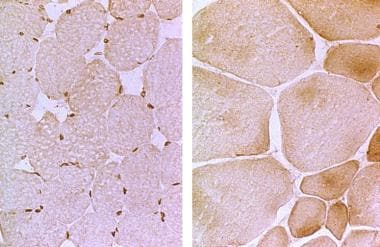
Muscular Dystrophy, Emery-Dreifuss
Thymopoietins
Muscular Dystrophies
Muscular Dystrophy, Duchenne
Muscular Dystrophy, Animal
Dystrophin
Myotonic Dystrophy
Muscular Dystrophies, Limb-Girdle
Muscular Dystrophy, Facioscapulohumeral
Mice, Inbred mdx
Sarcoglycans
Corneal Dystrophies, Hereditary
Dystroglycans
Muscular Dystrophy, Oculopharyngeal
Utrophin
Muscle, Skeletal
Fuchs' Endothelial Dystrophy
Retinal Dystrophies
Collagen Type VI
Pedigree
Dystrophin-Associated Proteins
Wisconsin
Distinct regions specify the nuclear membrane targeting of emerin, the responsible protein for Emery-Dreifuss muscular dystrophy. (1/82)
Emery-Dreifuss muscular dystrophy is a neuromuscular disorder that has three characteristics: (a) early contracture of the elbows, Achilles tendons and postcervical muscles; (b) slowly progressive wasting and weakness of skeletal muscle; and (c) cardiomyopathy with severe conduction block. The responsible gene for the X-linked recessive form of this disease encodes an inner nuclear membrane protein named emerin. Although emerin is absent in tissues from patients with this disorder, it remains obscure why the loss of this widely expressed protein affects selectively skeletal muscle, heart and joints. As the first step to address this question, we examined the molecular regions of emerin that are essential for nuclear membrane targeting and stability of the protein. We found that the C-terminal hydrophobic region was necessary, but not sufficient, for nuclear membrane anchoring and stability of the protein. In the absence of this transmembrane domain, the upstream nucleoplasmic domain showed no firm association with the nuclear rim, but showed the tendency to accumulate at the nucleolus-like structures. Furthermore, proper targeting of emerin to the nuclear membrane required the latter half of the nucleoplasmic domain. These characteristics are distinct from those of lamina-associated polypeptide 2. Our findings indicate that emerin has distinct interactions with the inner nuclear membrane components that may be required for the stability and function of rigorously moving nuclei in tissues such as skeletal muscle, heart and joints. (+info)Cardiac involvement in Emery Dreifuss muscular dystrophy: a case series. (2/82)
Three patients with Emery Dreifuss muscular dystrophy are reported. Emery Dreifuss muscular dystrophy is an X linked muscular dystrophy, in which locomotor involvement is characteristically mild and slowly progressive. The effect on the heart becomes apparent in the teenage years and is characterised by cardiac conduction defects and infiltration of the myocardium by fibrous and adipose tissue. It first affects the atria, which results in atrial paralysis; treatment with ventricular pacing is usually needed. Female carriers can develop heart problems and are at risk of sudden death. Relatives of affected patients should be offered screening with electrocardiography and echocardiography. (+info)The Emery-Dreifuss muscular dystrophy phenotype arises from aberrant targeting and binding of emerin at the inner nuclear membrane. (3/82)
The product of the X-linked Emery-Dreifuss muscular dystrophy gene is a single-membrane-spanning protein called emerin, which is localized to the inner nuclear membrane of all tissues studied. To examine whether a number of the mutant forms of emerin expressed in patients are mislocalized, we transfected GFP-emerin cDNA constructs reflecting these mutations into undifferentiated C2C12 myoblasts and showed that both wild type and all the mutant emerins are targeted to the nuclear membrane, but the mutants to a lesser extent. Mutant Del236-241 (deletion in transmembrane region) was mainly expressed as cytoplasmic aggregates, with only trace amounts at the nuclear envelope. Complete removal of the transmembrane region and C-terminal tail relocated emerin to the nucleoplasm. Mutations in emerin's N-terminal domain had a less severe effect on disrupting nuclear envelope targeting. This data suggests that emerin contains multiple non-overlapping nuclear-membrane-targeting determinants. Analysis of material immunoisolated using emerin antibodies, from either undifferentiated C2C12 myoblasts or purified hepatocyte nuclei, demonstrated that both A- and B-type lamins and nuclear actin interact with emerin. This is the first report of proteins interacting with emerin. The EDMD phenotype can thus arise by either the absence or a reduction in emerin at the nuclear envelope, and both of these disrupt its interactions with that of structural components of the nucleus. We propose that an emerin-nuclear protein complex exists at the nuclear envelope and that one of its primary roles is to stabilize the nuclear membrane against the mechanical stresses that are generated in muscle cells during contraction. (+info)Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. (4/82)
BACKGROUND: Inherited mutations cause approximately 35 percent of cases of dilated cardiomyopathy; however, few genes associated with this disease have been identified. Previously, we located a gene defect that was responsible for autosomal dominant dilated cardiomyopathy and conduction-system disease on chromosome 1p1-q21, where nuclear-envelope proteins lamin A and lamin C are encoded by the LMNA (lamin A/C) gene. Mutations in the head or tail domain of this gene cause Emery-Dreifuss muscular dystrophy, a childhood-onset disease characterized by joint contractures and in some cases by abnormalities of cardiac conduction during adulthood. METHODS: We evaluated 11 families with autosomal dominant dilated cardiomyopathy and conduction-system disease. Sequences of the lamin A/C exons were determined in probands from each family, and variants were confirmed by restriction-enzyme digestion. The genotypes of the family members were ascertained. RESULTS: Five novel missense mutations were identified: four in the alpha-helical-rod domain of the lamin A/C gene, and one in the lamin C tail domain. Each mutation caused heritable, progressive conduction-system disease (sinus bradycardia, atrioventricular conduction block, or atrial arrhythmias) and dilated cardiomyopathy. Heart failure and sudden death occurred frequently within these families. No family members with mutations had either joint contractures or skeletal myopathy. Serum creatine kinase levels were normal in family members with mutations of the lamin rod but mildly elevated in some family members with a defect in the tail domain of lamin C. CONCLUSIONS: Genetic defects in distinct domains of the nuclear-envelope proteins lamin A and lamin C selectively cause dilated cardiomyopathy with conduction-system disease or autosomal dominant Emery-Dreifuss muscular dystrophy. Missense mutations in the rod domain of the lamin A/C gene provide a genetic cause for dilated cardiomyopathy and indicate that this intermediate filament protein has an important role in cardiac conduction and contractility. (+info)Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. (5/82)
Emery-Dreifuss muscular dystrophy (EMD) is a condition characterized by the clinical triad of early-onset contractures, progressive weakness in humeroperoneal muscles, and cardiomyopathy with conduction block. The disease was described for the first time as an X-linked muscular dystrophy, but autosomal dominant and autosomal recessive forms were reported. The genes for X-linked EMD and autosomal dominant EMD (AD-EMD) were identified. We report here that heterozygote mutations in LMNA, the gene for AD-EMD, may cause diverse phenotypes ranging from typical EMD to no phenotypic effect. Our results show that LMNA mutations are also responsible for the recessive form of the disease. Our results give further support to the notion that different genetic forms of EMD have a common pathophysiological background. The distribution of the mutations in AD-EMD patients (in the tail and in the 2A rod domain) suggests that unique interactions between lamin A/C and other nuclear components exist that have an important role in cardiac and skeletal muscle function. (+info)Clinical variability and molecular diagnosis in a four-generation family with X-linked Emery-Dreifuss muscular dystrophy. (6/82)
AIM: To describe the clinical variability of X-linked Emery-Dreifuss muscular dystrophy (X-EDMD) with cardiac involvement in a four-generation family with a novel mutation in the STA gene. METHODS: Clinical data were provided for 4 affected males and a female carrier. The Western blot analysis of emerin was performed on lymphoblastoid cell lines and followed by sequencing of the emerin gene. RESULTS: A thymine insertion at nucleotide 417 in exon 2, resulting in a frameshift with a premature stop codon at position 62 and absence of functional protein, was found in one of the three available patients. In ten-year-old proband's dizygotic twin-nephews the intermittent first-degree A-V block, atrial and ventricular ectopy, atrial runs, and exit sinus block were found, although the echocardiographic findings were normal. One of the twins also had short episodes of atrial fibrillation, idioventricular rhythm, and junctional rhythm. CONCLUSION: Cardiac abnormalities in the proband's ten-year-old dizygotic twins without evident clinical features suggestive of EDMD were remarkable in contrast to the oldest patient in the family, who lived to the age of 63 without a pacemaker, and to the proband who had a very early onset of muscle wasting and weakness, and a pacemaker implantation at the age of 27. This striking intra-familial variability in cardiac involvement associated with specific null mutation (417 ins T) has practical early diagnostic and possibly preventive implications. It also points at genetic and environmental factors as causes of clinical features in X-EDMD. (+info)Emery-Dreifuss muscular dystrophy: anatomical-clinical correlation (case report). (7/82)
We report on a man that had weakness of humeroperoneal distribution associated with limited range of motion of the cervical spine and elbows since he was 5 years old. At age 26 he developed tachycardia episodes. A complex arrhythmia was discovered, and a nodal ablation was done with a cardiac pacemaker implanted. The patient had an arrhythmia and sudden death followed this. Emery-Dreifuss muscular dystrophy is a rare recessive X-linked muscular disorder where mixed patterns in electromyography and muscle histology (neurogenic and/or myopathic) have caused nosological confusion. The autopsy findings are here described and correlated to the clinical features in an attempt to better understand the ambiguous findings concerning the process etiology. (+info)The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. (8/82)
To fully understand genome function, the linear genome map must be integrated with a spatial map of chromosomes in the nucleus. Distinct nuclear addresses for a few human chromosomes have been described. Previously we have demonstrated that the gene-rich human chromosome 19 is located in a more central position in the nucleus than the similarly sized, but gene-poor, chromosome 18. To determine whether these two chromosomes are a paradigm for the organization of chromatin in the human nucleus, we have now analysed the nuclear organization of every human chromosome in diploid lymphoblasts and primary fibroblasts. We find that the most gene-rich chromosomes concentrate at the centre of the nucleus, whereas the more gene-poor chromosomes are located towards the nuclear periphery. In contrast, we find no significant relationship between chromosome size and position within the nucleus. Proteins of the nuclear membrane or lamina are candidates for molecules that might anchor regions of the genome at the nuclear periphery and it has been suggested that disruption of this organization may play a role in some disease pathologies. We show that the intranuclear organization of chromosomes is not altered in cells that lack the integral nuclear membrane protein emerin, from an individual with X-linked Emery--Dreifuss muscular dystrophy. This suggests that emerin is not necessary for localizing chromosomes at the nuclear periphery and that the muscular dystrophy phenotype in such individuals is not due to grossly altered nuclear organization of chromatin. (+info)Emery-Dreifuss muscular dystrophy (EDMD) is a genetic disorder characterized by the triad of 1) early contractures of the elbow and Achilles tendons, 2) slowly progressive muscle weakness and wasting, which begins in the muscles around the shoulder and pelvis and later involves the arms and legs, and 3) cardiac conduction defects that can lead to serious heart rhythm abnormalities.
EDMD is caused by mutations in one of several genes, including the EMD, LMNA, FHL1, and SYNE1/2 genes. These genes provide instructions for making proteins that are important for maintaining the structure and function of muscle cells, as well as the electrical activity of the heart.
The symptoms of EDMD can vary in severity and age of onset, even among family members with the same genetic mutation. Treatment typically focuses on managing the symptoms of the disease, including physical therapy to maintain mobility, bracing or surgery for contractures, and medications to manage cardiac arrhythmias. In some cases, a heart transplant may be necessary.
Thymopoietins are a group of hormone-like polypeptides that play a crucial role in the development and differentiation of T-lymphocytes (T-cells) within the thymus gland. The term "thymopoietin" is often used to refer specifically to a particular polypeptide called thymopoietin alpha, which was first identified in the 1970s. Thymopoietin alpha helps to promote the differentiation of immature T-cells into mature T-cells, and it also contributes to the process of negative selection, whereby self-reactive T-cells are eliminated to prevent autoimmune disorders.
Other factors that contribute to thymopoiesis (the production of T-cells in the thymus) may also be referred to as thymopoietins, including interleukin-7 (IL-7), which is produced by stromal cells in the thymus and helps to support the survival and proliferation of immature T-cells.
Overall, thymopoietins play a critical role in maintaining immune homeostasis and preventing the development of autoimmune diseases.
Muscular dystrophies are a group of genetic disorders that primarily affect skeletal muscles, causing progressive weakness and degeneration. They are characterized by the lack or deficiency of a protein called dystrophin, which is essential for maintaining the integrity of muscle fibers. The most common form is Duchenne muscular dystrophy (DMD), but there are many other types with varying symptoms and severity. Over time, muscle wasting and weakness can lead to disability and shortened lifespan, depending on the type and progression of the disease. Treatment typically focuses on managing symptoms, maintaining mobility, and supporting quality of life.
Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by progressive muscle weakness and degeneration. It is caused by the absence of dystrophin, a protein that helps keep muscle cells intact. Without dystrophin, the muscle cells break down and are replaced with scar tissue, leading to loss of muscle function over time.
DMD primarily affects boys, as it is inherited in an X-linked recessive pattern, meaning that females who carry one affected X chromosome typically do not show symptoms but can pass the gene on to their offspring. Symptoms usually begin in early childhood and include difficulty with motor skills such as walking, running, and climbing stairs. Over time, the muscle weakness progresses and can lead to loss of ambulation, respiratory and cardiac complications, and ultimately, premature death.
Currently, there is no cure for DMD, but various treatments such as corticosteroids, physical therapy, and assisted ventilation can help manage symptoms and improve quality of life. Gene therapy approaches are also being investigated as potential treatments for this disorder.
I'm sorry for any confusion, but "Muscular Dystrophy, Animal" is not a standard medical term. Muscular Dystrophy is a group of genetic disorders that cause progressive weakness and loss of muscle mass. They are primarily human diseases and there are no known animal models of muscular dystrophy that directly correspond to any type of muscular dystrophy in humans.
However, scientists often use animals (like mice, dogs, and cats) as models for human diseases, including various types of muscular dystrophies. These animal models are used to study the disease process and to test potential treatments. For example, the mdx mouse is a well-known model of Duchenne Muscular Dystrophy (DMD), which is caused by a mutation in the dystrophin gene. This mouse lacks the muscle protein dystrophin, similar to humans with DMD, and shows many of the same symptoms, making it a valuable tool for research.
Dystrophin is a protein that provides structural stability to muscle fibers. It is an essential component of the dystrophin-glycoprotein complex, which helps maintain the integrity of the sarcolemma (the membrane surrounding muscle cells) during muscle contraction and relaxation. Dystrophin plays a crucial role in connecting the cytoskeleton of the muscle fiber to the extracellular matrix, allowing for force transmission and protecting the muscle cell from damage.
Mutations in the DMD gene, which encodes dystrophin, can lead to various forms of muscular dystrophy, including Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). In DMD, a severe form of the disease, genetic alterations typically result in little or no production of functional dystrophin, causing progressive muscle weakness, wasting, and degeneration. In BMD, a milder form of the disorder, partially functional dystrophin is produced, leading to less severe symptoms and later onset of the disease.
Myotonic dystrophy is a genetic disorder characterized by progressive muscle weakness, myotonia (delayed relaxation of muscles after contraction), and other symptoms. It is caused by an expansion of repetitive DNA sequences in the DMPK gene on chromosome 19 (type 1) or the ZNF9 gene on chromosome 3 (type 2). These expansions result in abnormal protein production and accumulation, which disrupt muscle function and can also affect other organs such as the heart, eyes, and endocrine system. Myotonic dystrophy is a progressive disease, meaning that symptoms tend to worsen over time. It is typically divided into two types: myotonic dystrophy type 1 (DM1), which is more common and severe, and myotonic dystrophy type 2 (DM2), which tends to be milder with a later onset of symptoms.
Limb-girdle muscular dystrophy (LGMD) is a group of rare inherited disorders that cause progressive weakness and wasting of the muscles in the arms and legs, particularly those around the shoulders and hips (the limb-girdle region). The condition affects both males and females and presents at different ages, depending on the specific type of LGMD.
LGMD is caused by mutations in various genes that play a role in maintaining muscle integrity and function. These genetic defects lead to a deficiency or dysfunction of certain proteins necessary for muscle health, ultimately resulting in muscle degeneration and weakness. There are more than 30 different subtypes of LGMD, each with its own set of causative genes and inheritance patterns (autosomal dominant or autosomal recessive).
Symptoms of limb-girdle muscular dystrophy may include:
1. Progressive muscle weakness and wasting in the arms, legs, shoulders, and hips
2. Difficulty with activities such as climbing stairs, lifting objects, or getting up from a seated position
3. Enlarged calf muscles (pseudohypertrophy) due to muscle degeneration and fat replacement
4. Muscle contractures, joint stiffness, and limited range of motion
5. Difficulty walking, using wheelchair assistance in advanced stages
6. Respiratory complications due to weakened chest muscles in some cases
Diagnosis of LGMD typically involves a combination of clinical evaluation, family history, muscle biopsy, genetic testing, and blood tests for creatine kinase (CK) levels, which are often elevated in muscular dystrophies. Treatment is primarily supportive and focuses on maintaining mobility, preventing complications, and preserving quality of life through physical therapy, assistive devices, and orthopedic interventions as needed. No cure currently exists for limb-girdle muscular dystrophy, but ongoing research aims to develop targeted therapies based on the underlying genetic defects.
Facioscapulohumeral Muscular Dystrophy (FSHD) is a genetic muscle disorder characterized by the progressive weakness and wasting (atrophy) of muscles in the face, shoulders, arms, and legs. It is caused by the abnormal expression of a gene called DUX4, which is normally only active during early embryonic development. In FSHD, this gene becomes reactivated in muscle cells, leading to their degeneration and death.
The symptoms of FSHD typically begin in late childhood or adolescence, although they can also appear in adulthood. The first noticeable sign is often difficulty raising the arms above the head or a weakened grip. Over time, the muscles of the face may become affected, leading to problems with smiling, swallowing, and speaking. The muscle weakness in FSHD tends to progress slowly, but it can vary widely from person to person. Some people with FSHD may require wheelchair assistance, while others may continue to walk with only minor limitations.
FSHD is inherited in an autosomal dominant manner, which means that a child has a 50% chance of inheriting the disease-causing gene from an affected parent. However, about 30% of cases are the result of new mutations and occur in people with no family history of the disorder. Currently, there is no cure for FSHD, but various treatments can help manage its symptoms and improve quality of life. These may include physical therapy, orthotics, assistive devices, and medications to treat pain or other complications.
'Mice, Inbred mdx' is a genetic strain of laboratory mice that are widely used as a model to study Duchenne muscular dystrophy (DMD), a severe and progressive muscle-wasting disorder in humans. The 'mdx' designation refers to the specific genetic mutation present in these mice, which is a point mutation in the gene encoding for dystrophin, a crucial protein involved in maintaining the structural integrity of muscle fibers.
Inbred mdx mice carry a spontaneous mutation in exon 23 of the dystrophin gene, resulting in the production of a truncated and nonfunctional form of the protein. This leads to a phenotype that closely resembles DMD in humans, including muscle weakness, degeneration, and fibrosis. The inbred nature of these mice ensures consistent genetic backgrounds and disease manifestations, making them valuable tools for studying the pathophysiology of DMD and testing potential therapies.
It is important to note that while the inbred mdx mouse model has been instrumental in advancing our understanding of DMD, it does not fully recapitulate all aspects of the human disease. Therefore, findings from these mice should be carefully interpreted and validated in more complex models or human studies before translating them into clinical applications.
Sarcoglycans are a group of proteins that are part of the dystrophin-glycoprotein complex in muscle cells. This complex helps to maintain the structural integrity of the muscle fiber by forming a link between the cytoskeleton and the extracellular matrix. Sarcoglycans are located on the surface of the muscle fiber and play a critical role in protecting the muscle from damage during contraction.
There are four main sarcoglycans, known as alpha, beta, gamma, and delta-sarcoglycan. Mutations in any one of these proteins can lead to a group of genetic disorders known as the sarcoglycanopathies, which are characterized by progressive muscle weakness and wasting. The most severe form of this disorder is called limb-girdle muscular dystrophy type 2C (LGMD2C), which is caused by mutations in the gamma-sarcoglycan gene.
In addition to their role in muscle cells, sarcoglycans have also been found to be expressed in other tissues, including the brain and the lungs, suggesting that they may have additional functions beyond their structural role in muscle.
Corneal dystrophies, hereditary are a group of genetic disorders that affect the cornea, which is the clear, outermost layer at the front of the eye. These conditions are characterized by the buildup of abnormal material in the cornea, leading to decreased vision, pain, or cloudiness in the eye.
There are many different types of corneal dystrophies, each affecting a specific layer of the cornea and having its own pattern of inheritance. Some common types include:
1. Fuchs' endothelial dystrophy: This affects the inner lining of the cornea (endothelium) and causes swelling and cloudiness in the cornea. It is typically inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the condition if one parent has it.
2. Granular dystrophy: This affects the stroma, which is the middle layer of the cornea. It causes the formation of opaque, grayish-white deposits in the cornea that can affect vision. It is typically inherited in an autosomal dominant or recessive manner.
3. Lattice dystrophy: This also affects the stroma and is characterized by the formation of a lattice-like pattern of fine, whitish lines in the cornea. It is typically inherited in an autosomal dominant manner.
4. Macular dystrophy: This affects the central part of the cornea (macula) and can cause cloudiness, leading to decreased vision. It is typically inherited in an autosomal recessive manner.
Treatment for corneal dystrophies may include eyedrops, medications, or surgery, depending on the severity of the condition and its impact on vision. In some cases, a corneal transplant may be necessary to restore vision.
Dystroglycans are a type of protein that play a crucial role in the structure and function of the muscle membrane (sarcolemma). They are an essential component of the dystrophin-glycoprotein complex, which helps maintain the stability and integrity of the sarcolemma during muscle contraction and relaxation.
Dystroglycans consist of two subunits: alpha-dystroglycan and beta-dystroglycan. Alpha-dystroglycan is a large, heavily glycosylated protein that extends from the intracellular space to the extracellular matrix, where it interacts with various extracellular matrix proteins such as laminin and agrin. Beta-dystroglycan, on the other hand, spans the muscle membrane and binds to dystrophin, a cytoskeletal protein that helps maintain the structural integrity of the sarcolemma.
Mutations in genes encoding for proteins involved in the glycosylation of alpha-dystroglycan can lead to a group of genetic disorders known as congenital muscular dystrophies, which are characterized by muscle weakness, hypotonia, and developmental delays. These disorders include Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy, and Muscle-Eye-Brain disease, among others.
Oculopharyngeal Muscular Dystrophy (OPMD) is a genetic disorder that affects the muscles, particularly those around the eyes and throat. The medical definition of OPMD, as per the National Organization for Rare Disorders (NORD), is:
"Oculopharyngeal Muscular Dystrophy (OPMD) is an inherited neuromuscular disorder characterized by progressive weakness of specific muscle groups, particularly those around the eyes (ocular) and throat (pharyngeal). The symptoms may include drooping of the eyelids (ptosis), difficulty swallowing (dysphagia), and, in some cases, proximal limb weakness. Onset of the disorder usually occurs in adulthood, typically after age 40, but earlier onsets have been reported."
The underlying cause of OPMD is a genetic mutation that leads to the production of an abnormal protein in muscle cells, ultimately resulting in muscle degeneration and weakness.
Utrophin is a protein that is found in muscle cells. It is similar in structure and function to dystrophin, which is a protein that is deficient or abnormal in people with Duchenne and Becker muscular dystrophy. Utrophin is present in both fetal and adult muscle, but its expression is usually limited to the nerve endings of the muscle fibers. However, in certain conditions such as muscle injury or disease, utrophin can be upregulated and expressed more widely throughout the muscle fiber. Research has shown that increasing the levels of utrophin in muscle cells could potentially compensate for the lack of dystrophin and provide a therapeutic approach to treating muscular dystrophy.
Skeletal muscle, also known as striated or voluntary muscle, is a type of muscle that is attached to bones by tendons or aponeuroses and functions to produce movements and support the posture of the body. It is composed of long, multinucleated fibers that are arranged in parallel bundles and are characterized by alternating light and dark bands, giving them a striped appearance under a microscope. Skeletal muscle is under voluntary control, meaning that it is consciously activated through signals from the nervous system. It is responsible for activities such as walking, running, jumping, and lifting objects.
Fuchs' Endothelial Dystrophy is a medical condition that affects the eye's cornea. It is a slowly progressing disorder that causes the endothelium, a thin layer of cells lining the inner surface of the cornea, to deteriorate and eventually fail to function properly. This results in swelling of the cornea, leading to cloudy vision, distorted vision, and sensitivity to light.
The condition is typically inherited and tends to affect both eyes. It is more common in women than in men and usually becomes apparent after the age of 50. There is no cure for Fuchs' Endothelial Dystrophy, but treatments such as corneal transplantation can help improve vision and alleviate symptoms.
Retinal dystrophies are a group of genetic eye disorders that primarily affect the retina, a light-sensitive layer at the back of the eye. These conditions are characterized by progressive degeneration and death of photoreceptor cells (rods and cones) in the retina, leading to vision loss.
The term "dystrophy" refers to a condition that results from the abnormal or defective development and function of tissues or organs. In the case of retinal dystrophies, the photoreceptor cells do not develop or function properly, resulting in visual impairment.
Retinal dystrophies can present at any age, from infancy to adulthood, and can have varying degrees of severity. Some common symptoms include night blindness, decreased visual acuity, loss of peripheral vision, light sensitivity, and color vision abnormalities.
Examples of retinal dystrophies include retinitis pigmentosa, Stargardt disease, Usher syndrome, and Leber congenital amaurosis, among others. These conditions are typically inherited and can be caused by mutations in various genes that play a role in the development and function of the retina.
There is currently no cure for retinal dystrophies, but research is ongoing to develop treatments that may slow or halt the progression of these conditions, such as gene therapy and stem cell transplantation.
Collagen Type VI is a type of collagen that is widely expressed in various tissues, including skeletal muscle, skin, and blood vessels. It is a major component of the extracellular matrix and plays important roles in maintaining tissue structure and function. Collagen Type VI forms microfilaments that provide structural support to the basement membrane and regulate cell-matrix interactions. Mutations in the genes encoding collagen Type VI can lead to several inherited connective tissue disorders, such as Bethlem myopathy and Ullrich congenital muscular dystrophy.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Dystrophin-associated proteins (DAPs) are a group of structural and functional proteins that interact with dystrophin, a cytoskeletal protein found in muscle cells. Dystrophin helps to maintain the integrity of the muscle fiber membrane, or sarcolemma, during contractions. The dystrophin-associated protein complex (DAPC) includes dystroglycans, sarcoglycans, syntrophins, and dystrobrevins, among others.
Mutations in genes encoding DAPs can lead to various forms of muscular dystrophy, a group of genetic disorders characterized by progressive muscle weakness and degeneration. For example, mutations in the sarcoglycan gene can cause limb-girdle muscular dystrophy type 2C (LGMD2C), while defects in dystroglycan can result in congenital muscular dystrophy with mental retardation and structural brain abnormalities.
In summary, DAPs are a group of proteins that interact with dystrophin to maintain the stability and function of muscle fibers. Defects in these proteins can lead to various forms of muscular dystrophy.
I'm sorry for any confusion, but "motorcycles" are not a medical term. Motorcycles are vehicles with two wheels and an engine, typically operated by a rider. They are not a medical condition or procedure. If you have any questions about motorcycle safety or injuries related to motorcycle accidents, I would be happy to provide information on those topics from a medical perspective.
I'm sorry for any confusion, but "Wisconsin" is a U.S. state located in the Midwest and is not a medical term or condition. If you have any medical questions or terms you would like defined, I'd be happy to help with those!
 Emery-Dreifuss muscular dystrophy
Emery-Dreifuss muscular dystrophy Emery-Dreifuss muscular dystrophy: MedlinePlus Genetics
Emery-Dreifuss muscular dystrophy: MedlinePlus Genetics Emery-Dreifuss Muscular Dystrophy Medication
Emery-Dreifuss Muscular Dystrophy Medication Emery-Dreifuss Muscular Dystrophy - Remember The Girls
Emery-Dreifuss Muscular Dystrophy - Remember The Girls Peter Wiley - Search Results - PubMed
Peter Wiley - Search Results - PubMed![TMEM43 transmembrane protein 43 [Homo sapiens (human)] - Gene - NCBI](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAYAAAAf8/9hAAAB1ElEQVQ4jaWSPWgTcRjGf/ehuWhobO2JxGJRY3taTTRV2yoqSpW6iIWO4iAoUsRBioNDKUWKLU7i4KA4OfhVREQnETRia03k7IdiS0LaQYKJQg3mLtfc30GySNUDn/V5nx/vy/vAf0pqad3db2xquiBJku93s2Tb2eEHdw1rTcsxol23sObTjN7oIp9KVmaU9kMdTxcLAyiqGtA0bfms+XKQULSdQG2EmnUx0q9ughAA8p/CFW0IN3Sv0vUI5p2zIMpUrd5JeP/Jii//80ZJUlrb9lyV8qn3zI5dB8A4MoBWtcITAKBmZe3eRmPzccYf9uIUsyzx6zQd7fMMAIjFdgxpkuPy4clFANbu6qa6fouybXtznxeAoqoBn0/zz5kvBqVQ5DBasJ5gXaPnDQAWFpwCkiwLZekyAMp2wTPAsqy5d8nEZcIHThPQo7jlIua9854BibdvekqKX8PouARAOn6F+c8pT4Bc7svz6U8f77O1cwDVV439PcPU4yHw8AUhhDPyOn4OfWOMuuZfBZp41INTLACorhC2/Jc2zsxMX8vl8lMcPBUHFL5mnpEZGa748sS42esKYS8WLtl2NjE22s/6fScIhtr48W2S5O0zIFwvp3vST6Z+myCvkaonAAAAAElFTkSuQmCC) TMEM43 transmembrane protein 43 [Homo sapiens (human)] - Gene - NCBI
TMEM43 transmembrane protein 43 [Homo sapiens (human)] - Gene - NCBI The DIY Scientist, the Olympian, and the Mutated Gene - ProPublica
The DIY Scientist, the Olympian, and the Mutated Gene - ProPublica Muscular Dystrophy Ireland | EASTER EGG HUNT - UNDER 12's
Muscular Dystrophy Ireland | EASTER EGG HUNT - UNDER 12's Medisinsk beskrivelse av Emery-Dreifuss' muskeldystrofi - Frambu
Medisinsk beskrivelse av Emery-Dreifuss' muskeldystrofi - Frambu Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US Feedback on Ortho Tri-Cyclen, page 163 - AskDocWeb.com
Feedback on Ortho Tri-Cyclen, page 163 - AskDocWeb.com![Recombinant Anti-Nesprin1/Syne-1 antibody [EPR14196] (ab192234) | Abcam](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAYAAAAf8/9hAAABm0lEQVQ4jaWTv0tbURTHP/cl75lqTIiNRFyEJIiUxNB2qf+D6NIuDg7WwcXFxU2yOznYte2klFIqpXVqoXQqgTYZKhURUURTlajJy++Xdx1eeJrmTc/vcuHc8/3cc869V8ileBZkClcSOcW9GUCmFPdmS16n4PfjKp/2KxQbJsmwxnwywOs/RUoNCcB0rI+xAbUb0DQls9tnbO7qHcC3OyWOigbn1RYA0aDXGbD8o8Dmro6qCOYS/Tx6qPHztMbGXx3Zzll5FuJppKe7hULNZD17jQA+TA0xGe21Nh4HmRj2sfjtAoDno36iQdUG2EPM5Gs0WpJ4SL01t7UwHqBPdZ63HTVMaxWOaWBK6Ri3AU8iPSgC9i6bfDmodCS9yhWpGs4AT3piIA3Qrykc6y1+ndV5v1cmX25xWGqy9vua1cyVbRh84GEk4CXk81gVy6WYja4YkpnP/9jaL3eckghrnOgGhZrV57vJCC9G/cB/19jrFXycHuLrUZXtgwpXdZPUoMbLZIA3dx5SMnx7jR0VuNG9/4ICIufaLWT2BlLHjkWr+SchAAAAAElFTkSuQmCC) Recombinant Anti-Nesprin1/Syne-1 antibody [EPR14196] (ab192234) | Abcam
Recombinant Anti-Nesprin1/Syne-1 antibody [EPR14196] (ab192234) | Abcam Genes | Free Full-Text | Multisystem Proteinopathy Due to VCP Mutations: A Review of Clinical Heterogeneity and Genetic...
Genes | Free Full-Text | Multisystem Proteinopathy Due to VCP Mutations: A Review of Clinical Heterogeneity and Genetic... RFA-AR-23-001: Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Centers (MDSRC) (P50 Clinical Trial Optional)
RFA-AR-23-001: Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Centers (MDSRC) (P50 Clinical Trial Optional) Common Conditions -- Respiratory Muscle Aids
Common Conditions -- Respiratory Muscle Aids Likely genetic source of muscle weakness found in six previously undiagnosed children | ScienceDaily
Likely genetic source of muscle weakness found in six previously undiagnosed children | ScienceDaily Quest - Article - The Great Trach Escape: Is it for You? | Muscular Dystrophy Association
Quest - Article - The Great Trach Escape: Is it for You? | Muscular Dystrophy Association aging<...
aging<...