unusual and often spontaneous milk production or leakage from the nipples, which can occur in both men and women who do not have a recent history of pregnancy or breastfeeding.
2. Menstrual irregularities: In women, high prolactin levels can interfere with the normal functioning of other hormones, leading to menstrual irregularities such as infrequent periods (oligomenorrhea) or absent periods (amenorrhea), and sometimes infertility.
3. Sexual dysfunction: In both men and women, high prolactin levels can cause decreased libido and sexual desire. Men may also experience erectile dysfunction and reduced sperm production.
4. Bone loss: Over time, high prolactin levels can lead to decreased bone density and an increased risk of osteoporosis due to the disruption of other hormones that regulate bone health.
5. Headaches and visual disturbances: As the tumor grows, it may put pressure on surrounding structures in the brain, leading to headaches and potential vision problems such as blurred vision or decreased peripheral vision.
Diagnosis typically involves measuring prolactin levels in the blood and performing imaging tests like an MRI (magnetic resonance imaging) scan to assess the size of the tumor. Treatment usually consists of medication to lower prolactin levels, such as dopamine agonists (e.g., bromocriptine or cabergoline), which can also help shrink the tumor. In some cases, surgery may be necessary if medication is ineffective or if the tumor is large and causing severe symptoms.
Paraganglioma is a rare type of tumor that develops in the nervous system, specifically in the paraganglia. Paraganglia are clusters of specialized nerve cells throughout the body that release hormones in response to stress or physical activity. Most paragangliomas are benign (noncancerous), but some can be malignant (cancerous) and may spread to other parts of the body.
Paragangliomas can occur in various locations, including the head and neck region (called "head and neck paragangliomas") or near the spine, abdomen, or chest (called "extra-adrenal paragangliomas"). When they develop in the adrenal glands, which are located on top of each kidney, they are called pheochromocytomas.
Paragangliomas can produce and release hormones such as epinephrine (adrenaline) and norepinephrine, leading to symptoms like high blood pressure, rapid heart rate, sweating, anxiety, and headaches. Treatment typically involves surgical removal of the tumor, along with medications to manage symptoms and control hormone levels before and after surgery.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Proto-oncogenes are normal genes that are present in all cells and play crucial roles in regulating cell growth, division, and death. They code for proteins that are involved in signal transduction pathways that control various cellular processes such as proliferation, differentiation, and survival. When these genes undergo mutations or are activated abnormally, they can become oncogenes, which have the potential to cause uncontrolled cell growth and lead to cancer. Oncogenes can contribute to tumor formation through various mechanisms, including promoting cell division, inhibiting programmed cell death (apoptosis), and stimulating blood vessel growth (angiogenesis).
Loss of Heterozygosity (LOH) is a term used in genetics to describe the loss of one copy of a gene or a segment of a chromosome, where there was previously a pair of different genes or chromosomal segments (heterozygous). This can occur due to various genetic events such as mutation, deletion, or mitotic recombination.
LOH is often associated with the development of cancer, as it can lead to the loss of tumor suppressor genes, which normally help to regulate cell growth and division. When both copies of a tumor suppressor gene are lost or inactivated, it can result in uncontrolled cell growth and the formation of a tumor.
In medical terms, LOH is used as a biomarker for cancer susceptibility, progression, and prognosis. It can also be used to identify individuals who may be at increased risk for certain types of cancer, or to monitor patients for signs of cancer recurrence.
Cyclin-Dependent Kinase Inhibitor p18, also known as CDKN2C or INK4c, is a protein that regulates the cell cycle. It inhibits the activity of cyclin-dependent kinases (CDKs), specifically the CDK4 and CDK6 proteins, which play crucial roles in regulating the progression of the cell cycle.
The p18 protein functions as a tumor suppressor by preventing the phosphorylation and activation of the retinoblastoma protein (pRb) by CDK4/6. When pRb is not phosphorylated, it remains bound to E2F transcription factors, inhibiting their ability to promote the expression of genes required for cell cycle progression.
Mutations or deletions in the CDKN2C gene can lead to uncontrolled cell growth and contribute to tumor development, making p18 an important factor in cancer biology and potential therapeutic target.
A vipoma, also known as a verner morrison syndrome or a non-insulin-secreting pancreatic tumor, is a rare medical condition characterized by the excessive production and secretion of vasoactive intestinal peptides (VIP) from a functional neuroendocrine tumor in the pancreas. This leads to a series of symptoms known as watery diarrhea, hypokalemia, and acidosis (WDHA) syndrome due to the effects of VIP on the gastrointestinal system. Symptoms include severe watery diarrhea, dehydration, electrolyte imbalances, and low blood pressure. Treatment typically involves surgical removal of the tumor, along with supportive care to manage symptoms and correct electrolyte abnormalities.
Human chromosome pair 10 refers to a group of genetic materials that are present in every cell of the human body. Chromosomes are thread-like structures that carry our genes and are located in the nucleus of most cells. They come in pairs, with one set inherited from each parent.
Chromosome pair 10 is one of the 22 autosomal chromosome pairs, meaning they contain genes that are not related to sex determination. Each member of chromosome pair 10 is a single, long DNA molecule that contains thousands of genes and other genetic material.
Chromosome pair 10 is responsible for carrying genetic information that influences various traits and functions in the human body. Some of the genes located on chromosome pair 10 are associated with certain medical conditions, such as hereditary breast and ovarian cancer syndrome, neurofibromatosis type 1, and Waardenburg syndrome type 2A.
It's important to note that while chromosomes carry genetic information, not all variations in the DNA sequence will result in a change in phenotype or function. Some variations may have no effect at all, while others may lead to changes in how proteins are made and function, potentially leading to disease or other health issues.
Normetanephrine is defined as a major metabolite of epinephrine (adrenaline), which is formed by the action of catechol-O-methyltransferase (COMT) on metanephrine. It is primarily produced in the adrenal gland and is also found in the sympathetic nervous system. Normetanephrine is often measured in clinical testing to help diagnose pheochromocytoma, a rare tumor of the adrenal glands that can cause high blood pressure and other symptoms due to excessive production of catecholamines. Increased levels of normetanephrine in the urine or plasma may indicate the presence of a pheochromocytoma or other conditions associated with increased catecholamine release.
DNA Mutational Analysis is a laboratory test used to identify genetic variations or changes (mutations) in the DNA sequence of a gene. This type of analysis can be used to diagnose genetic disorders, predict the risk of developing certain diseases, determine the most effective treatment for cancer, or assess the likelihood of passing on an inherited condition to offspring.
The test involves extracting DNA from a patient's sample (such as blood, saliva, or tissue), amplifying specific regions of interest using polymerase chain reaction (PCR), and then sequencing those regions to determine the precise order of nucleotide bases in the DNA molecule. The resulting sequence is then compared to reference sequences to identify any variations or mutations that may be present.
DNA Mutational Analysis can detect a wide range of genetic changes, including single-nucleotide polymorphisms (SNPs), insertions, deletions, duplications, and rearrangements. The test is often used in conjunction with other diagnostic tests and clinical evaluations to provide a comprehensive assessment of a patient's genetic profile.
It is important to note that not all mutations are pathogenic or associated with disease, and the interpretation of DNA Mutational Analysis results requires careful consideration of the patient's medical history, family history, and other relevant factors.
Hereditary neoplastic syndromes refer to genetic disorders that predispose affected individuals to develop tumors or cancers. These syndromes are caused by inherited mutations in specific genes that regulate cell growth and division. As a result, cells may divide and grow uncontrollably, leading to the formation of benign or malignant tumors.
Examples of hereditary neoplastic syndromes include:
1. Hereditary breast and ovarian cancer syndrome (HBOC): This syndrome is caused by mutations in the BRCA1 or BRCA2 genes, which increase the risk of developing breast, ovarian, and other cancers.
2. Lynch syndrome: Also known as hereditary non-polyposis colorectal cancer (HNPCC), this syndrome is caused by mutations in DNA mismatch repair genes, leading to an increased risk of colon, endometrial, and other cancers.
3. Li-Fraumeni syndrome: This syndrome is caused by mutations in the TP53 gene, which increases the risk of developing a wide range of cancers, including breast, brain, and soft tissue sarcomas.
4. Familial adenomatous polyposis (FAP): This syndrome is caused by mutations in the APC gene, leading to the development of numerous colon polyps that can become cancerous if not removed.
5. Neurofibromatosis type 1 (NF1): This syndrome is caused by mutations in the NF1 gene and is characterized by the development of benign tumors called neurofibromas on the nerves and skin.
6. Von Hippel-Lindau disease (VHL): This syndrome is caused by mutations in the VHL gene, leading to an increased risk of developing various types of tumors, including kidney, pancreas, and adrenal gland tumors.
Individuals with hereditary neoplastic syndromes often have a higher risk of developing cancer than the general population, and they may require more frequent screening and surveillance to detect cancers at an early stage when they are more treatable.
A missense mutation is a type of point mutation in which a single nucleotide change results in the substitution of a different amino acid in the protein that is encoded by the affected gene. This occurs when the altered codon (a sequence of three nucleotides that corresponds to a specific amino acid) specifies a different amino acid than the original one. The function and/or stability of the resulting protein may be affected, depending on the type and location of the missense mutation. Missense mutations can have various effects, ranging from benign to severe, depending on the importance of the changed amino acid for the protein's structure or function.
A lipoma is a common, benign (non-cancerous) soft tissue growth. It is composed of adipose or fatty tissue and typically found just beneath the skin, but they can also occur deeper within the body. Lipomas are usually round, moveable, and painless, although they may cause discomfort if they grow large enough to put pressure on nearby nerves or if they're located in a sensitive area. They generally grow slowly over time. Surgical removal is an option if the lipoma becomes bothersome or grows significantly in size. It's important to note that while lipomas are typically harmless, any new lumps or bumps should be evaluated by a healthcare professional to confirm the diagnosis and rule out other more serious conditions.
Acromegaly is a rare hormonal disorder that typically occurs in middle-aged adults. It results from the pituitary gland producing too much growth hormone (GH) during adulthood. The excessive production of GH leads to abnormal growth of body tissues, particularly in the hands, feet, and face.
The term "acromegaly" is derived from two Greek words: "akros," meaning extremities, and "megaly," meaning enlargement. In most cases, acromegaly is caused by a benign tumor (adenoma) of the pituitary gland, which results in overproduction of GH.
Common symptoms include enlarged hands and feet, coarse facial features, deepened voice, joint pain, and sweating. If left untreated, acromegaly can lead to serious complications such as diabetes, hypertension, heart disease, and arthritis. Treatment usually involves surgical removal of the tumor, radiation therapy, or medication to control GH production.
Parathyroid diseases refer to conditions that affect the parathyroid glands, which are small endocrine glands located in the neck, near or attached to the back surface of the thyroid gland. The primary function of the parathyroid glands is to produce and secrete parathyroid hormone (PTH), a crucial hormone that helps regulate calcium and phosphorus levels in the blood and bones.
There are four parathyroid glands, and they can develop various diseases, including:
1. Hyperparathyroidism: A condition where one or more parathyroid glands produce excessive amounts of PTH. This can lead to an imbalance in calcium and phosphorus levels, resulting in symptoms such as fatigue, weakness, bone pain, kidney stones, and increased risk of osteoporosis. Hyperparathyroidism can be primary (caused by a benign or malignant tumor in the parathyroid gland), secondary (due to chronic kidney disease or vitamin D deficiency), or tertiary (when secondary hyperparathyroidism becomes autonomous and continues even after correcting the underlying cause).
2. Hypoparathyroidism: A condition where the parathyroid glands do not produce enough PTH, leading to low calcium levels in the blood (hypocalcemia) and high phosphorus levels (hyperphosphatemia). Symptoms of hypoparathyroidism may include muscle spasms, tingling sensations in the fingers, toes, or lips, anxiety, cataracts, and seizures. Hypoparathyroidism can be caused by surgical removal of the parathyroid glands, autoimmune disorders, radiation therapy, or genetic conditions.
3. Parathyroid tumors: Abnormal growths in the parathyroid glands can lead to hyperparathyroidism. Benign tumors (adenomas) are the most common cause of primary hyperparathyroidism. Malignant tumors (carcinomas) are rare but can also occur, leading to more severe symptoms and a worse prognosis.
4. Parathyroid dysfunction in genetic disorders: Some genetic syndromes, such as multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2A (MEN2A), and hyperparathyroidism-jaw tumor syndrome (HPT-JT), can involve parathyroid gland abnormalities, leading to hyperparathyroidism or other related conditions.
Proper diagnosis and management of parathyroid disorders are crucial for maintaining optimal calcium homeostasis and preventing complications associated with hypocalcemia or hypercalcemia. Treatment options may include surgery, medication, dietary modifications, and monitoring hormone levels.
Single-Stranded Conformational Polymorphism (SSCP) is not a medical condition but rather a laboratory technique used in molecular biology and genetics. It refers to the phenomenon where a single-stranded DNA or RNA molecule can adopt different conformations or shapes based on its nucleotide sequence, even if the difference in the sequence is as small as a single base pair change. This property is used in SSCP analysis to detect mutations or variations in DNA or RNA sequences.
In SSCP analysis, the denatured single-stranded DNA or RNA sample is subjected to electrophoresis on a non-denaturing polyacrylamide gel. The different conformations of the single-stranded molecules migrate at different rates in the gel, creating multiple bands that can be visualized by staining or other detection methods. The presence of additional bands or shifts in band patterns can indicate the presence of a sequence variant or mutation.
SSCP analysis is often used as a screening tool for genetic diseases, cancer, and infectious diseases to identify genetic variations associated with these conditions. However, it has largely been replaced by more sensitive and accurate methods such as next-generation sequencing.
A neoplasm is a tumor or growth that is formed by an abnormal and excessive proliferation of cells, which can be benign or malignant. Neoplasm proteins are therefore any proteins that are expressed or produced in these neoplastic cells. These proteins can play various roles in the development, progression, and maintenance of neoplasms.
Some neoplasm proteins may contribute to the uncontrolled cell growth and division seen in cancer, such as oncogenic proteins that promote cell cycle progression or inhibit apoptosis (programmed cell death). Others may help the neoplastic cells evade the immune system, allowing them to proliferate undetected. Still others may be involved in angiogenesis, the formation of new blood vessels that supply the tumor with nutrients and oxygen.
Neoplasm proteins can also serve as biomarkers for cancer diagnosis, prognosis, or treatment response. For example, the presence or level of certain neoplasm proteins in biological samples such as blood or tissue may indicate the presence of a specific type of cancer, help predict the likelihood of cancer recurrence, or suggest whether a particular therapy will be effective.
Overall, understanding the roles and behaviors of neoplasm proteins can provide valuable insights into the biology of cancer and inform the development of new diagnostic and therapeutic strategies.
A pancreatectomy is a surgical procedure in which all or part of the pancreas is removed. There are several types of pancreatectomies, including:
* **Total pancreatectomy:** Removal of the entire pancreas, as well as the spleen and nearby lymph nodes. This type of pancreatectomy is usually done for patients with cancer that has spread throughout the pancreas or for those who have had multiple surgeries to remove pancreatic tumors.
* **Distal pancreatectomy:** Removal of the body and tail of the pancreas, as well as nearby lymph nodes. This type of pancreatectomy is often done for patients with tumors in the body or tail of the pancreas.
* **Partial (or segmental) pancreatectomy:** Removal of a portion of the head or body of the pancreas, as well as nearby lymph nodes. This type of pancreatectomy is often done for patients with tumors in the head or body of the pancreas that can be removed without removing the entire organ.
* **Pylorus-preserving pancreaticoduodenectomy (PPPD):** A type of surgery used to treat tumors in the head of the pancreas, as well as other conditions such as chronic pancreatitis. In this procedure, the head of the pancreas, duodenum, gallbladder, and bile duct are removed, but the stomach and lower portion of the esophagus (pylorus) are left in place.
After a pancreatectomy, patients may experience problems with digestion and blood sugar regulation, as the pancreas plays an important role in these functions. Patients may need to take enzyme supplements to help with digestion and may require insulin therapy to manage their blood sugar levels.
A point mutation is a type of genetic mutation where a single nucleotide base (A, T, C, or G) in DNA is altered, deleted, or substituted with another nucleotide. Point mutations can have various effects on the organism, depending on the location of the mutation and whether it affects the function of any genes. Some point mutations may not have any noticeable effect, while others might lead to changes in the amino acids that make up proteins, potentially causing diseases or altering traits. Point mutations can occur spontaneously due to errors during DNA replication or be inherited from parents.
Gastrins are a group of hormones that are produced by G cells in the stomach lining. These hormones play an essential role in regulating gastric acid secretion and motor functions of the gastrointestinal tract. The most well-known gastrin is known as "gastrin-17," which is released into the bloodstream and stimulates the release of hydrochloric acid from parietal cells in the stomach lining.
Gastrins are stored in secretory granules within G cells, and their release is triggered by several factors, including the presence of food in the stomach, gastrin-releasing peptide (GRP), and vagus nerve stimulation. Once released, gastrins bind to specific receptors on parietal cells, leading to an increase in intracellular calcium levels and the activation of enzymes that promote hydrochloric acid secretion.
Abnormalities in gastrin production can lead to several gastrointestinal disorders, including gastrinomas (tumors that produce excessive amounts of gastrin), which can cause severe gastric acid hypersecretion and ulcers. Conversely, a deficiency in gastrin production can result in hypochlorhydria (low stomach acid levels) and impaired digestion.
Adrenalectomy is a surgical procedure in which one or both adrenal glands are removed. The adrenal glands are small, triangular-shaped glands located on top of each kidney that produce hormones such as cortisol, aldosterone, and adrenaline (epinephrine).
There are several reasons why an adrenalectomy may be necessary. For example, the procedure may be performed to treat tumors or growths on the adrenal glands, such as pheochromocytomas, which can cause high blood pressure and other symptoms. Adrenalectomy may also be recommended for patients with Cushing's syndrome, a condition in which the body is exposed to too much cortisol, or for those with adrenal cancer.
During an adrenalectomy, the surgeon makes an incision in the abdomen or back and removes the affected gland or glands. In some cases, laparoscopic surgery may be used, which involves making several small incisions and using specialized instruments to remove the gland. After the procedure, patients may need to take hormone replacement therapy to compensate for the loss of adrenal gland function.
A heterozygote is an individual who has inherited two different alleles (versions) of a particular gene, one from each parent. This means that the individual's genotype for that gene contains both a dominant and a recessive allele. The dominant allele will be expressed phenotypically (outwardly visible), while the recessive allele may or may not have any effect on the individual's observable traits, depending on the specific gene and its function. Heterozygotes are often represented as 'Aa', where 'A' is the dominant allele and 'a' is the recessive allele.
Multiple primary neoplasms refer to the occurrence of more than one primary malignant tumor in an individual, where each tumor is unrelated to the other and originates from separate cells or organs. This differs from metastatic cancer, where a single malignancy spreads to multiple sites in the body. Multiple primary neoplasms can be synchronous (occurring at the same time) or metachronous (occurring at different times). The risk of developing multiple primary neoplasms increases with age and is associated with certain genetic predispositions, environmental factors, and lifestyle choices such as smoking and alcohol consumption.
Hyperplasia is a medical term that refers to an abnormal increase in the number of cells in an organ or tissue, leading to an enlargement of the affected area. It's a response to various stimuli such as hormones, chronic irritation, or inflammation. Hyperplasia can be physiological, like the growth of breast tissue during pregnancy, or pathological, like in the case of benign or malignant tumors. The process is generally reversible if the stimulus is removed. It's important to note that hyperplasia itself is not cancerous, but some forms of hyperplasia can increase the risk of developing cancer over time.
Thymus neoplasms are abnormal growths in the thymus gland that result from uncontrolled cell division. The term "neoplasm" refers to any new and abnormal growth of tissue, also known as a tumor. Thymus neoplasms can be benign or malignant (cancerous).
Malignant thymus neoplasms are called thymomas or thymic carcinomas. Thymomas are the most common type and tend to grow slowly, invading nearby tissues and organs. They can also spread (metastasize) to other parts of the body. Thymic carcinomas are rarer and more aggressive, growing and spreading more quickly than thymomas.
Symptoms of thymus neoplasms may include coughing, chest pain, difficulty breathing, or swelling in the neck or upper chest. Treatment options for thymus neoplasms depend on the type, size, location, and stage of the tumor, as well as the patient's overall health. Treatment may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Tumor suppressor genes are a type of gene that helps to regulate and prevent cells from growing and dividing too rapidly or in an uncontrolled manner. They play a critical role in preventing the formation of tumors and cancer. When functioning properly, tumor suppressor genes help to repair damaged DNA, control the cell cycle, and trigger programmed cell death (apoptosis) when necessary. However, when these genes are mutated or altered, they can lose their ability to function correctly, leading to uncontrolled cell growth and the development of tumors. Examples of tumor suppressor genes include TP53, BRCA1, and BRCA2.
A Growth Hormone-Secreting Pituitary Adenoma (GH-secreting pituitary adenoma, or GHoma) is a type of benign tumor that develops in the pituitary gland and results in excessive production of growth hormone (GH). This leads to a condition known as acromegaly if it occurs in adults, or gigantism if it occurs in children before the closure of the growth plates.
Symptoms of GH-secreting pituitary adenoma may include:
1. Coarsening of facial features
2. Enlargement of hands and feet
3. Deepened voice due to thickening of vocal cords
4. Increased sweating and body odor
5. Joint pain and stiffness
6. Sleep apnea
7. Fatigue, weakness, or muscle wasting
8. Headaches
9. Vision problems
10. Irregular menstrual periods in women
11. Erectile dysfunction in men
Diagnosis typically involves measuring the levels of GH and insulin-like growth factor 1 (IGF-1) in the blood, along with imaging tests like MRI or CT scans to locate and characterize the tumor. Treatment options include surgical removal of the tumor, radiation therapy, and medication to control GH production. Regular follow-ups are necessary to monitor for potential recurrence.
Exons are the coding regions of DNA that remain in the mature, processed mRNA after the removal of non-coding intronic sequences during RNA splicing. These exons contain the information necessary to encode proteins, as they specify the sequence of amino acids within a polypeptide chain. The arrangement and order of exons can vary between different genes and even between different versions of the same gene (alternative splicing), allowing for the generation of multiple protein isoforms from a single gene. This complexity in exon structure and usage significantly contributes to the diversity and functionality of the proteome.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
Pancreaticoduodenectomy, also known as the Whipple procedure, is a complex surgical operation that involves the removal of the head of the pancreas, the duodenum (the first part of the small intestine), the gallbladder, and the distal common bile duct. In some cases, a portion of the stomach may also be removed. The remaining parts of the pancreas, bile duct, and intestines are then reconnected to allow for the digestion of food and drainage of bile.
This procedure is typically performed as a treatment for various conditions affecting the pancreas, such as tumors (including pancreatic cancer), chronic pancreatitis, or traumatic injuries. It is a major surgical operation that requires significant expertise and experience to perform safely and effectively.
Hypercalcemia is a medical condition characterized by an excess of calcium ( Ca2+ ) in the blood. While the normal range for serum calcium levels is typically between 8.5 to 10.2 mg/dL (milligrams per deciliter) or 2.14 to 2.55 mmol/L (millimoles per liter), hypercalcemia is generally defined as a serum calcium level greater than 10.5 mg/dL or 2.6 mmol/L.
Hypercalcemia can result from various underlying medical disorders, including primary hyperparathyroidism, malignancy (cancer), certain medications, granulomatous diseases, and excessive vitamin D intake or production. Symptoms of hypercalcemia may include fatigue, weakness, confusion, memory loss, depression, constipation, nausea, vomiting, increased thirst, frequent urination, bone pain, and kidney stones. Severe or prolonged hypercalcemia can lead to serious complications such as kidney failure, cardiac arrhythmias, and calcification of soft tissues. Treatment depends on the underlying cause and severity of the condition.
Polymerase Chain Reaction (PCR) is a laboratory technique used to amplify specific regions of DNA. It enables the production of thousands to millions of copies of a particular DNA sequence in a rapid and efficient manner, making it an essential tool in various fields such as molecular biology, medical diagnostics, forensic science, and research.
The PCR process involves repeated cycles of heating and cooling to separate the DNA strands, allow primers (short sequences of single-stranded DNA) to attach to the target regions, and extend these primers using an enzyme called Taq polymerase, resulting in the exponential amplification of the desired DNA segment.
In a medical context, PCR is often used for detecting and quantifying specific pathogens (viruses, bacteria, fungi, or parasites) in clinical samples, identifying genetic mutations or polymorphisms associated with diseases, monitoring disease progression, and evaluating treatment effectiveness.
The term "DNA, neoplasm" is not a standard medical term or concept. DNA refers to deoxyribonucleic acid, which is the genetic material present in the cells of living organisms. A neoplasm, on the other hand, is a tumor or growth of abnormal tissue that can be benign (non-cancerous) or malignant (cancerous).
In some contexts, "DNA, neoplasm" may refer to genetic alterations found in cancer cells. These genetic changes can include mutations, amplifications, deletions, or rearrangements of DNA sequences that contribute to the development and progression of cancer. Identifying these genetic abnormalities can help doctors diagnose and treat certain types of cancer more effectively.
However, it's important to note that "DNA, neoplasm" is not a term that would typically be used in medical reports or research papers without further clarification. If you have any specific questions about DNA changes in cancer cells or neoplasms, I would recommend consulting with a healthcare professional or conducting further research on the topic.
A codon is a sequence of three adjacent nucleotides in DNA or RNA that specifies the insertion of a particular amino acid during protein synthesis, or signals the beginning or end of translation. In DNA, these triplets are read during transcription to produce a complementary mRNA molecule, which is then translated into a polypeptide chain during translation. There are 64 possible codons in the standard genetic code, with 61 encoding for specific amino acids and three serving as stop codons that signal the termination of protein synthesis.
Parathyroid hormone (PTH) is a polypeptide hormone that plays a crucial role in the regulation of calcium and phosphate levels in the body. It is produced and secreted by the parathyroid glands, which are four small endocrine glands located on the back surface of the thyroid gland.
The primary function of PTH is to maintain normal calcium levels in the blood by increasing calcium absorption from the gut, mobilizing calcium from bones, and decreasing calcium excretion by the kidneys. PTH also increases phosphate excretion by the kidneys, which helps to lower serum phosphate levels.
In addition to its role in calcium and phosphate homeostasis, PTH has been shown to have anabolic effects on bone tissue, stimulating bone formation and preventing bone loss. However, chronic elevations in PTH levels can lead to excessive bone resorption and osteoporosis.
Overall, Parathyroid Hormone is a critical hormone that helps maintain mineral homeostasis and supports healthy bone metabolism.
The thyroid gland is a major endocrine gland located in the neck, anterior to the trachea and extends from the lower third of the Adams apple to the suprasternal notch. It has two lateral lobes, connected by an isthmus, and sometimes a pyramidal lobe. This gland plays a crucial role in the metabolism, growth, and development of the human body through the production of thyroid hormones (triiodothyronine/T3 and thyroxine/T4) and calcitonin. The thyroid hormones regulate body temperature, heart rate, and the production of protein, while calcitonin helps in controlling calcium levels in the blood. The function of the thyroid gland is controlled by the hypothalamus and pituitary gland through the thyroid-stimulating hormone (TSH).
Thymectomy is a surgical procedure that involves the removal of the thymus gland. The thymus gland is a part of the immune system located in the upper chest, behind the sternum (breastbone), and above the heart. It is responsible for producing white blood cells called T-lymphocytes, which help fight infections.
Thymectomy is often performed as a treatment option for patients with certain medical conditions, such as:
* Myasthenia gravis: an autoimmune disorder that causes muscle weakness and fatigue. In some cases, the thymus gland may contain abnormal cells that contribute to the development of myasthenia gravis. Removing the thymus gland can help improve symptoms in some patients with this condition.
* Thymomas: tumors that develop in the thymus gland. While most thymomas are benign (non-cancerous), some can be malignant (cancerous) and may require surgical removal.
* Myasthenic syndrome: a group of disorders characterized by muscle weakness and fatigue, similar to myasthenia gravis. In some cases, the thymus gland may be abnormal and contribute to the development of these conditions. Removing the thymus gland can help improve symptoms in some patients.
Thymectomy can be performed using various surgical approaches, including open surgery (through a large incision in the chest), video-assisted thoracoscopic surgery (VATS, using small incisions and a camera to guide the procedure), or robotic-assisted surgery (using a robot to perform the procedure through small incisions). The choice of surgical approach depends on several factors, including the size and location of the thymus gland, the patient's overall health, and the surgeon's expertise.
Heterozygote detection is a method used in genetics to identify individuals who carry one normal and one mutated copy of a gene. These individuals are known as heterozygotes and they do not typically show symptoms of the genetic disorder associated with the mutation, but they can pass the mutated gene on to their offspring, who may then be affected.
Heterozygote detection is often used in genetic counseling and screening programs for recessive disorders such as cystic fibrosis or sickle cell anemia. By identifying heterozygotes, individuals can be informed of their carrier status and the potential risks to their offspring. This information can help them make informed decisions about family planning and reproductive options.
Various methods can be used for heterozygote detection, including polymerase chain reaction (PCR) based tests, DNA sequencing, and genetic linkage analysis. The choice of method depends on the specific gene or mutation being tested, as well as the availability and cost of the testing technology.
The endocrine system is a complex network of glands and organs that produce, store, and secrete hormones. It plays a crucial role in regulating various functions in the body, including metabolism, growth and development, tissue function, sexual function, reproduction, sleep, and mood.
Endocrine system diseases or disorders occur when there is a problem with the production or regulation of hormones. This can result from:
1. Overproduction or underproduction of hormones by the endocrine glands.
2. Impaired response of target cells to hormones.
3. Disruption in the feedback mechanisms that regulate hormone production.
Examples of endocrine system diseases include:
1. Diabetes Mellitus - a group of metabolic disorders characterized by high blood sugar levels due to insulin deficiency or resistance.
2. Hypothyroidism - underactive thyroid gland leading to slow metabolism, weight gain, fatigue, and depression.
3. Hyperthyroidism - overactive thyroid gland causing rapid heartbeat, anxiety, weight loss, and heat intolerance.
4. Cushing's Syndrome - excess cortisol production resulting in obesity, high blood pressure, and weak muscles.
5. Addison's Disease - insufficient adrenal hormone production leading to weakness, fatigue, and low blood pressure.
6. Acromegaly - overproduction of growth hormone after puberty causing enlargement of bones, organs, and soft tissues.
7. Gigantism - similar to acromegaly but occurs before puberty resulting in excessive height and body size.
8. Hypopituitarism - underactive pituitary gland leading to deficiencies in various hormones.
9. Hyperparathyroidism - overactivity of the parathyroid glands causing calcium imbalances and kidney stones.
10. Precocious Puberty - early onset of puberty due to premature activation of the pituitary gland.
Treatment for endocrine system diseases varies depending on the specific disorder and may involve medication, surgery, lifestyle changes, or a combination of these approaches.
Octreotide is a synthetic analogue of the natural hormone somatostatin, which is used in medical treatment. It is a octapeptide with similar effects to somatostatin, but with a longer duration of action. Octreotide is primarily used in the management of acromegaly, gastroenteropancreatic neuroendocrine tumors (GEP-NETs), and diarrhea and flushing associated with carcinoid syndrome.
It works by inhibiting the release of several hormones, including growth hormone, insulin, glucagon, and gastrin. This results in a decrease in symptoms caused by excessive hormone secretion, such as reduced growth hormone levels in acromegaly, decreased tumor size in some GEP-NETs, and improved diarrhea and flushing in carcinoid syndrome.
Octreotide is available in several forms, including short-acting subcutaneous injections (Sandostatin®), long-acting depot intramuscular injections (Sandostatin LAR®), and a slow-release formulation for the treatment of diarrhea associated with AIDS (Mycapssa™).
The medical definition of Octreotide is:
A synthetic octapeptide analogue of somatostatin, used in the management of acromegaly, gastroenteropancreatic neuroendocrine tumors (GEP-NETs), and diarrhea and flushing associated with carcinoid syndrome. Octreotide inhibits the release of several hormones, including growth hormone, insulin, glucagon, and gastrin, leading to symptomatic improvement in these conditions. It is available as short-acting subcutaneous injections, long-acting depot intramuscular injections, and a slow-release formulation for diarrhea associated with AIDS.
Endocrine glands are ductless glands in the human body that release hormones directly into the bloodstream, which then carry the hormones to various tissues and organs in the body. These glands play a crucial role in regulating many of the body's functions, including metabolism, growth and development, tissue function, sexual function, reproduction, sleep, and mood.
Examples of endocrine glands include the pituitary gland, thyroid gland, parathyroid glands, adrenal glands, pineal gland, pancreas, ovaries, and testes. Each of these glands produces specific hormones that have unique effects on various target tissues in the body.
The endocrine system works closely with the nervous system to regulate many bodily functions through a complex network of feedback mechanisms. Disorders of the endocrine system can result in a wide range of symptoms and health problems, including diabetes, thyroid disease, growth disorders, and sexual dysfunction.
The endocrine system is a complex network of glands and organs that produce, store, and secrete hormones. It plays a crucial role in regulating various functions and processes in the body, including metabolism, growth and development, tissue function, sexual function, reproduction, sleep, and mood.
The major endocrine glands include:
1. Pituitary gland: located at the base of the brain, it is often referred to as the "master gland" because it controls other glands' functions. It produces and releases several hormones that regulate growth, development, and reproduction.
2. Thyroid gland: located in the neck, it produces hormones that regulate metabolism, growth, and development.
3. Parathyroid glands: located near the thyroid gland, they produce parathyroid hormone, which regulates calcium levels in the blood.
4. Adrenal glands: located on top of the kidneys, they produce hormones that regulate stress response, metabolism, and blood pressure.
5. Pancreas: located in the abdomen, it produces hormones such as insulin and glucagon that regulate blood sugar levels.
6. Sex glands (ovaries and testes): they produce sex hormones such as estrogen, progesterone, and testosterone that regulate sexual development and reproduction.
7. Pineal gland: located in the brain, it produces melatonin, a hormone that regulates sleep-wake cycles.
The endocrine system works closely with the nervous system to maintain homeostasis or balance in the body's internal environment. Hormones are chemical messengers that travel through the bloodstream to target cells or organs, where they bind to specific receptors and elicit a response. Disorders of the endocrine system can result from overproduction or underproduction of hormones, leading to various health problems such as diabetes, thyroid disorders, growth disorders, and sexual dysfunction.
Cyclin-Dependent Kinase Inhibitor p27, also known as CDKN1B or p27Kip1, is a protein that regulates the cell cycle. It inhibits the activity of certain cyclin-dependent kinases (CDKs), which are enzymes that play key roles in regulating the progression of the cell cycle.
The cell cycle is a series of events that cells undergo as they grow and divide. Cyclins and CDKs help to control the different stages of the cell cycle by activating and deactivating various proteins at specific times. The p27 protein acts as a brake on the cell cycle, preventing cells from dividing too quickly or abnormally.
When p27 binds to a CDK-cyclin complex, it prevents the complex from phosphorylating its target proteins, which are necessary for the progression of the cell cycle. By inhibiting CDK activity, p27 helps to ensure that cells divide only when the proper conditions are met.
Mutations in the CDKN1B gene, which encodes p27, have been associated with several types of cancer, including breast, lung, and prostate cancer. These mutations can lead to decreased levels of p27 or impaired function, allowing cells to divide uncontrollably and form tumors.
A frameshift mutation is a type of genetic mutation that occurs when the addition or deletion of nucleotides in a DNA sequence is not divisible by three. Since DNA is read in groups of three nucleotides (codons), which each specify an amino acid, this can shift the "reading frame," leading to the insertion or deletion of one or more amino acids in the resulting protein. This can cause a protein to be significantly different from the normal protein, often resulting in a nonfunctional protein and potentially causing disease. Frameshift mutations are typically caused by insertions or deletions of nucleotides, but they can also result from more complex genetic rearrangements.
Genetic markers are specific segments of DNA that are used in genetic mapping and genotyping to identify specific genetic locations, diseases, or traits. They can be composed of short tandem repeats (STRs), single nucleotide polymorphisms (SNPs), restriction fragment length polymorphisms (RFLPs), or variable number tandem repeats (VNTRs). These markers are useful in various fields such as genetic research, medical diagnostics, forensic science, and breeding programs. They can help to track inheritance patterns, identify genetic predispositions to diseases, and solve crimes by linking biological evidence to suspects or victims.
The adrenal glands are a pair of endocrine glands that are located on top of the kidneys. Each gland has two parts: the outer cortex and the inner medulla. The adrenal cortex produces hormones such as cortisol, aldosterone, and androgens, which regulate metabolism, blood pressure, and other vital functions. The adrenal medulla produces catecholamines, including epinephrine (adrenaline) and norepinephrine (noradrenaline), which help the body respond to stress by increasing heart rate, blood pressure, and alertness.
Chromosome mapping, also known as physical mapping, is the process of determining the location and order of specific genes or genetic markers on a chromosome. This is typically done by using various laboratory techniques to identify landmarks along the chromosome, such as restriction enzyme cutting sites or patterns of DNA sequence repeats. The resulting map provides important information about the organization and structure of the genome, and can be used for a variety of purposes, including identifying the location of genes associated with genetic diseases, studying evolutionary relationships between organisms, and developing genetic markers for use in breeding or forensic applications.
Gene deletion is a type of mutation where a segment of DNA, containing one or more genes, is permanently lost or removed from a chromosome. This can occur due to various genetic mechanisms such as homologous recombination, non-homologous end joining, or other types of genomic rearrangements.
The deletion of a gene can have varying effects on the organism, depending on the function of the deleted gene and its importance for normal physiological processes. If the deleted gene is essential for survival, the deletion may result in embryonic lethality or developmental abnormalities. However, if the gene is non-essential or has redundant functions, the deletion may not have any noticeable effects on the organism's phenotype.
Gene deletions can also be used as a tool in genetic research to study the function of specific genes and their role in various biological processes. For example, researchers may use gene deletion techniques to create genetically modified animal models to investigate the impact of gene deletion on disease progression or development.
Carcinoma is a type of cancer that develops from epithelial cells, which are the cells that line the inner and outer surfaces of the body. These cells cover organs, glands, and other structures within the body. Carcinomas can occur in various parts of the body, including the skin, lungs, breasts, prostate, colon, and pancreas. They are often characterized by the uncontrolled growth and division of abnormal cells that can invade surrounding tissues and spread to other parts of the body through a process called metastasis. Carcinomas can be further classified based on their appearance under a microscope, such as adenocarcinoma, squamous cell carcinoma, and basal cell carcinoma.
X-ray computed tomography (CT or CAT scan) is a medical imaging method that uses computer-processed combinations of many X-ray images taken from different angles to produce cross-sectional (tomographic) images (virtual "slices") of the body. These cross-sectional images can then be used to display detailed internal views of organs, bones, and soft tissues in the body.
The term "computed tomography" is used instead of "CT scan" or "CAT scan" because the machines take a series of X-ray measurements from different angles around the body and then use a computer to process these data to create detailed images of internal structures within the body.
CT scanning is a noninvasive, painless medical test that helps physicians diagnose and treat medical conditions. CT imaging provides detailed information about many types of tissue including lung, bone, soft tissue and blood vessels. CT examinations can be performed on every part of the body for a variety of reasons including diagnosis, surgical planning, and monitoring of therapeutic responses.
In computed tomography (CT), an X-ray source and detector rotate around the patient, measuring the X-ray attenuation at many different angles. A computer uses this data to construct a cross-sectional image by the process of reconstruction. This technique is called "tomography". The term "computed" refers to the use of a computer to reconstruct the images.
CT has become an important tool in medical imaging and diagnosis, allowing radiologists and other physicians to view detailed internal images of the body. It can help identify many different medical conditions including cancer, heart disease, lung nodules, liver tumors, and internal injuries from trauma. CT is also commonly used for guiding biopsies and other minimally invasive procedures.
In summary, X-ray computed tomography (CT or CAT scan) is a medical imaging technique that uses computer-processed combinations of many X-ray images taken from different angles to produce cross-sectional images of the body. It provides detailed internal views of organs, bones, and soft tissues in the body, allowing physicians to diagnose and treat medical conditions.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
I'm sorry for any confusion, but "Japan" is not a medical term. Japan is the name of a country, officially known as Nippon-koku or Nihon-koku in Japanese, and is located in East Asia. It is an island nation in the Pacific Ocean with a population of about 126 million people.
If you have any medical questions or terms that you would like me to define, please let me know!
An allele is a variant form of a gene that is located at a specific position on a specific chromosome. Alleles are alternative forms of the same gene that arise by mutation and are found at the same locus or position on homologous chromosomes.
Each person typically inherits two copies of each gene, one from each parent. If the two alleles are identical, a person is said to be homozygous for that trait. If the alleles are different, the person is heterozygous.
For example, the ABO blood group system has three alleles, A, B, and O, which determine a person's blood type. If a person inherits two A alleles, they will have type A blood; if they inherit one A and one B allele, they will have type AB blood; if they inherit two B alleles, they will have type B blood; and if they inherit two O alleles, they will have type O blood.
Alleles can also influence traits such as eye color, hair color, height, and other physical characteristics. Some alleles are dominant, meaning that only one copy of the allele is needed to express the trait, while others are recessive, meaning that two copies of the allele are needed to express the trait.
"Family Health" is not a term that has a single, widely accepted medical definition. However, in the context of healthcare and public health, "family health" often refers to the physical, mental, and social well-being of all members of a family unit. It includes the assessment, promotion, and prevention of health conditions that affect individual family members as well as the family as a whole.
Family health may also encompass interventions and programs that aim to strengthen family relationships, communication, and functioning, as these factors can have a significant impact on overall health outcomes. Additionally, family health may involve addressing social determinants of health, such as poverty, housing, and access to healthcare, which can affect the health of families and communities.
Overall, family health is a holistic approach to healthcare that recognizes the importance of considering the needs and experiences of all family members in promoting and maintaining good health.
Somatostatin is a hormone that inhibits the release of several hormones and also has a role in slowing down digestion. It is produced by the body in various parts of the body, including the hypothalamus (a part of the brain), the pancreas, and the gastrointestinal tract.
Somatostatin exists in two forms: somatostatin-14 and somatostatin-28, which differ in their length. Somatostatin-14 is the predominant form found in the brain, while somatostatin-28 is the major form found in the gastrointestinal tract.
Somatostatin has a wide range of effects on various physiological processes, including:
* Inhibiting the release of several hormones such as growth hormone, insulin, glucagon, and gastrin
* Slowing down digestion by inhibiting the release of digestive enzymes from the pancreas and reducing blood flow to the gastrointestinal tract
* Regulating neurotransmission in the brain
Somatostatin is used clinically as a diagnostic tool for detecting certain types of tumors that overproduce growth hormone or other hormones, and it is also used as a treatment for some conditions such as acromegaly (a condition characterized by excessive growth hormone production) and gastrointestinal disorders.
Genetic linkage is the phenomenon where two or more genetic loci (locations on a chromosome) tend to be inherited together because they are close to each other on the same chromosome. This occurs during the process of sexual reproduction, where homologous chromosomes pair up and exchange genetic material through a process called crossing over.
The closer two loci are to each other on a chromosome, the lower the probability that they will be separated by a crossover event. As a result, they are more likely to be inherited together and are said to be linked. The degree of linkage between two loci can be measured by their recombination frequency, which is the percentage of meiotic events in which a crossover occurs between them.
Linkage analysis is an important tool in genetic research, as it allows researchers to identify and map genes that are associated with specific traits or diseases. By analyzing patterns of linkage between markers (identifiable DNA sequences) and phenotypes (observable traits), researchers can infer the location of genes that contribute to those traits or diseases on chromosomes.
3T3 cells are a type of cell line that is commonly used in scientific research. The name "3T3" is derived from the fact that these cells were developed by treating mouse embryo cells with a chemical called trypsin and then culturing them in a flask at a temperature of 37 degrees Celsius.
Specifically, 3T3 cells are a type of fibroblast, which is a type of cell that is responsible for producing connective tissue in the body. They are often used in studies involving cell growth and proliferation, as well as in toxicity tests and drug screening assays.
One particularly well-known use of 3T3 cells is in the 3T3-L1 cell line, which is a subtype of 3T3 cells that can be differentiated into adipocytes (fat cells) under certain conditions. These cells are often used in studies of adipose tissue biology and obesity.
It's important to note that because 3T3 cells are a type of immortalized cell line, they do not always behave exactly the same way as primary cells (cells that are taken directly from a living organism). As such, researchers must be careful when interpreting results obtained using 3T3 cells and consider any potential limitations or artifacts that may arise due to their use.
Neoplastic cell transformation is a process in which a normal cell undergoes genetic alterations that cause it to become cancerous or malignant. This process involves changes in the cell's DNA that result in uncontrolled cell growth and division, loss of contact inhibition, and the ability to invade surrounding tissues and metastasize (spread) to other parts of the body.
Neoplastic transformation can occur as a result of various factors, including genetic mutations, exposure to carcinogens, viral infections, chronic inflammation, and aging. These changes can lead to the activation of oncogenes or the inactivation of tumor suppressor genes, which regulate cell growth and division.
The transformation of normal cells into cancerous cells is a complex and multi-step process that involves multiple genetic and epigenetic alterations. It is characterized by several hallmarks, including sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, enabling replicative immortality, induction of angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism, and evading immune destruction.
Neoplastic cell transformation is a fundamental concept in cancer biology and is critical for understanding the molecular mechanisms underlying cancer development and progression. It also has important implications for cancer diagnosis, prognosis, and treatment, as identifying the specific genetic alterations that underlie neoplastic transformation can help guide targeted therapies and personalized medicine approaches.
Follow-up studies are a type of longitudinal research that involve repeated observations or measurements of the same variables over a period of time, in order to understand their long-term effects or outcomes. In medical context, follow-up studies are often used to evaluate the safety and efficacy of medical treatments, interventions, or procedures.
In a typical follow-up study, a group of individuals (called a cohort) who have received a particular treatment or intervention are identified and then followed over time through periodic assessments or data collection. The data collected may include information on clinical outcomes, adverse events, changes in symptoms or functional status, and other relevant measures.
The results of follow-up studies can provide important insights into the long-term benefits and risks of medical interventions, as well as help to identify factors that may influence treatment effectiveness or patient outcomes. However, it is important to note that follow-up studies can be subject to various biases and limitations, such as loss to follow-up, recall bias, and changes in clinical practice over time, which must be carefully considered when interpreting the results.
DNA Sequence Analysis is the systematic determination of the order of nucleotides in a DNA molecule. It is a critical component of modern molecular biology, genetics, and genetic engineering. The process involves determining the exact order of the four nucleotide bases - adenine (A), guanine (G), cytosine (C), and thymine (T) - in a DNA molecule or fragment. This information is used in various applications such as identifying gene mutations, studying evolutionary relationships, developing molecular markers for breeding, and diagnosing genetic diseases.
The process of DNA Sequence Analysis typically involves several steps, including DNA extraction, PCR amplification (if necessary), purification, sequencing reaction, and electrophoresis. The resulting data is then analyzed using specialized software to determine the exact sequence of nucleotides.
In recent years, high-throughput DNA sequencing technologies have revolutionized the field of genomics, enabling the rapid and cost-effective sequencing of entire genomes. This has led to an explosion of genomic data and new insights into the genetic basis of many diseases and traits.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
The pancreas is a glandular organ located in the abdomen, posterior to the stomach. It has both exocrine and endocrine functions. The exocrine portion of the pancreas consists of acinar cells that produce and secrete digestive enzymes into the duodenum via the pancreatic duct. These enzymes help in the breakdown of proteins, carbohydrates, and fats in food.
The endocrine portion of the pancreas consists of clusters of cells called islets of Langerhans, which include alpha, beta, delta, and F cells. These cells produce and secrete hormones directly into the bloodstream, including insulin, glucagon, somatostatin, and pancreatic polypeptide. Insulin and glucagon are critical regulators of blood sugar levels, with insulin promoting glucose uptake and storage in tissues and glucagon stimulating glycogenolysis and gluconeogenesis to raise blood glucose when it is low.
Restriction Fragment Length Polymorphism (RFLP) is a term used in molecular biology and genetics. It refers to the presence of variations in DNA sequences among individuals, which can be detected by restriction enzymes. These enzymes cut DNA at specific sites, creating fragments of different lengths.
In RFLP analysis, DNA is isolated from an individual and treated with a specific restriction enzyme that cuts the DNA at particular recognition sites. The resulting fragments are then separated by size using gel electrophoresis, creating a pattern unique to that individual's DNA. If there are variations in the DNA sequence between individuals, the restriction enzyme may cut the DNA at different sites, leading to differences in the length of the fragments and thus, a different pattern on the gel.
These variations can be used for various purposes, such as identifying individuals, diagnosing genetic diseases, or studying evolutionary relationships between species. However, RFLP analysis has largely been replaced by more modern techniques like polymerase chain reaction (PCR)-based methods and DNA sequencing, which offer higher resolution and throughput.
X-ray therapy, also known as radiation therapy, is a medical treatment that uses high-energy radiation to destroy cancer cells and shrink or control the growth of tumors. The radiation used in x-ray therapy can come from a machine outside the body (external beam radiation) or from radioactive material placed in or near the tumor (internal radiation or brachytherapy).
The goal of x-ray therapy is to kill cancer cells while minimizing harm to normal cells. The treatment is carefully planned and tailored to the size, shape, and location of the tumor, as well as the patient's overall health. X-ray therapy can be used alone or in combination with other cancer treatments, such as surgery and chemotherapy.
It is important to note that x-ray therapy itself does not cause cancer, but it can increase the risk of developing secondary cancers in the future. This risk is generally low and will be weighed against the potential benefits of treatment. Patients should discuss any concerns about this risk with their healthcare provider.
Genotype, in genetics, refers to the complete heritable genetic makeup of an individual organism, including all of its genes. It is the set of instructions contained in an organism's DNA for the development and function of that organism. The genotype is the basis for an individual's inherited traits, and it can be contrasted with an individual's phenotype, which refers to the observable physical or biochemical characteristics of an organism that result from the expression of its genes in combination with environmental influences.
It is important to note that an individual's genotype is not necessarily identical to their genetic sequence. Some genes have multiple forms called alleles, and an individual may inherit different alleles for a given gene from each parent. The combination of alleles that an individual inherits for a particular gene is known as their genotype for that gene.
Understanding an individual's genotype can provide important information about their susceptibility to certain diseases, their response to drugs and other treatments, and their risk of passing on inherited genetic disorders to their offspring.
PC12 cells are a type of rat pheochromocytoma cell line, which are commonly used in scientific research. Pheochromocytomas are tumors that develop from the chromaffin cells of the adrenal gland, and PC12 cells are a subtype of these cells.
PC12 cells have several characteristics that make them useful for research purposes. They can be grown in culture and can be differentiated into a neuron-like phenotype when treated with nerve growth factor (NGF). This makes them a popular choice for studies involving neuroscience, neurotoxicity, and neurodegenerative disorders.
PC12 cells are also known to express various neurotransmitter receptors, ion channels, and other proteins that are relevant to neuronal function, making them useful for studying the mechanisms of drug action and toxicity. Additionally, PC12 cells can be used to study the regulation of cell growth and differentiation, as well as the molecular basis of cancer.
Genetic association studies are a type of epidemiological research that aims to identify statistical associations between genetic variations and particular traits or diseases. These studies typically compare the frequency of specific genetic markers, such as single nucleotide polymorphisms (SNPs), in individuals with a given trait or disease to those without it.
The goal of genetic association studies is to identify genetic factors that contribute to the risk of developing common complex diseases, such as diabetes, heart disease, or cancer. By identifying these genetic associations, researchers hope to gain insights into the underlying biological mechanisms of these diseases and develop new strategies for prevention, diagnosis, and treatment.
It's important to note that while genetic association studies can identify statistical associations between genetic markers and traits or diseases, they cannot prove causality. Further research is needed to confirm and validate these findings and to understand the functional consequences of the identified genetic variants.
Immunohistochemistry (IHC) is a technique used in pathology and laboratory medicine to identify specific proteins or antigens in tissue sections. It combines the principles of immunology and histology to detect the presence and location of these target molecules within cells and tissues. This technique utilizes antibodies that are specific to the protein or antigen of interest, which are then tagged with a detection system such as a chromogen or fluorophore. The stained tissue sections can be examined under a microscope, allowing for the visualization and analysis of the distribution and expression patterns of the target molecule in the context of the tissue architecture. Immunohistochemistry is widely used in diagnostic pathology to help identify various diseases, including cancer, infectious diseases, and immune-mediated disorders.
Medical Definition:
Magnetic Resonance Imaging (MRI) is a non-invasive diagnostic imaging technique that uses a strong magnetic field and radio waves to create detailed cross-sectional or three-dimensional images of the internal structures of the body. The patient lies within a large, cylindrical magnet, and the scanner detects changes in the direction of the magnetic field caused by protons in the body. These changes are then converted into detailed images that help medical professionals to diagnose and monitor various medical conditions, such as tumors, injuries, or diseases affecting the brain, spinal cord, heart, blood vessels, joints, and other internal organs. MRI does not use radiation like computed tomography (CT) scans.
Paraganglioma, extra-adrenal, is a type of rare tumor that develops in the nervous system's paraganglia, which are groups of specialized cells that are responsible for regulating blood pressure and other bodily functions. Unlike adrenal paragangliomas, which form in the adrenal glands located on top of the kidneys, extra-adrenal paragangliomas develop outside of the adrenal glands, in various locations along the sympathetic and parasympathetic nervous systems. These tumors can be functional or nonfunctional, meaning they may or may not produce hormones such as catecholamines (epinephrine, norepinephrine, and dopamine). Functional extra-adrenal paragangliomas can cause symptoms related to excessive hormone production, including hypertension, sweating, headaches, and rapid heartbeat. Treatment typically involves surgical removal of the tumor, along with preoperative preparation to manage potential hormonal imbalances.
Genetic predisposition to disease refers to an increased susceptibility or vulnerability to develop a particular illness or condition due to inheriting specific genetic variations or mutations from one's parents. These genetic factors can make it more likely for an individual to develop a certain disease, but it does not guarantee that the person will definitely get the disease. Environmental factors, lifestyle choices, and interactions between genes also play crucial roles in determining if a genetically predisposed person will actually develop the disease. It is essential to understand that having a genetic predisposition only implies a higher risk, not an inevitable outcome.
The term "Asian Continental Ancestry Group" is a medical/ethnic classification used to describe a person's genetic background and ancestry. According to this categorization, individuals with origins in the Asian continent are grouped together. This includes populations from regions such as East Asia (e.g., China, Japan, Korea), South Asia (e.g., India, Pakistan, Bangladesh), Southeast Asia (e.g., Philippines, Indonesia, Thailand), and Central Asia (e.g., Kazakhstan, Uzbekistan, Tajikistan). It is important to note that this broad categorization may not fully capture the genetic diversity within these regions or accurately reflect an individual's specific ancestral origins.
Endocrine disruptors are defined as exogenous (external) substances or mixtures that interfere with the way hormones work in the body, leading to negative health effects. They can mimic, block, or alter the normal synthesis, secretion, transport, binding, action, or elimination of natural hormones in the body responsible for maintaining homeostasis, reproduction, development, and/or behavior.
Endocrine disruptors can be found in various sources, including industrial chemicals, pesticides, pharmaceuticals, and personal care products. They have been linked to a range of health problems, such as cancer, reproductive issues, developmental disorders, neurological impairments, and immune system dysfunction.
Examples of endocrine disruptors include bisphenol A (BPA), phthalates, dioxins, polychlorinated biphenyls (PCBs), perfluoroalkyl substances (PFAS), and certain pesticides like dichlorodiphenyltrichloroethane (DDT) and vinclozolin.
It is important to note that endocrine disruptors can have effects at very low doses, and their impact may depend on the timing of exposure, particularly during critical windows of development such as fetal growth and early childhood.
Neoplastic gene expression regulation refers to the processes that control the production of proteins and other molecules from genes in neoplastic cells, or cells that are part of a tumor or cancer. In a normal cell, gene expression is tightly regulated to ensure that the right genes are turned on or off at the right time. However, in cancer cells, this regulation can be disrupted, leading to the overexpression or underexpression of certain genes.
Neoplastic gene expression regulation can be affected by a variety of factors, including genetic mutations, epigenetic changes, and signals from the tumor microenvironment. These changes can lead to the activation of oncogenes (genes that promote cancer growth and development) or the inactivation of tumor suppressor genes (genes that prevent cancer).
Understanding neoplastic gene expression regulation is important for developing new therapies for cancer, as targeting specific genes or pathways involved in this process can help to inhibit cancer growth and progression.
Carcinoma, neuroendocrine is a type of cancer that arises from the neuroendocrine cells, which are specialized cells that have both nerve and hormone-producing functions. These cells are found throughout the body, but neuroendocrine tumors (NETs) most commonly occur in the lungs, gastrointestinal tract, pancreas, and thyroid gland.
Neuroendocrine carcinomas can be classified as well-differentiated or poorly differentiated based on how closely they resemble normal neuroendocrine cells under a microscope. Well-differentiated tumors tend to grow more slowly and are less aggressive than poorly differentiated tumors.
Neuroendocrine carcinomas can produce and release hormones and other substances that can cause a variety of symptoms, such as flushing, diarrhea, wheezing, and heart palpitations. Treatment for neuroendocrine carcinoma depends on the location and extent of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches.
Cervical intraepithelial neoplasia (CIN) is a term used to describe the abnormal growth and development of cells on the surface of the cervix. These changes are usually caused by human papillomavirus (HPV) infection, which is a common sexually transmitted infection. CIN is not cancer, but it can develop into cancer if left untreated.
The term "intraepithelial" refers to the fact that the abnormal cells are found in the epithelium, or the lining of the cervix. The term "neoplasia" means abnormal growth or development of cells. CIN is further classified into three grades based on the severity of the cell changes:
* CIN 1: Mild dysplasia (abnormal cell growth) affecting the lower third of the epithelium.
* CIN 2: Moderate dysplasia affecting the lower two-thirds of the epithelium.
* CIN 3: Severe dysplasia or carcinoma in situ, which means that the abnormal cells are found in the full thickness of the epithelium and have a high risk of progressing to invasive cancer if not treated.
It's important to note that CIN can regress on its own without treatment, especially in younger women. However, some cases may progress to invasive cervical cancer if left untreated. Regular Pap testing is recommended to detect and monitor any abnormal cell changes in the cervix. If CIN is detected, further diagnostic procedures such as a colposcopy or biopsy may be performed to determine the extent of the abnormality and guide treatment decisions.
Genetic polymorphism refers to the occurrence of multiple forms (called alleles) of a particular gene within a population. These variations in the DNA sequence do not generally affect the function or survival of the organism, but they can contribute to differences in traits among individuals. Genetic polymorphisms can be caused by single nucleotide changes (SNPs), insertions or deletions of DNA segments, or other types of genetic rearrangements. They are important for understanding genetic diversity and evolution, as well as for identifying genetic factors that may contribute to disease susceptibility in humans.
 Multiple endocrine neoplasia type 2B - Wikipedia
Multiple endocrine neoplasia type 2B - Wikipedia
Multiple Endocrine Neoplasia Type 2b | Profiles RNS
 Multiple endocrine neoplasia, type 2b (Concept Id: C0025269)
- MedGen - NCBI
Multiple endocrine neoplasia, type 2b (Concept Id: C0025269)
- MedGen - NCBI
 Multiple Endocrine Neoplasia, Type 2B (MEN 2B) - Endocrine and Metabolic Disorders - MSD Manual Professional Edition
Multiple Endocrine Neoplasia, Type 2B (MEN 2B) - Endocrine and Metabolic Disorders - MSD Manual Professional Edition
 Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918...
Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918...
 Multiple endocrine neoplasia type 2: A review
Multiple endocrine neoplasia type 2: A review
 SelectedWorks - Helen Donis-Keller
SelectedWorks - Helen Donis-Keller
 Chronic Megacolon: Background, Pathophysiology, Etiology
Chronic Megacolon: Background, Pathophysiology, Etiology
Multiple Endocrine Neoplasia, Type 4 (MEN 4) - Endocrine and Metabolic Disorders - Merck Manuals Professional Edition
Internet Scientific Publications
![Thyroid Cancer Treatment (PDQ®): Treatment - Patient Information [NCI] | Michigan Medicine](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAMAAAAoLQ9TAAAADFBMVEUCJUr/zDMHKEoIKUkHBCdKAAAAMklEQVQYlWNgIAcwMjIyMDEwMIMZUAFGZBrGQvDBTAjALQBRjywAx3ABVBrNWkx3kAgALh0AUuCGqxsAAAAASUVORK5CYII=) Thyroid Cancer Treatment (PDQ®): Treatment - Patient Information [NCI] | Michigan Medicine
Thyroid Cancer Treatment (PDQ®): Treatment - Patient Information [NCI] | Michigan Medicine
Parental Age and Retinoblastoma - A Retrospective Study of Demographic Data and Genetic Analysis
 SKIN SIGNS OF SYSTEMIC CANCERS | Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology, 8e | AccessMedicine | McGraw...
SKIN SIGNS OF SYSTEMIC CANCERS | Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology, 8e | AccessMedicine | McGraw...
 "Conjunctival and Lingual Mucosal Neuromas Without Multiple Endocrine N" by Talia N. Shoshany, Christopher J. Rapuano et al.
"Conjunctival and Lingual Mucosal Neuromas Without Multiple Endocrine N" by Talia N. Shoshany, Christopher J. Rapuano et al.
 Shailendra B. Patel
Shailendra B. Patel
 Multiple endocrine neoplasia: MedlinePlus Genetics
Multiple endocrine neoplasia: MedlinePlus Genetics
 How DNA Evidence Works | HowStuffWorks
How DNA Evidence Works | HowStuffWorks
 Thyroid cancer - Doctors and departments - Mayo Clinic
Thyroid cancer - Doctors and departments - Mayo Clinic
Thyroid Cancer Guidelines: Guidelines Summary, Thyroid Nodule Evaluation, Diagnosis
 Amani Alameer, MD| Endocrinology | MedStar Health
Amani Alameer, MD| Endocrinology | MedStar Health
 Gene Table | Myriad Genetics
Gene Table | Myriad Genetics
 Cytopathologic diagnosis of fine needle aspiration biopsies of thyroid nodules
Cytopathologic diagnosis of fine needle aspiration biopsies of thyroid nodules
Skin Signs of Systemic Cancers | Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology, 9e | AccessEmergency Medicine ...
 Diabetes: total risk - benefit of SGLT2 inhibitors and GLP1 agonists
Diabetes: total risk - benefit of SGLT2 inhibitors and GLP1 agonists
 Neuronal and mixed neuronal-glial
- Brain Tumour Research
Neuronal and mixed neuronal-glial
- Brain Tumour Research
 Most recent papers in the journal Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics | Read by QxMD
Most recent papers in the journal Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics | Read by QxMD
 Department of Genetics - Research output
- Research Profiles at Washington University School of Medicine
Department of Genetics - Research output
- Research Profiles at Washington University School of Medicine
Specific PHGKB|Rare Diseases PHGKB|PHGKB
 Utah Medical Home Portal - Constipation
Utah Medical Home Portal - Constipation
High, narrow palate (Concept Id: C1837404)
- MedGen - NCBI
Tumors32
- Multiple endocrine neoplasia type 2B (MEN 2B) is a genetic disease that causes multiple tumors on the mouth, eyes, and endocrine glands. (wikipedia.org)
- It is the most severe type of multiple endocrine neoplasia, differentiated by the presence of benign oral and submucosal tumors in addition to endocrine malignancies. (wikipedia.org)
- Overview of Multiple Endocrine Neoplasias (MEN) The multiple endocrine neoplasia (MEN) syndromes comprise 4 genetically distinct familial diseases involving adenomatous hyperplasia and malignant tumors in several endocrine glands. (msdmanuals.com)
- Inherited multiple tumors and mosaic pleiotropism in man. (medscape.com)
- Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. (medscape.com)
- As in multiple endocrine neoplasia type 2, the mutations likely result in an overactive RET protein that can trigger cells to grow and divide uncontrollably and can lead to the formation of tumors. (medlineplus.gov)
- Multiple endocrine neoplasia typically involves the development of tumors in two or more of the body's hormone-producing glands, called endocrine glands. (medlineplus.gov)
- The overactive protein likely triggers cells to grow and divide abnormally, which can lead to the formation of tumors in the endocrine system and other tissues. (medlineplus.gov)
- Multiple endocrine neoplasia type 2 (MEN2) is a rare genetic polyglandular cancer syndrome, characterized by the 100% prevalence of medullary thyroid carcinoma (MTC) and an increased risk of develop other specific tumors affecting additional glands of the endocrine system. (rarediseases.org)
- MEN2 endocrine tumors may secrete excessive amounts of hormones into the bloodstream, which can result in a variety of symptoms. (rarediseases.org)
- Multiple endocrine neoplasias type 2 (MEN2) is an inherited disorder characterized by the development of medullary thyroid cancer (MTC), parathyroid tumors, and pheochromocytoma . (medscape.com)
- The combination of FMTC and tumors of other endocrine glands is called multiple endocrine neoplasia type 2 (MEN 2). (cancer.org)
- Many different types of tumors are associated with multiple endocrine neoplasia. (nih.gov)
- Hyperparathyroidism is the most common feature, followed by tumors of the pituitary gland, additional endocrine glands, and other organs. (nih.gov)
- The loss of functional menin allows cells to divide too frequently, leading to the formation of tumors characteristic of multiple endocrine neoplasia type 1. (nih.gov)
- It is unclear why these tumors preferentially affect endocrine tissues. (nih.gov)
- This unchecked cell division can lead to the formation of tumors in endocrine glands and other tissues. (nih.gov)
- Depending on their localization and grade of malignancy, neuroendocrine tumors differ in expression of various SSR types. (almclinmed.ru)
- MEN type 1 is a rare genetic disorder in which benign (noncancerous) tumors arise from the cells of various glands of the endocrine system. (rarediseases.org)
- Multiple endocrine neoplasia (MEN) type 2 is a rare genetic disorder characterized by an increased risk of developing a specific form of thyroid cancer (medullary thyroid carcinoma) and benign tumors affecting additional glands of the endocrine system. (rarediseases.org)
- Pancreatic endocrine tumors (e.g. (allthingsmedicine.com)
- Expertise in the surgical management of endocrine disorders and malignancies such as thyroid cancer, Graves' disease, hyperparathyroidism, adrenal tumors etc. (mayoclinic.org)
- A group of autosomal dominant diseases characterized by the combined occurrence of tumors involving two or more ENDOCRINE GLANDS that secrete PEPTIDE HORMONES or AMINES. (uchicago.edu)
- Association between neuroendocrine tumors biomarkers and primary tumor site and disease type based on total 68Ga-DOTATATE-Avid tumor volume measurements. (uchicago.edu)
- Normal structural dopamine type 2 receptor gene in prolactin-secreting and other pituitary tumors. (uchicago.edu)
- Type 2A (MEN2A): tumors or excessive growths form on at least two of the following three glands: thyroid, adrenal, or parathyroid. (endocrinology-centers.com)
- Type 2B (MEN2B): formerly called multiple endocrine neoplasia type 3, this type can cause adrenal gland tumors, medullary thyroid cancer, and painful growths around nerves in your mucus membranes (neuromas). (endocrinology-centers.com)
- Familial medullary thyroid carcinoma (FMTC): patients with this condition have an 80% chance of developing medullary thyroid cancer, but less than a 5% chance of developing other endocrine tumors. (endocrinology-centers.com)
- It affects approximately 1 in every 40,000 people, and can lead to tumors in the endocrine system, including pheochromocytoma. (nih.gov)
- Squamous cell carcinoma, the most frequent type of cancer in these areas, must be distinguished from benign (noncancerous) tumors of the throat and neck. (marystolfacancerfoundation.org)
- Renal tumors include about 3% of all malignant neoplasia of adulthood. (biopticka.cz)
- Renal epithelial tumors can be, according to Heidelberg and UICC97 classification, divided into several types. (biopticka.cz)
Syndrome16
- A variety of eponyms have been proposed for MEN 2B, such as Williams-Pollock syndrome, Gorlin-Vickers syndrome, and Wagenmann-Froboese syndrome. (wikipedia.org)
- reported cases of a difference disease-the "multiple mucosal neuroma syndrome"-having the physical phenotype of MEN2B, but without variations in the RET gene and without malignancy. (wikipedia.org)
- Multiple endocrine neoplasia, type 2B (MEN 2B) is an autosomal dominant syndrome characterized by medullary thyroid carcinoma, pheochromocytoma, multiple mucosal neuromas and intestinal ganglioneuromas, and often a marfanoid habitus and other skeletal abnormalities. (msdmanuals.com)
- Multiple Endocrine Neoplasia, Type 2A (MEN 2A) Multiple endocrine neoplasia, type 2A (MEN 2A) is an autosomal dominant syndrome characterized by medullary carcinoma of the thyroid, pheochromocytoma, parathyroid hyperplasia or adenomas (causing. (msdmanuals.com)
- Pheochromocytomas are a feature of multiple endocrine neoplasia type 2 (described below), but they can also be nonsyndromic, which means they occur without the other signs and symptoms of the syndrome. (medlineplus.gov)
- Three are the endocrine glands most often affected in MEN2 syndrome: the thyroid, the adrenal glands and, only in the MEN2A variants, the parathyroids. (rarediseases.org)
- A major component of the syndrome of multiple endocrine neoplasia, type 2b. (nih.gov)
- MTC is most aggressive in the MEN 2b syndrome. (cancer.org)
- C C6432 Multiple Endocrine Neoplasia Multiple Endocrine Neoplasia Syndrome(s) MEN Syndromes An autosomal dominant inherited neoplastic syndrome characterized by the development of various endocrine neoplasms and abnormalities in various anatomic sites. (nih.gov)
- Carcinoid tumor of the pancreas causing the diarrheogenic syndrome: report of a case combined with multiple endocrine neoplasia, type I. Surgery. (uchicago.edu)
- Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays. (exeterlaboratory.com)
- The different types of multiple endocrine neoplasia syndrome affect these different glands in different ways. (endocrinology-centers.com)
- Also called multiple endocrine adenomatosis or Wermer's syndrome, multiple endocrine neoplasia type 1 is the result of mutations of the MEN1 gene, causing tumor growth on the parathyroid gland, and then on the pancreas or pituitary gland. (endocrinology-centers.com)
- VHL syndrome is classified into two types, type 1 and type 2. (nih.gov)
- Medullary thyroid carcinoma occurs in sporadic or hereditary (25% of cases) forms, as part of multiple endocrine neoplasia type 2 (MEN 2) syndromes or as the familial medullary thyroid carcinoma-only syndrome (FMTC). (shifrinmd.com)
- If this is a familial cancer syndrome, such as MEN 2A (Sipple's syndrome), familial medullary thyroid carcinoma (FMTC), or MEN 2B, genetic testing of the blood for the RET proto-oncogene is indicated. (shifrinmd.com)
MEN2B5
- Multiple endocrine neoplasia type 2B (MEN2B) is a genetic disease in which one or more of the endocrine glands are overactive and form a tumor (neoplasia). (nih.gov)
- People with MEN2B typically have a body type with long arms, legs, and fingers. (nih.gov)
- Multiple endocrine neoplasia type 2 (MEN2) includes the following phenotypes: MEN2A, familial medullary thyroid carcinoma (FMTC, which may be a variant of MEN2A), and MEN2B. (nih.gov)
- familial medullary thyroid cancer (FMTC), multiple endocrine neoplasia type 2A (MEN2A) and multiple endocrine neoplasia type 2B (MEN2B). (omicsonline.org)
- Linkage of the multiple endocrine neoplasia type 2B gene (MEN2B) to chromosome 10 markers linked to MEN2A. (uchicago.edu)
Pheochromocytomas6
- The role of RET proto-oncogene mutation analysis in the diagnosis of multiple endocrine neoplasia type 2 (MEN 2) gene carriers and in the discrimination of sporadic and familial medullary thyroid carcinomas and pheochromocytomas. (nih.gov)
- Pheochromocytomas occur earlier than in patients with type 2A MEN. (medscape.com)
- 15. Bilateral subtotal adrenal resection for bilateral pheochromocytomas in multiple endocrine neoplasia, type IIa: a case report. (nih.gov)
- In MEN 2b, MTC is associated with pheochromocytomas and with benign growths of nerve tissue on the tongue and elsewhere called neuromas . (cancer.org)
- Another is MEN-2B associated with bilateral MTC, mucosal neuromas mainly involving the gastrointestinal (GI) tract and eyelids, bilateral pheochromocytomas, and a marfanoid habitus and is also sometimes associated with Hirschsprung disease. (touchendocrinology.com)
- Patients with VHL type 1 do not develop pheochromocytomas. (nih.gov)
Tumours5
- Introduction: The autosomal dominant multiple endocrine neoplasia type 1 (MEN1), characterized by parathyroid hyperplasia (PH), neuroendocrine digestive tumours (NET) and pituitary adenomas (PA), is due to mutations in the tumor suppressor gene MEN1 encoding a 610-amino acid protein, menin. (endocrine-abstracts.org)
- Somatic mutations of c-ret are responsible for a large proportion of thyroid papillary carcinomas, while germ-line mutations are responsible for multiple endocrine neoplasia types 2A and 2B, dominantly inherited cancer syndromes characterized by multiple tumours of neuroectodermal origin. (umn.edu)
- Multiple endocrine neoplasia (MEN) is distinguished by the development of tumours involving two or more endocrine glands within the same patient. (ukdiss.com)
- There are four major recognised types of MEN (MEN1-MEN4), and each type is characterised by the occurence of tumours within particular endocrine glands as seen in the picture above. (ukdiss.com)
- Multiple endocrine neoplasia type 1 is a rare inherited disease, which can result in tumours in the pituitary and parathyroid glands, and pancreas. (yourhormones.info)
Pheochromocytoma8
- Multiple endocrine neoplasia (MEN) type 2B is a heritable endocrine disorder characterized by medullary thyroid carcinoma (MTC), pheochromocytoma, multiple mucosal neuromas, and a marfanoid habitus. (nih.gov)
- Mutations in the RET gene increase the risk of developing a type of paraganglioma called pheochromocytoma. (medlineplus.gov)
- 2. Mucosal neuroma, pheochromocytoma and medullary thyroid carcinoma: multiple endocrine neoplasia type 3. (nih.gov)
- 3. Multiple endocrine neoplasia (MEN)--an overview and case report--patient with sporadic bilateral pheochromocytoma, hyperparathyroidism and marfanoid habitus. (nih.gov)
- 11. Pheochromocytoma in multiple endocrine neoplasia type II: an example of the two-hit theory of neoplasia. (nih.gov)
- 17. [Composite pheochromocytoma associated with multiple endocrine neoplasia type 2B]. (nih.gov)
- Multiple endocrine neoplasia (MEN) types 2A and 2B are rare genetic diseases, which lead to the development of medullary thyroid cancer and pheochromocytoma. (nih.gov)
- MEN 2B is defined as the presence of medullary thyroid carcinoma, marfanoid habitus, medullated corneal nerve fibers, ganglioneuromatosis of the gut and oral mucosa, and pheochromocytoma associated with a germline RET mutation. (shifrinmd.com)
Form of multiple endocrine2
- More than 25 mutations in the RET gene are known to cause a form of multiple endocrine neoplasia called type 2. (medlineplus.gov)
- MEN type 4 is the rarest form of multiple endocrine neoplasia. (endocrinology-centers.com)
FMTC7
- If MEN 2a, MEN 2b, or isolated FMTC runs in your family, you may be at very high risk of developing MTC. (cancer.org)
- Multiple endocrine neoplasia type 2 is divided into three subtypes: type 2A, type 2B (formerly called type 3), and familial medullary thyroid carcinoma (FMTC). (nih.gov)
- for example, hyperparathyroidism occurs only in type 2A, and medullary thyroid carcinoma is the only feature of FMTC. (nih.gov)
- Among the subtypes of type 2, type 2A is the most common form, followed by FMTC. (nih.gov)
- Gain-of-function RET mutations are responsible for multiple endocrine neoplasia syndromes (MEN) 2A and 2B and familial medullary thyroid carcinoma (FMTC), whereas loss-of-function mutations are found in Hirschsprung disease. (unica.it)
- Familial history and clinical and biochemical evaluation of first-degree relatives were negative for FMTC, MEN 2A and 2B, and Hirschsprung disease. (unica.it)
- The severity of medullary thyroid carcinoma could be classified as following from most favorable to most aggressive: FMTC, following by MEN 2A, following by sporadic medullary thyroid carcinoma, and the worst one is MEN 2B related medullary thyroid carcinoma. (shifrinmd.com)
Adenomatosis1
- Wermer P. Endocrine adenomatosis and peptic ulcer in a large kindred. (medscape.com)
Mutations10
- Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. (medscape.com)
- Most of the RET gene mutations that cause multiple endocrine neoplasia type 2 change single protein building blocks (amino acids) in the RET protein. (medlineplus.gov)
- The mutations responsible for multiple endocrine neoplasia type 2 result in an overactive RET protein that can transmit signals without first attaching to growth factors outside the cell. (medlineplus.gov)
- There are 2 subtypes, MEN 2a and MEN 2b, both of which are caused by mutations (defects) in a gene called RET . (cancer.org)
- Multiple endocrine neoplasia type 4 appears to have signs and symptoms similar to those of type 1, although it is caused by mutations in a different gene. (nih.gov)
- Mutations in the MEN1 , RET , and CDKN1B genes can cause multiple endocrine neoplasia. (nih.gov)
- Mutations in the MEN1 gene cause multiple endocrine neoplasia type 1. (nih.gov)
- Mutations in the RET gene cause multiple endocrine neoplasia type 2. (nih.gov)
- Caused by mutations to the RET (ret proto-oncogene) gene, there are three subtypes of multiple endocrine neoplasia type 2. (endocrinology-centers.com)
- Different genetic mutations have been associated with the pathogenesis and tumor progression of various types of NETs. (statpearls.com)
MEN21
- A total of 55 complete self-reported responses have been received (MEN1 = 27, MEN2 = 21, MEN3 = 7), including 8 people who needed to visit hospital (6 for severe breathing difficulties: x4 MEN1, x2 MEN2, and x2 for problems relating to their endocrine condition). (amend.org.uk)
Tumor4
- There are four types recognized: type 1 (MEN 1), caused by inactivation of the tumor suppressor gene MEN-1, type 2A (MEN 2A), caused by mutation of the RET gene, type 2B (MEN 2B) also caused by mutation of the RET gene, and type 4 (MEN 4) caused by mutation of the CDKN1B gene. (nih.gov)
- C3010 Endocrine Neoplasm C118467 Pediatric Endocrine Terminology C C3878 Thyroid Gland Anaplastic Carcinoma Anaplastic Thyroid Carcinoma Undifferentiated Thyroid Tumor A primary carcinoma of the thyroid gland composed of undifferentiated cells. (nih.gov)
- The Pharmacology and Experimental Therapeutics Section (PETS) focuses on the clinical development of new agents for the treatment of refractory childhood cancers and genetic tumor predisposition syndromes (GTPS) such as neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2), and hereditary medullary thyroid carcinoma (MTC). (nih.gov)
- A total thyroidectomy (complete surgical removal of the thyroid) or removal of a portion of the thyroid is recommended, depending on the type of tumor. (marystolfacancerfoundation.org)
Gene1
- Defects in this gene are a cause of maturity onset diabetes of the young type 3 (MODY3) and also can result in the appearance of hepatic adenomas. (cancerindex.org)
Neuromas1
- Patients with MEN 2B develop medullary thyroid carcinomas and numerous neural defects including neuromas. (nih.gov)
Familial multiple1
- Germ line mutation in the RET proto-oncogene associated with familial multiple endocrine neoplasia type 2B: a case report. (nih.gov)
MeSH1
- Multiple Endocrine Neoplasia" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (uchicago.edu)
Subtypes1
- Multiple endocrine neoplasia type 2 is divided into three subtypes: type 2A, type 2B, and familial medullary thyroid carcinoma. (medlineplus.gov)
Benign1
- These neoplasias are often benign but can be malignant. (uchicago.edu)
Syndromes2
- A genetically heterogenous group of autosomal dominant neoplastic syndromes characterized by the development of neoplasms in various endocrine organs. (nih.gov)
- Approximately 25% of the cases are inherited as part of the multiple endocrine neoplasia type-2 (MEN-2) syndromes. (touchendocrinology.com)
Hyperparathyroidism2
- Hyperparathyroidism is most common initial clinical manifestation of type 1 multiple endocrine neoplasia (MEN). (medscape.com)
- The most common sign of multiple endocrine neoplasia type 1 is overactivity of the parathyroid glands (hyperparathyroidism). (nih.gov)
NONINSULIN-DEPENDEN1
- Diabetes mellitus type 2 (formerly noninsulin-dependent diabetes mellitus (NIDDM) or adult-onset diabetes ) is a metabolic disorder that is characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency. (thediabeticpharmacist.com)
NEOPLASMS1
- Patients with MEN 4 develop endocrine neoplasms, particularly in the parathyroid glands, pituitary, and pancreas. (nih.gov)
Pancreas3
- DM develops due to either a lack of insulin production (type 1 DM), as a result of destroyed beta cells of pancreas due to an autoimmune reaction, or resistance to insulin (type 2 DM), where insulin is being produced at least in the early stages of disease because beta cells are still present, but tissues do not respond to insulin [1]. (escardio.org)
- b) latent autoimmune diabetes of adults (LADA), usually occurring in men over 40 years of age, in whom a progressive autoimmune process destroys the beta cells of pancreas, which is similar to type 1 DM. (escardio.org)
- [ 2 ] This is in contrast to diabetes mellitus type 1 , in which there is an absolute insulin deficiency due to destruction of islet cells in the pancreas . (thediabeticpharmacist.com)
Mutation4
- Type 2A most often results from a mutation that substitutes the amino acid arginine for the amino acid cysteine at position 634 (written as Cys634Arg or C634R). (medlineplus.gov)
- More than 90 percent of cases of type 2B are caused by a mutation that replaces the amino acid methionine with the amino acid threonine at position 918 (written as Met918Thr or M918T). (medlineplus.gov)
- A germ-line activating mutation in the RET ( REarranged during Transfection ) protooncogene is reported in nearly all cases of hereditary forms of MTC associated with multiple endocrine neoplasia type 2 (MEN 2), and a somatic RET mutation can be present in up to 50% of sporadic forms of MTC [ 3 , 4 ]. (hindawi.com)
- This type of mutation is present throughout a person's life in virtually every cell in the body. (nih.gov)
Clinical1
- In the clinical trials conducted thus far, the use of SGLT2 inhibitors was shown to improve the quality of life of patients with type 2 DM, have benefit in treatment of HF, either with reduced or preserved ejection fraction of the left ventricle. (escardio.org)
Phenotype2
- Thymic carcinoid in multiple endocrine neoplasia 1: genotype-phenotype correlation and prevention. (medscape.com)
- This requires that at least two affected individuals present with a familial MTC, MEN 2A or MEN 2B phenotype [ 9 ]. (hindawi.com)
Symptoms5
- When Do Symptoms of Multiple endocrine neoplasia type 2B Begin? (nih.gov)
- These types are distinguished by the genes involved, the types of hormones made, and the characteristic signs and symptoms. (nih.gov)
- The signs and symptoms of multiple endocrine neoplasia type 2 are relatively consistent within any one family. (nih.gov)
- Multiple endocrine neoplasia symptoms vary depending on the type and the glands that are affected. (endocrinology-centers.com)
- What are the signs and symptoms of multiple endocrine neoplasia type 1? (yourhormones.info)
Patients6
- Patients at risk for MEN 2B should undergo genetic screening in infancy. (nih.gov)
- Maintenance of stable glycemic control is an important prerequisite of effective treatment of patients with type 1 diabetes mellitus (DM). (almclinmed.ru)
- To study changes of glycated hemoglobin (HbA1c), rates of hypoglycemia, diabetes-dependent quality of life and treatment satisfaction in patients with type 1 DM, who have been switched to insulin degludec. (almclinmed.ru)
- Despite the prevalence of type 2 DM, there are few effective long-term treatments for the patients. (escardio.org)
- The American Multiple Endocrine Neoplasia Support is a voluntary organization whose mission is to provide education and support to patients, their families and medical personnel regarding multiple endocrine neoplasia (MEN) type 1, MEN type 2a, MEN type 2b. (rarediseases.org)
- Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: an international, multicentre, retrospective study. (bvsalud.org)
Body's2
- Multiple endocrine neoplasia is a group of disorders that affect the body's network of hormone-producing glands called the endocrine system. (nih.gov)
- Your body's endocrine system is a series of glands that release hormones that control important functions like growth and metabolism. (endocrinology-centers.com)
Diagnosis2
- DNA testing is now the preferred method of establishing a diagnosis for MEN 2B, and is thought to be almost 100% sensitive and specific. (wikipedia.org)
- 6,10,11 These fundamental biologic mechanisms have all been exploited in the diagnosis and treatment of thyroid cancer 12 and theoretically offer promise for the treatment of breast and other types of cancer. (touchendocrinology.com)
Pancreatic1
- SSR2A expression was associated with the degree of malignancy and is higher in pancreatic NET Grade 2A (Ki67 to 10%), Grade 2B (Ki67 10-19%) and in neuroendocrine carcinomas Grade 3, compared to Grade 1 (16/50 (32%), 37/61 (60.6%), 8/12 (66.7%) and 20/24 (83.3%), respectively). (almclinmed.ru)
Cancers1
- 25.**The most common type of thyroid cancer is papillary cancer, which accounts for approximately 70% of all thyroid cancers. (medicosrepublic.com)
Organs2
- The endocrine system is the network of glands that secrete hormones into the bloodstream to reach distant target tissues and organs within the body. (rarediseases.org)
- [2] This was then disproven by the concept of the origin of APUD cells from multipotent stem cells before being implanted in different body organs, hence the diffuse endocrine system. (statpearls.com)
Malignancy1
- Thyroid cancer is the most common endocrine malignancy. (touchendocrinology.com)
Disorder1
- Neurofibromatosis type 1, or von Recklinghausen's disease, is a fairly common disorder, affecting approximately 1 in 4000 people. (nih.gov)
Thyroid cancer4
- Medullary Thyroid Carcinoma There are 4 general types of thyroid cancer. (msdmanuals.com)
- Several inherited conditions have been linked to different types of thyroid cancer, as has family history. (cancer.org)
- The most common sign of multiple endocrine neoplasia type 2 is a form of thyroid cancer called medullary thyroid carcinoma. (nih.gov)
- It is more aggressive type of the thyroid cancer. (shifrinmd.com)
Genetic disease1
- Multiple endocrine neoplasia type 2B is a genetic disease. (nih.gov)
Glands involved1
- They are classified by the endocrine glands involved and the degree of aggressiveness. (uchicago.edu)
Cancer2
- Endocrine-related cancer 2016 Dec 23 (12): 909-920. (cdc.gov)
- Patricia A. Cronin M.D., FRCS, is an endocrine surgeon and breast surgical oncologist in Mayo Clinic Arizona and the Mayo Clinic Comprehensive Cancer Center. (mayoclinic.org)
Hereditary1
- It may be multiple endocrine neoplasia (MEN), a group of hereditary disorders that affect the endocrine system. (endocrinology-centers.com)
Diagnostic1
- Objectives: We evaluated the diagnostic power of different used evaluation criteria in ultrasound 2B, Doppler and elastography.Method: Retrospective data analyses of all cases with breast lumps, that were evaluated and also operated during the study follow-up period. (endocrine-abstracts.org)
Diabetes mellitus2
- Type 2 diabetes makes up about 90% of cases of diabetes with the other 10% due primarily to diabetes mellitus type 1 and gestational diabetes . (thediabeticpharmacist.com)
- People with type 2 diabetes mellitus may rarely present with nonketotic hyperosmolar coma (a condition of very high blood sugar associated with a decreased level of consciousness and low blood pressure ). (thediabeticpharmacist.com)