Muscular Dystrophy, Oculopharyngeal
Poly(A)-Binding Protein II
Muscular Dystrophies
Poly(A)-Binding Protein I
Muscular Dystrophy, Duchenne
Muscular Dystrophy, Animal
Pharyngeal Muscles
Blepharoptosis
Health Level Seven
Dystrophin
Trinucleotide Repeat Expansion
Intranuclear Inclusion Bodies
Myotonic Dystrophy
Muscular Dystrophies, Limb-Girdle
Muscular Dystrophy, Facioscapulohumeral
Mice, Inbred mdx
Muscular Dystrophy, Emery-Dreifuss
Sarcoglycans
Corneal Dystrophies, Hereditary
Dystroglycans
Utrophin
Muscle, Skeletal
Fuchs' Endothelial Dystrophy
Thymopoietins
Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. (1/37)
Oculopharyngeal muscular dystrophy (OPMD) is a late-onset autosomal dominant muscular dystrophy that results from small expansions of a polyalanine tract in the PABPN1 gene. Intranuclear inclusions are the pathological hallmark of OPMD. The mechanism by which protein aggregation in OPMD might relate to a toxic gain-of-function has so far remained elusive. Whether protein aggregates themselves are pathogenic or are the consequence of an unidentified underlying molecular mechanism is still unclear. Here, we report that protein aggregation in a cell model of OPMD directly impaires the function of the ubiquitin-proteasome pathway (UPP) as well as molecular chaperone functions. The proteasome inhibitor lactacystin causes significant increase of protein aggregation and toxicity. Moreover, overexpression of molecular chaperones (HSP40 and HSP70) suppressed protein aggregation and toxicity. We also provide evidence that mPABPN1-ala17 protein aggregation proportionally correlates with toxicity. Furthermore, we show that co-expression of chaperones in our OPMD cell model increases the solubility of mPABPN1-ala17 and transfected cell survival rate. Our studies suggest that molecular regulators of polyalanine protein solubility and degradation may provide insights into new mechanisms in OPMD pathogenesis. Further analysis of the cellular and molecular mechanisms by which UPP and molecular chaperones influence the degradation of misfolded proteins could provide novel concepts and targets for the treatment and understanding of the pathogenesis of OPMD and neurodegenerative diseases. (+info)Trinucleotide expansions leading to an extended poly-L-alanine segment in the poly (A) binding protein PABPN1 cause fibril formation. (2/37)
The nuclear poly(A) binding protein (PABPN1) stimulates poly(A) polymerase and controls the lengths of poly(A) tails during pre-mRNA processing. The wild-type protein possesses 10 consecutive Ala residues immediately after the start methionine. Trinucleotide expansions in the coding sequence result in an extension of the Ala stretch to maximal 17 Ala residues in total. Individuals carrying the trinucleotide expansions suffer from oculopharyngeal muscular dystrophy (OPMD). Intranuclear inclusions consisting predominantly of PABPN1 have been recognized as a pathological hallmark of the genetic disorder. To elucidate the molecular events that lead to disease, recombinant PABPN1, and N-terminal fragments of the protein with varying poly-L-alanine stretches were analyzed. As the full-length protein displayed a strong tendency to aggregate into amorphous deposits, soluble N-terminal fragments were also studied. Expansion of the poly-L-alanine sequence to the maximal length observed in OPMD patients led to an increase of alpha-helical structure. Upon prolonged incubation the protein was found in fibrils that showed all characteristics of amyloid-like fibers. The lag-phase of fibril formation could be reduced by seeding. Structural analysis of the fibrils indicated antiparallel beta-sheets. (+info)Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy. (3/37)
Autosomal dominant oculopharyngeal muscular dystrophy (OPMD) is a late-onset disorder characterized clinically by progressive ptosis, dysphagia and limb weakness, and by unique intranuclear inclusions in the skeletal muscle fibers. The disease is caused by the expansion of a 10-alanine stretch to 12-17 alanine residues in the poly(A)-binding protein, nuclear 1 (PABPN1; PABP2). While PABPN1 is a major component of the inclusions in OPMD, the exact cause of the disease is unknown. To elucidate the molecular mechanism and to construct a useful model for therapeutic trials, we have generated transgenic mice expressing the hPABPN1. Transgenic mice lines expressing a normal hPABPN1 with 10-alanine stretch did not reveal myopathic changes, whereas lines expressing high levels of expanded hPABPN1 with a 13-alanine stretch showed an apparent myopathy phenotype, especially in old age. Pathological studies in the latter mice disclosed intranuclear inclusions consisting of aggregated mutant hPABPN1 product. Furthermore, some TUNEL positive nuclei were shown around degenerating fibers and a cluster of it in the lesion in necrotic muscle fibers. Interestingly, the degree of myopathic changes was more prominent in the eyelid and pharyngeal muscles. Further, muscle weakness in the limbs was apparent as shown by the fatigability test. Nuclear inclusions seemed to develop gradually with aging, at least after 1 week of age, in model mouse muscles. We established the first transgenic mouse model of OPMD by expressing mutated PABPN1, and our model mice appear to have more dramatic alternations in myofiber viability. (+info)Oculopharyngeal muscular dystrophy-like nuclear inclusions are present in normal magnocellular neurosecretory neurons of the hypothalamus. (4/37)
Intranuclear inclusions composed of tubular filaments constitute a pathological hallmark of oculopharyngeal muscular dystrophy (OPMD). Autosomal dominant OPMD is caused by (GCG) repeat expansions in the gene that encodes for poly(A) binding protein nuclear 1 (PABPN1). The mutation results in the expansion of a polyalanine stretch in the N-terminus of the protein. It has been proposed that mutated PABPN1 induces protein aggregation, which in turn causes the formation of the filamentous nuclear inclusions. Here we report the presence of intranuclear inclusions composed of tubular filaments in oxytocin-producing neurons from normal rat hypothalamus. Like OPMD inclusions, the filamentous structures in neurosecretory neurons accumulate PABPN1, poly(A) RNA, ubiquitin and proteasomes. These inclusions do not contain members of Hsp40 and HDJ-2/DNAJ families of chaperones. The proportion of oxytocin-producing neurons that contain inclusions decreases during parturition and lactation (when synthesis and release of oxytocin is maximal) and increases at 1 day post-weaning (when occurs a drastic reduction in the production of the hormone). Thus, PABPN1 filaments in normal neurons are dynamic structures, the appearance of which correlate with changes in cellular activity. These data provide the first physiological evidence that polyalanine expansions are not essential to induce polymerization of PABPN1 into filamentous nuclear inclusions. (+info)Autosomal recessive oculopharyngodistal myopathy: a distinct phenotypical, histological, and genetic entity. (5/37)
We present a 25 year follow up of two siblings with autosomal recessive (AR) oculopharyngodistal myopathy. Remarkable in these patients, in comparison with patients with oculopharyngeal muscular dystrophy (OPMD), are the earlier age of onset, severe facial weakness, external ophthalmoplegia early in the course of the disease, and distal weakness in the limbs. Histological features included basophilic-rimmed vacuoles, but the typical OPMD intranuclear filaments were absent. These clinical and histological characteristics are comparable with those of two Japanese patients with AR oculopharyngodistal myopathy. This myopathy has usually been described as an autosomal dominant (AD) muscle disorder. It shares some clinical and histological characteristics with OPMD, but most patients with AD oculopharyngodistal myopathy are genetically different. Here we exclude an expansion of the GCG repeat or any other mutation in the coding region of the PABPN1 gene (responsible for OPMD) in patients with AR oculopharyngodistal myopathy. From this we conclude that AR oculopharyngodistal myopathy is a distinct phenotypical, histological, and genetic entity. (+info)Induction of HSP70 expression and recruitment of HSC70 and HSP70 in the nucleus reduce aggregation of a polyalanine expansion mutant of PABPN1 in HeLa cells. (6/37)
Nuclear inclusions formed by the aggregation of a polyalanine expansion mutant of the nuclear poly(A)-binding protein (PABPN1) is a hallmark of oculopharyngeal muscular dystrophy (OPMD). OPMD is a dominant autosomal disease in which patients exhibit progressive difficulty of swallowing and eyelid elevation, starting around the age of 50. At present, there is no specific treatment to reduce the aggregate burden in patients. However, in cell culture models of OPMD, reduction of protein aggregation can be achieved by ectopic expression of HSP70. As gene transfer may not be the most effective means to elevate HSP70 levels, we tested four pharmacological agents for their ability to induce HSP70, recruit both HSP70 and HSC70 into the cell nucleus and reduce mutant PABPN1 aggregation in a HeLa cell culture model. We show here that exposure to moderate levels of ZnSO4, 8-hydroxyquinoline, ibuprofen and indomethacin produced a robust stress response resulting in the induction of HSP70 in HeLa cells expressing the mutant PABPN1 as a green fluorescent protein (GFP) fusion protein. Both HSP70 and the constitutive chaperone HSC70 localized in the nucleus of cells treated with any one of the four agents. This stress response was similar to what was observed following hyperthermia. All four agents also caused a significant reduction in the cellular burden of protein aggregates, as was judged by confocal microscopy and solubility changes of the aggregates. A concomitant reduction of cell death in drug-treated mutant PABPN1 expressing cells was also observed. (+info)Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. (7/37)
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant disease that presents in the fifth or sixth decade with dysphagia, ptosis and proximal limb weakness. OPMD is caused by the abnormal expansion of a polyalanine tract within the coding region of polyA binding protein nuclear 1 (PABPN1). The resultant mutant PABPN1 forms aggregates within the nuclei of skeletal muscle fibres. We have previously described a transgenic mouse model of OPMD that recapitulates the human disease and develops progressive muscle weakness accompanied by the formation of aggregates in skeletal muscle nuclei. The chemical chaperone trehalose has been used effectively to alleviate symptoms in a mouse model of Huntington's disease and is thought to elicit its effect by binding and stabilizing partially folded polyglutamine proteins and inhibiting the formation of aggregates. Here, we show that trehalose reduces aggregate formation and toxicity of mutant PABPN1 in cell models. Furthermore, oral administration of trehalose attenuated muscle weakness, reduced aggregate formation and decreased the number of TUNEL-labelled nuclei in skeletal muscle in an OPMD transgenic mouse model. Thus, anti-aggregation therapy may prove effective in the treatment of human OPMD. (+info)Prevention of oculopharyngeal muscular dystrophy-associated aggregation of nuclear polyA-binding protein with a single-domain intracellular antibody. (8/37)
Oculopharyngeal muscular dystrophy (OPMD) belongs to the group of protein aggregation disorders and is caused by extensions of the N-terminal polyalanine stretch of the nuclear polyA-binding protein 1 (PABPN1). The presence of PABPN1-containing intranuclear aggregates in skeletal muscle is unique for OPMD and is also observed in transgenic mouse and cell models for OPMD. These models consistently support a direct role for the protein aggregation in OPMD pathogenesis. We have isolated and characterized a diverse panel of single-domain antibody reagents (VHH), recognizing different epitopes in PABPN1. The antibody reagents specifically detect endogenous PABPN1 in cell lysates on western blot and label PABPN1 in cultured cells and muscle sections. When expressed intracellularly as intrabodies in a cellular model for OPMD, aggregation of PABPN1 was prevented in a dose-dependent manner. More importantly yet, these intrabodies could also reduce the presence of already existing aggregates. Given the domain specificity of VHH-mediated aggregation interference, this approach at least allows the definition of the nucleation kernel in aggregation-prone proteins, thus facilitating etiological insight into this and other protein aggregation disorders, and ultimately, it may well provide useful therapeutic agents. (+info)Oculopharyngeal Muscular Dystrophy (OPMD) is a genetic disorder that affects the muscles, particularly those around the eyes and throat. The medical definition of OPMD, as per the National Organization for Rare Disorders (NORD), is:
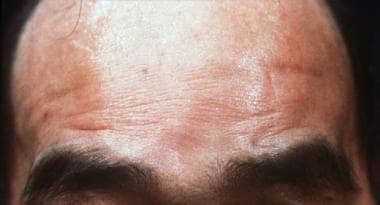
"Oculopharyngeal Muscular Dystrophy (OPMD) is an inherited neuromuscular disorder characterized by progressive weakness of specific muscle groups, particularly those around the eyes (ocular) and throat (pharyngeal). The symptoms may include drooping of the eyelids (ptosis), difficulty swallowing (dysphagia), and, in some cases, proximal limb weakness. Onset of the disorder usually occurs in adulthood, typically after age 40, but earlier onsets have been reported."
The underlying cause of OPMD is a genetic mutation that leads to the production of an abnormal protein in muscle cells, ultimately resulting in muscle degeneration and weakness.
Muscular dystrophies are a group of genetic disorders that primarily affect skeletal muscles, causing progressive weakness and degeneration. They are characterized by the lack or deficiency of a protein called dystrophin, which is essential for maintaining the integrity of muscle fibers. The most common form is Duchenne muscular dystrophy (DMD), but there are many other types with varying symptoms and severity. Over time, muscle wasting and weakness can lead to disability and shortened lifespan, depending on the type and progression of the disease. Treatment typically focuses on managing symptoms, maintaining mobility, and supporting quality of life.
Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by progressive muscle weakness and degeneration. It is caused by the absence of dystrophin, a protein that helps keep muscle cells intact. Without dystrophin, the muscle cells break down and are replaced with scar tissue, leading to loss of muscle function over time.
DMD primarily affects boys, as it is inherited in an X-linked recessive pattern, meaning that females who carry one affected X chromosome typically do not show symptoms but can pass the gene on to their offspring. Symptoms usually begin in early childhood and include difficulty with motor skills such as walking, running, and climbing stairs. Over time, the muscle weakness progresses and can lead to loss of ambulation, respiratory and cardiac complications, and ultimately, premature death.
Currently, there is no cure for DMD, but various treatments such as corticosteroids, physical therapy, and assisted ventilation can help manage symptoms and improve quality of life. Gene therapy approaches are also being investigated as potential treatments for this disorder.
I'm sorry for any confusion, but "Muscular Dystrophy, Animal" is not a standard medical term. Muscular Dystrophy is a group of genetic disorders that cause progressive weakness and loss of muscle mass. They are primarily human diseases and there are no known animal models of muscular dystrophy that directly correspond to any type of muscular dystrophy in humans.
However, scientists often use animals (like mice, dogs, and cats) as models for human diseases, including various types of muscular dystrophies. These animal models are used to study the disease process and to test potential treatments. For example, the mdx mouse is a well-known model of Duchenne Muscular Dystrophy (DMD), which is caused by a mutation in the dystrophin gene. This mouse lacks the muscle protein dystrophin, similar to humans with DMD, and shows many of the same symptoms, making it a valuable tool for research.
The pharyngeal muscles, also known as the musculature of the pharynx, are a group of skeletal muscles that make up the walls of the pharynx, which is the part of the throat located just above the esophagus and behind the nasal and oral cavities. These muscles play a crucial role in several vital functions, including:
1. Swallowing (deglutition): The pharyngeal muscles contract in a coordinated sequence to propel food or liquids from the mouth through the pharynx and into the esophagus during swallowing.
2. Speech: The contraction and relaxation of these muscles help shape the sounds produced by the vocal cords, contributing to the production of speech.
3. Respiration: The pharyngeal muscles assist in maintaining an open airway during breathing, especially during sleep and when the upper airways are obstructed.
The pharyngeal muscles consist of three layers: the outer circular muscle layer, the middle longitudinal muscle layer, and the inner inferior constrictor muscle layer. The specific muscles that make up these layers include:
1. Superior constrictor muscle (outer circular layer)
2. Middle constrictor muscle (middle longitudinal layer)
3. Inferior constrictor muscle (inner inferior constrictor layer)
4. Stylopharyngeus muscle
5. Salpingopharyngeus muscle
6. Palatopharyngeus muscle
7. Buccinator muscle (partially contributes to the middle longitudinal layer)
These muscles work together to perform their various functions, and any dysfunction in these muscles can lead to problems like swallowing difficulties (dysphagia), speech impairments, or respiratory issues.
Blepharoptosis is a medical term that refers to the drooping or falling of the upper eyelid. It is usually caused by weakness or paralysis of the muscle that raises the eyelid, known as the levator palpebrae superioris. This condition can be present at birth or acquired later in life due to various factors such as aging, nerve damage, eye surgery complications, or certain medical conditions like myasthenia gravis or brain tumors. Blepharoptosis may obstruct vision and cause difficulty with daily activities, and treatment options include eyedrops, eye patches, or surgical correction.
Health Level Seven (HL7) is a set of international standards for the transfer of clinical and administrative data between software applications used by various healthcare providers. The standards are developed and maintained by Health Level Seven International, an organization accredited by the American National Standards Institute.
The HL7 standards define the structure and format of the messages that are exchanged between different systems, such as electronic health records (EHRs), laboratory information systems, and radiology information systems. The messages contain clinical data, such as patient demographics, medication orders, and test results, as well as administrative data, such as billing information.
The HL7 standards are designed to be flexible and extensible, allowing for the integration of new data elements and message types as needed. They support a wide range of communication protocols, including file-based exchange, messaging using TCP/IP, and web services.
By providing a standardized way of exchanging healthcare data, HL7 helps to improve the efficiency and accuracy of care delivery, reduce costs, and enhance patient safety. It also facilitates the integration of disparate systems and enables the sharing of clinical data across different healthcare organizations.
Dystrophin is a protein that provides structural stability to muscle fibers. It is an essential component of the dystrophin-glycoprotein complex, which helps maintain the integrity of the sarcolemma (the membrane surrounding muscle cells) during muscle contraction and relaxation. Dystrophin plays a crucial role in connecting the cytoskeleton of the muscle fiber to the extracellular matrix, allowing for force transmission and protecting the muscle cell from damage.
Mutations in the DMD gene, which encodes dystrophin, can lead to various forms of muscular dystrophy, including Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). In DMD, a severe form of the disease, genetic alterations typically result in little or no production of functional dystrophin, causing progressive muscle weakness, wasting, and degeneration. In BMD, a milder form of the disorder, partially functional dystrophin is produced, leading to less severe symptoms and later onset of the disease.
Trinucleotide Repeat Expansion is a genetic mutation where a sequence of three DNA nucleotides is repeated more frequently than what is typically found in the general population. In this type of mutation, the number of repeats can expand or increase from one generation to the next, leading to an increased risk of developing certain genetic disorders.
These disorders are often neurological and include conditions such as Huntington's disease, myotonic dystrophy, fragile X syndrome, and Friedreich's ataxia. The severity of these diseases can be related to the number of repeats present in the affected gene, with a higher number of repeats leading to more severe symptoms or an earlier age of onset.
It is important to note that not all trinucleotide repeat expansions will result in disease, and some people may carry these mutations without ever developing any symptoms. However, if the number of repeats crosses a certain threshold, it can lead to genetic instability and an increased risk of disease development.
Intranuclear inclusion bodies are abnormal, rounded structures found within the nucleus of a cell. They are composed of aggregated proteins or other cellular components and can be associated with various viral infections and certain genetic disorders. These inclusion bodies can interfere with normal nuclear functions, leading to cell damage and contributing to the pathogenesis of diseases such as cytomegalovirus infection, rabies, and some forms of neurodegenerative disorders like polyglutamine diseases. The presence of intranuclear inclusion bodies is often used in diagnostic pathology to help identify specific underlying conditions.
Myotonic dystrophy is a genetic disorder characterized by progressive muscle weakness, myotonia (delayed relaxation of muscles after contraction), and other symptoms. It is caused by an expansion of repetitive DNA sequences in the DMPK gene on chromosome 19 (type 1) or the ZNF9 gene on chromosome 3 (type 2). These expansions result in abnormal protein production and accumulation, which disrupt muscle function and can also affect other organs such as the heart, eyes, and endocrine system. Myotonic dystrophy is a progressive disease, meaning that symptoms tend to worsen over time. It is typically divided into two types: myotonic dystrophy type 1 (DM1), which is more common and severe, and myotonic dystrophy type 2 (DM2), which tends to be milder with a later onset of symptoms.
Limb-girdle muscular dystrophy (LGMD) is a group of rare inherited disorders that cause progressive weakness and wasting of the muscles in the arms and legs, particularly those around the shoulders and hips (the limb-girdle region). The condition affects both males and females and presents at different ages, depending on the specific type of LGMD.
LGMD is caused by mutations in various genes that play a role in maintaining muscle integrity and function. These genetic defects lead to a deficiency or dysfunction of certain proteins necessary for muscle health, ultimately resulting in muscle degeneration and weakness. There are more than 30 different subtypes of LGMD, each with its own set of causative genes and inheritance patterns (autosomal dominant or autosomal recessive).
Symptoms of limb-girdle muscular dystrophy may include:
1. Progressive muscle weakness and wasting in the arms, legs, shoulders, and hips
2. Difficulty with activities such as climbing stairs, lifting objects, or getting up from a seated position
3. Enlarged calf muscles (pseudohypertrophy) due to muscle degeneration and fat replacement
4. Muscle contractures, joint stiffness, and limited range of motion
5. Difficulty walking, using wheelchair assistance in advanced stages
6. Respiratory complications due to weakened chest muscles in some cases
Diagnosis of LGMD typically involves a combination of clinical evaluation, family history, muscle biopsy, genetic testing, and blood tests for creatine kinase (CK) levels, which are often elevated in muscular dystrophies. Treatment is primarily supportive and focuses on maintaining mobility, preventing complications, and preserving quality of life through physical therapy, assistive devices, and orthopedic interventions as needed. No cure currently exists for limb-girdle muscular dystrophy, but ongoing research aims to develop targeted therapies based on the underlying genetic defects.
Facioscapulohumeral Muscular Dystrophy (FSHD) is a genetic muscle disorder characterized by the progressive weakness and wasting (atrophy) of muscles in the face, shoulders, arms, and legs. It is caused by the abnormal expression of a gene called DUX4, which is normally only active during early embryonic development. In FSHD, this gene becomes reactivated in muscle cells, leading to their degeneration and death.
The symptoms of FSHD typically begin in late childhood or adolescence, although they can also appear in adulthood. The first noticeable sign is often difficulty raising the arms above the head or a weakened grip. Over time, the muscles of the face may become affected, leading to problems with smiling, swallowing, and speaking. The muscle weakness in FSHD tends to progress slowly, but it can vary widely from person to person. Some people with FSHD may require wheelchair assistance, while others may continue to walk with only minor limitations.
FSHD is inherited in an autosomal dominant manner, which means that a child has a 50% chance of inheriting the disease-causing gene from an affected parent. However, about 30% of cases are the result of new mutations and occur in people with no family history of the disorder. Currently, there is no cure for FSHD, but various treatments can help manage its symptoms and improve quality of life. These may include physical therapy, orthotics, assistive devices, and medications to treat pain or other complications.
'Mice, Inbred mdx' is a genetic strain of laboratory mice that are widely used as a model to study Duchenne muscular dystrophy (DMD), a severe and progressive muscle-wasting disorder in humans. The 'mdx' designation refers to the specific genetic mutation present in these mice, which is a point mutation in the gene encoding for dystrophin, a crucial protein involved in maintaining the structural integrity of muscle fibers.
Inbred mdx mice carry a spontaneous mutation in exon 23 of the dystrophin gene, resulting in the production of a truncated and nonfunctional form of the protein. This leads to a phenotype that closely resembles DMD in humans, including muscle weakness, degeneration, and fibrosis. The inbred nature of these mice ensures consistent genetic backgrounds and disease manifestations, making them valuable tools for studying the pathophysiology of DMD and testing potential therapies.
It is important to note that while the inbred mdx mouse model has been instrumental in advancing our understanding of DMD, it does not fully recapitulate all aspects of the human disease. Therefore, findings from these mice should be carefully interpreted and validated in more complex models or human studies before translating them into clinical applications.
Emery-Dreifuss muscular dystrophy (EDMD) is a genetic disorder characterized by the triad of 1) early contractures of the elbow and Achilles tendons, 2) slowly progressive muscle weakness and wasting, which begins in the muscles around the shoulder and pelvis and later involves the arms and legs, and 3) cardiac conduction defects that can lead to serious heart rhythm abnormalities.
EDMD is caused by mutations in one of several genes, including the EMD, LMNA, FHL1, and SYNE1/2 genes. These genes provide instructions for making proteins that are important for maintaining the structure and function of muscle cells, as well as the electrical activity of the heart.
The symptoms of EDMD can vary in severity and age of onset, even among family members with the same genetic mutation. Treatment typically focuses on managing the symptoms of the disease, including physical therapy to maintain mobility, bracing or surgery for contractures, and medications to manage cardiac arrhythmias. In some cases, a heart transplant may be necessary.
Sarcoglycans are a group of proteins that are part of the dystrophin-glycoprotein complex in muscle cells. This complex helps to maintain the structural integrity of the muscle fiber by forming a link between the cytoskeleton and the extracellular matrix. Sarcoglycans are located on the surface of the muscle fiber and play a critical role in protecting the muscle from damage during contraction.
There are four main sarcoglycans, known as alpha, beta, gamma, and delta-sarcoglycan. Mutations in any one of these proteins can lead to a group of genetic disorders known as the sarcoglycanopathies, which are characterized by progressive muscle weakness and wasting. The most severe form of this disorder is called limb-girdle muscular dystrophy type 2C (LGMD2C), which is caused by mutations in the gamma-sarcoglycan gene.
In addition to their role in muscle cells, sarcoglycans have also been found to be expressed in other tissues, including the brain and the lungs, suggesting that they may have additional functions beyond their structural role in muscle.
Corneal dystrophies, hereditary are a group of genetic disorders that affect the cornea, which is the clear, outermost layer at the front of the eye. These conditions are characterized by the buildup of abnormal material in the cornea, leading to decreased vision, pain, or cloudiness in the eye.
There are many different types of corneal dystrophies, each affecting a specific layer of the cornea and having its own pattern of inheritance. Some common types include:
1. Fuchs' endothelial dystrophy: This affects the inner lining of the cornea (endothelium) and causes swelling and cloudiness in the cornea. It is typically inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the condition if one parent has it.
2. Granular dystrophy: This affects the stroma, which is the middle layer of the cornea. It causes the formation of opaque, grayish-white deposits in the cornea that can affect vision. It is typically inherited in an autosomal dominant or recessive manner.
3. Lattice dystrophy: This also affects the stroma and is characterized by the formation of a lattice-like pattern of fine, whitish lines in the cornea. It is typically inherited in an autosomal dominant manner.
4. Macular dystrophy: This affects the central part of the cornea (macula) and can cause cloudiness, leading to decreased vision. It is typically inherited in an autosomal recessive manner.
Treatment for corneal dystrophies may include eyedrops, medications, or surgery, depending on the severity of the condition and its impact on vision. In some cases, a corneal transplant may be necessary to restore vision.
Dystroglycans are a type of protein that play a crucial role in the structure and function of the muscle membrane (sarcolemma). They are an essential component of the dystrophin-glycoprotein complex, which helps maintain the stability and integrity of the sarcolemma during muscle contraction and relaxation.
Dystroglycans consist of two subunits: alpha-dystroglycan and beta-dystroglycan. Alpha-dystroglycan is a large, heavily glycosylated protein that extends from the intracellular space to the extracellular matrix, where it interacts with various extracellular matrix proteins such as laminin and agrin. Beta-dystroglycan, on the other hand, spans the muscle membrane and binds to dystrophin, a cytoskeletal protein that helps maintain the structural integrity of the sarcolemma.
Mutations in genes encoding for proteins involved in the glycosylation of alpha-dystroglycan can lead to a group of genetic disorders known as congenital muscular dystrophies, which are characterized by muscle weakness, hypotonia, and developmental delays. These disorders include Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy, and Muscle-Eye-Brain disease, among others.
Utrophin is a protein that is found in muscle cells. It is similar in structure and function to dystrophin, which is a protein that is deficient or abnormal in people with Duchenne and Becker muscular dystrophy. Utrophin is present in both fetal and adult muscle, but its expression is usually limited to the nerve endings of the muscle fibers. However, in certain conditions such as muscle injury or disease, utrophin can be upregulated and expressed more widely throughout the muscle fiber. Research has shown that increasing the levels of utrophin in muscle cells could potentially compensate for the lack of dystrophin and provide a therapeutic approach to treating muscular dystrophy.
Skeletal muscle, also known as striated or voluntary muscle, is a type of muscle that is attached to bones by tendons or aponeuroses and functions to produce movements and support the posture of the body. It is composed of long, multinucleated fibers that are arranged in parallel bundles and are characterized by alternating light and dark bands, giving them a striped appearance under a microscope. Skeletal muscle is under voluntary control, meaning that it is consciously activated through signals from the nervous system. It is responsible for activities such as walking, running, jumping, and lifting objects.
Fuchs' Endothelial Dystrophy is a medical condition that affects the eye's cornea. It is a slowly progressing disorder that causes the endothelium, a thin layer of cells lining the inner surface of the cornea, to deteriorate and eventually fail to function properly. This results in swelling of the cornea, leading to cloudy vision, distorted vision, and sensitivity to light.
The condition is typically inherited and tends to affect both eyes. It is more common in women than in men and usually becomes apparent after the age of 50. There is no cure for Fuchs' Endothelial Dystrophy, but treatments such as corneal transplantation can help improve vision and alleviate symptoms.
Thymopoietins are a group of hormone-like polypeptides that play a crucial role in the development and differentiation of T-lymphocytes (T-cells) within the thymus gland. The term "thymopoietin" is often used to refer specifically to a particular polypeptide called thymopoietin alpha, which was first identified in the 1970s. Thymopoietin alpha helps to promote the differentiation of immature T-cells into mature T-cells, and it also contributes to the process of negative selection, whereby self-reactive T-cells are eliminated to prevent autoimmune disorders.
Other factors that contribute to thymopoiesis (the production of T-cells in the thymus) may also be referred to as thymopoietins, including interleukin-7 (IL-7), which is produced by stromal cells in the thymus and helps to support the survival and proliferation of immature T-cells.
Overall, thymopoietins play a critical role in maintaining immune homeostasis and preventing the development of autoimmune diseases.
 Oculopharyngeal muscular dystrophy
Oculopharyngeal muscular dystrophy
Poly(A)-binding protein
PABPN1
David C. Rubinsztein
Oropharyngeal dysphagia
Ptosis (eyelid)
DNAJA1
DNA-directed RNA interference
SNW1
Benitec Biopharma
William Powers Jr.
List of diseases (O)
List of MeSH codes (C05)
Michel Fardeau
Muscular Dystrophy Community Assistance Research and Education Amendments of 2001
Paul D. Wellstone Muscular Dystrophy Community Assistance, Research and Education Amendments of 2013
Muscular system
List of MeSH codes (C10)
List of MeSH codes (C16)
List of OMIM disorder codes
Oculopharyngeal muscular dystrophy - Wikipedia
 Oculopharyngeal muscular dystrophy: MedlinePlus Genetics
Oculopharyngeal muscular dystrophy: MedlinePlus Genetics
 Muscle Ageing and Oculopharyngeal Muscular Dystrophy (OPMD) | LUMC
Muscle Ageing and Oculopharyngeal Muscular Dystrophy (OPMD) | LUMC
 Oculopharyngeal muscular dystrophy (OPMD)
Oculopharyngeal muscular dystrophy (OPMD)
 Oculopharyngeal Muscular Dystrophy (OPMD) | Neuro Notes
Oculopharyngeal Muscular Dystrophy (OPMD) | Neuro Notes
 Sporadic Inclusion Body Myositis: Practice Essentials, Background, Pathophysiology
Sporadic Inclusion Body Myositis: Practice Essentials, Background, Pathophysiology
 Multiple sclerosis - Annals Singapore
Multiple sclerosis - Annals Singapore
 Donders Thesis Series - Donders Institute
Donders Thesis Series - Donders Institute
 Trials and treatments developed with our support | AFM Téléthon
Trials and treatments developed with our support | AFM Téléthon
Jocelyn LAPORTE | IGBMC
Muscular Dystrophy Differential Diagnoses
Muscular Dystrophy | MD | MedlinePlus
Shorter Lab: Publications
 Ptosis Correction - StatPearls - NCBI Bookshelf
Ptosis Correction - StatPearls - NCBI Bookshelf
Michael Antoniou - Research output - King's College London
 Poly(A)-Binding Protein II | Harvard Catalyst Profiles | Harvard Catalyst
Poly(A)-Binding Protein II | Harvard Catalyst Profiles | Harvard Catalyst
 RFA-AR-23-001: Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Centers (MDSRC) (P50 Clinical Trial Optional)
RFA-AR-23-001: Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Centers (MDSRC) (P50 Clinical Trial Optional)
Ophthalmologic Manifestations of Myasthenia Gravis: Overview, Patient History, Physical Examination
 WikiGenes - Adoxa - (2Z,4S,4aR,5S,5aS,6R,12aS)-2-(amino...
WikiGenes - Adoxa - (2Z,4S,4aR,5S,5aS,6R,12aS)-2-(amino...
 Team 3- Cell and Molecular Orchestration in Muscle Regeneration, Ageing and Diseases - Institut de Myologie
Team 3- Cell and Molecular Orchestration in Muscle Regeneration, Ageing and Diseases - Institut de Myologie
 Browse by UCL Departments and Centres
-
UCL Discovery
Browse by UCL Departments and Centres
-
UCL Discovery
 UIC Anesthesiology at MARC 2023 | Chicago Medicine
UIC Anesthesiology at MARC 2023 | Chicago Medicine
So you'd like to open a restaurant in the Dominican Republic?
 muscular dystrophy - 국내최대의 영어사전, 전문용어, 의학 용어도 OK
muscular dystrophy - 국내최대의 영어사전, 전문용어, 의학 용어도 OK
 הרפואה - עיתון ההסתדרות הרפואית בישראל | דצמבר 2000
הרפואה - עיתון ההסתדרות הרפואית בישראל | דצמבר 2000
Find Research outputs - The University of Brighton
 Genes | Free Full-Text | Multisystem Proteinopathy Due to VCP Mutations: A Review of Clinical Heterogeneity and Genetic...
Genes | Free Full-Text | Multisystem Proteinopathy Due to VCP Mutations: A Review of Clinical Heterogeneity and Genetic...
 Helping make dysphagia less hard to swallow - McGill Reporter
Helping make dysphagia less hard to swallow - McGill Reporter
 Muscular dystrophy | Healthify
Muscular dystrophy | Healthify
Voermans N Catalogue en ligne
OPMD9
- Oculopharyngeal muscular dystrophy (OPMD) is a rare form of muscular dystrophy with symptoms generally starting when an individual is 40 to 50 years old. (wikipedia.org)
- The PABPN1 mutation contains a GCG trinucleotide repeat at the 5' end of the coding region, and expansion of this repeat which then leads to autosomal dominant oculopharyngeal muscular dystrophy (OPMD) disease. (wikipedia.org)
- Oculopharyngeal muscular dystrophy (OPMD) is a rare, autosomal dominant, late-onset myopathy. (lumc.nl)
- Oculopharyngeal muscular dystrophy (OPMD) is a rare genetic condition that causes weakness in the muscles around the upper eyelids and part of the throat called the pharynx. (digitaldiagnosticsolutions.com)
- The ultrastructural images demonstrate the presence of finely fibrillary (~8.5nm thickness) material within the nucleus characteristic of Oculopharyngeal Muscular Dystrophy (OPMD) due to trinucleotide repeat expansion (poly-alanine) in the PABPN1 gene. (arkanalabs.com)
- Our team is working on the molecular and cellular actors involved in human muscle regeneration , in muscle ageing and in muscular dystrophies including oculopharyngeal muscular dystrophy (OPMD) and Duchenne muscular dystrophy (DMD). (institut-myologie.org)
- J-P has a neuro-muscular condition known as Oculopharyngeal muscular dystrophy or OPMD, a debilitating but non-life-threatening form of muscular dystrophy. (dominicantoday.com)
- I collaborated on the development of a gene therapy AAV vector for Oculopharyngeal Muscular Dystrophy (OPMD), a rare muscle disease. (royalholloway.ac.uk)
- We use gene therapy technologies to target molecular defects in pre-clinical models of rare neuromuscular diseases such as Duchenne muscular dystrophy (DMD), Oculopharyngeal muscular dystrophy (OPMD) and Facioscapulohumeral muscular dystrophy (FSHD). (royalholloway.ac.uk)
Types of muscular dystrophy2
- What are the types of muscular dystrophy (MD)? (medlineplus.gov)
- There are several recognised types of muscular dystrophy (MD). These are described below. (healthify.nz)
Myotonic dystrophy3
- Metformin (N,N-dimethyl-biguanide) for Steinert myotonic dystrophy. (afm-telethon.fr)
- Methods We analysed 104 in-laboratory sleep studies of 73 patients with MD with five common types (DMD-Duchenne, Becker MD, CMD-congenital, LGMD-limb-girdle and DM-myotonic dystrophy). (bmj.com)
- This study looks into five major types of MD (Duchenne MD, Becker MD, congenital MD, LGMD-limb-girdle and DM-myotonic dystrophy). (bmj.com)
Facioscapulohumeral muscular3
- Facioscapulohumeral muscular dystrophy, which often starts in the teenage years. (medlineplus.gov)
- Journal of medical genetics SMCHD1 mutation spectrum for facioscapulohumeral muscular dystrophy type 2 (FSHD2) and Bosma arhinia microphthalmia syndrome (BAMS) reveals disease-specific localisation of variants in the ATPase domain. (myobase.org)
- Facioscapulohumeral muscular dystrophy (FSH, FSHD). (cvs.com)
Distal1
- Muscular atrophy affecting muscles in the distal portions of the extremities. (nih.gov)
Becker8
- Becker muscular dystrophy, which is similar to Duchenne but is less severe and gets worse more slowly. (medlineplus.gov)
- Like Duchenne muscular dystrophy, Becker muscular dystrophy typically affects only males (1 in 30,000) and causes heart problems. (healthnbeautytips.co)
- Duchenne dystrophy and Becker dystrophy are more common in boys. (cvs.com)
- Duchenne and Becker muscular dystrophies. (cvs.com)
- Available at: https://www.dynamed.com/condition/duchenne-and-becker-muscular-dystrophies. (cvs.com)
- М'язова дистрофія Дюшенна та м'язова дистрофія Беккера Duchenne muscular dystrophy and Becker muscular dystrophy are X-linked recessive disorders characterized by progressive proximal muscle weakness caused by muscle fiber degeneration. (msdmanuals.com)
- Becker dystrophy. (msdmanuals.com)
- Becker dystrophy, although closely related to Duchenne, has a later onset and causes milder symptoms. (msdmanuals.com)
Rare form of muscular dystrophy1
- This rare form of muscular dystrophy appears from childhood to early teens and affects mainly males. (healthnbeautytips.co)
Autosomal2
- In most cases oculopharyngeal muscular dystrophy is inherited via autosomal dominance. (wikipedia.org)
- Emery-Dreifuss muscular dystrophy with autosomal dominant transmission. (medscape.com)
Limb-girdle-mu1
- In its most common form, Limb-girdle muscular dystrophy causes progressive weakness that begins in the hips and moves to the shoulders, arms, and legs. (healthnbeautytips.co)
Emery-dreifuss-1
- Emery-Dreifuss muscular dystrophy. (medscape.com)
Duchenne Muscular18
- Gene therapy trial to assess a microdystrophin (abbreviated version of the Duchenne muscular dystrophy gene) associated with an AVV vector. (afm-telethon.fr)
- This drug, used in oncology for almost 40 years, is assessed in Duchenne muscular dystrophy, following preclinical studies supported by the AFM-Telethon. (afm-telethon.fr)
- Dystrophin: the protein product of the Duchenne muscular dystrophy locus. (medscape.com)
- Emery AEH, Muntoni F, Quinlivan R. Duchenne Muscular Dystrophy (Oxford Monographs on Medical Genetics) . (medscape.com)
- Donders J, Taneja C. Neurobehavioral characteristics of children with Duchenne muscular dystrophy. (medscape.com)
- Intelligence and the gene for Duchenne muscular dystrophy. (medscape.com)
- Leibowitz D, Dubowitz V. Intellect and behaviour in Duchenne muscular dystrophy. (medscape.com)
- Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. (medscape.com)
- Sussman MD. Advantage of early spinal stabilization and fusion in patients with Duchenne muscular dystrophy. (medscape.com)
- Surgical stabilization of the spine in Duchenne muscular dystrophy. (medscape.com)
- Management of scoliosis in Duchenne muscular dystrophy: a large 10-year retrospective study. (medscape.com)
- Duchenne muscular dystrophy, which is the most common childhood form. (medlineplus.gov)
- T Cell Responses to Dystrophin in a Natural History Study of Duchenne Muscular Dystrophy. (ucl.ac.uk)
- My initial work was focused on developing antisense reagents for exon skipping of Duchenne muscular dystrophy. (royalholloway.ac.uk)
- The most common form of muscular dystrophy in children, Duchenne muscular dystrophy typically affects only males. (healthnbeautytips.co)
- This form is similar to Duchenne muscular dystrophy, but the disease is much milder: symptoms appear later and progress more slowly. (healthnbeautytips.co)
- In 1986, researchers discovered the gene that, when defective or flawed, causes Duchenne muscular dystrophy. (healthnbeautytips.co)
- Duchenne muscular dystrophy occurs when that gene fails to make dystrophin. (healthnbeautytips.co)
Forms of muscular dystrophy2
- It is generally less severe, progresses more slowly, and affects fewer muscles than other forms of muscular dystrophy. (healthnbeautytips.co)
- Congenital muscular dystrophy is not a single disorder but instead refers to muscular dystrophy evident at birth or in infancy, occurring from any of several rare forms of muscular dystrophy. (msdmanuals.com)
PABPN16
- The genetics of this type of muscular dystrophy revolve around the PABPN1 gene. (wikipedia.org)
- The diagnosis of oculopharyngeal muscular dystrophy can be done via two methods, a muscle biopsy or a blood draw with genetic testing for GCG trinucleotide expansions in the PABPN1 gene. (wikipedia.org)
- Mutations in the PABPN1 gene cause oculopharyngeal muscular dystrophy. (medlineplus.gov)
- Mutations in the PABPN1 gene that cause oculopharyngeal muscular dystrophy result in a PABPN1 protein with an abnormally long (extended) polyalanine tract that includes between 11 and 18 alanines. (medlineplus.gov)
- Correlation between PABPN1 genotype and disease severity in oculopharyngeal muscular dystrophy. (arkanalabs.com)
- With the exception of oculopharyngeal muscular dystrophy* (whose main pathogenic mechanism is the expansion of triplets in the PABPN1 gene, a technique performed in our laboratory which must be specifically requested), the rest of the pathologies have a specific panel for their analysis. (digitis.net)
Treatments for muscular dystrophy2
- What are the treatments for muscular dystrophy (MD)? (medlineplus.gov)
- our work focuses on developing new genetic treatments for muscular dystrophy and muscle diseases. (royalholloway.ac.uk)
Weakness10
- Oculopharyngeal muscular dystrophy is a genetic condition characterized by muscle weakness that begins in adulthood, typically after age 40. (medlineplus.gov)
- Many people with oculopharyngeal muscular dystrophy also have weakness and wasting (atrophy) of the tongue. (medlineplus.gov)
- Individuals with oculopharyngeal muscular dystrophy frequently have weakness in the muscles near the center of the body (proximal muscles), particularly muscles in the shoulders, upper legs, and hips (limb-girdle muscles). (medlineplus.gov)
- Rarely, individuals have a severe form of oculopharyngeal muscular dystrophy with muscle weakness that begins before age 45, and have trouble walking independently by age 60. (medlineplus.gov)
- The resulting loss of muscle cells over time most likely causes the muscle weakness seen in people with oculopharyngeal muscular dystrophy. (medlineplus.gov)
- Although it is a commonly held belief that carriers merely pass on the disease and are unaffected, female carriers can have similar muscular weakness as affected males. (healthify.nz)
- The two forms that have been identified - Fukuyama and congenital muscular dystrophy with myosin deficiency - cause muscle weakness at birth or in the first few months of life, along with severe and early contractures. (healthnbeautytips.co)
- [ 1 ] This leads to muscular weakness with easy 'fatiguability', which is worse on exercise and improves with rest. (patient.info)
- Muscular dystrophy is a group of disorders that cause muscle weakness over time. (cvs.com)
- Muscular dystrophies are distinguished by the selective distribution of weakness and the specific nature of the genetic abnormality involved. (msdmanuals.com)
Amyotrophic lateral s1
- MDA advocated for priority COVID-19 vaccine access for people living with muscular dystrophy, amyotrophic lateral sclerosis (ALS), and other neuromuscular diseases. (mdaquest.org)
Diagnosis1
- Shapiro F, Specht L. The diagnosis and orthopaedic treatment of inherited muscular diseases of childhood. (medscape.com)
20231
- June 21, 2023 - Notice of Intent to Publish a Funding Opportunity Announcement for Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Centers (MDSRC) (P50 Clinical Trial Optional). (nih.gov)
Genetics1
- Bushby K. Genetics and the muscular dystrophies. (medscape.com)
Spinal muscula1
- Zolgensma® (onasemnogene abeparvovec: first gene therapy treatment derived partly from research conducted at Genethon ) for spinal muscular atrophy linked to SMN1. (afm-telethon.fr)
Mitochondrial1
- Oculopharyngeal muscular dystrophy: recent ultrastructural evidence for mitochondrial abnormalities. (medscape.com)
Muscle4
- Afterwards, I joined the gene therapy laboratory at Royal Holloway, and since then I contributed to the development of novel gene therapy agents and antisense therapeutics for the treatment of muscular dystrophies and other muscle conditions. (royalholloway.ac.uk)
- Structural diseases during infancy and adult age comprise the rest of muscular dystrophies: a group of hereditary diseases that affect the skeletal muscle, with the characteristic progressive degeneration of muscle fibers which causes loss of strength. (digitis.net)
- Oculopharyngeal muscle dystrophy is a progressive disease. (icliniq.com)
- All such dystrophies are genetically recessive and result from mutations in a variety of different genes including those that encode for structural proteins of the basal membrane or the extracellular matrix of skeletal muscle fibers. (msdmanuals.com)
Muscles1
- Muscular dystrophy (MD) is a group of inherited diseases in which the muscles that control movement (called voluntary muscles) progressively weaken. (healthnbeautytips.co)
Therapies1
- Oculopharyngeal muscular dystrophy: potential therapies for an aggregate-associated disorder. (harvard.edu)
Disorders1
- Available at: https://www.ninds.nih.gov/health-information/disorders/muscular-dystrophy. (cvs.com)
Centers5
- The purpose of this Funding Opportunity Announcement (FOA) is to publicize a competition for Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Centers (MDSRCs). (nih.gov)
- These Centers promote collaborative basic, translational, and clinical research and provide important resources that can be used by the national muscular dystrophy research communities. (nih.gov)
- The Centers also provide outstanding environments for the training of new researchers capable of addressing high priority objectives in muscular dystrophy research. (nih.gov)
- A goal of this Centers program is to support important and innovative research in the muscular dystrophies that is best pursued through this interdisciplinary and collaborative center environment, and projects that may not be as effective if supported by "stand-alone" research project grants. (nih.gov)
- The Centers also provide outstanding environments for the training of new scientists electing to pursue careers conducting research in high priority areas of muscular dystrophy. (nih.gov)
Cardiac symptoms1
- Long-term complications such as muscular and cardiac symptoms as well as liver fibrosis/cirrhosis and hepatocellular carcinoma may have a severe impact on prognosis and quality of life. (nih.gov)
Infancy1
- Muscular dystrophy can appear in infancy up to middle age or later, and its form and severity are determined in part by the age at which it occurs. (healthnbeautytips.co)
Disease2
- Mutations within a GCG repeat region in the gene for poly(A) binding protein II have been shown to cause the disease MUSCULAR DYSTROPHY, OCULOPHARYNGEAL. (harvard.edu)
- Available at: http://www.mda.org/disease/congenital-muscular-dystrophy. (cvs.com)
Prevalence1
- In Europe, the prevalence of oculopharyngeal muscular dystrophy is estimated to be 1 in 100,000 people. (medlineplus.gov)
Adulthood2
- The most common form of muscular dystrophy in adults, myotonic muscular dystrophy affects both men and women, and it usually appears at any time from early childhood to adulthood. (healthnbeautytips.co)
- This form of muscular dystrophy appears in teens to early adulthood and affects males and females. (healthnbeautytips.co)
Correlation1
- Comment in: Genotype-phenotype correlation: The ultimate challenge in facioscapolohumeral muscular dystrophy. (myobase.org)
Dystrophin1
- González-Herrera L, Gamas-Trujillo PA, García-Escalante MG, Castillo-Zapata I, Pinto-Escalante D. [Identifying deletions in the dystrophin gene and detecting carriers in families with Duchenne's/Becker's muscular dystrophy]. (medscape.com)
Abnormalities1
- POMT2 intragenic deletions and splicing abnormalities causing congenital muscular dystrophy with mental retardation. (medscape.com)
Form2
- This condition can occur with no known family history of BMD or DMD, so all females who are suspected of having any form of muscular dystrophy should be tested to determine if they could be a manifesting carrier because of the genetic implications. (healthify.nz)
- This form of muscular dystrophy appears in men and women in their 40s, 50s, and 60s. (healthnbeautytips.co)
Type2
- Muscular dystrophy is caused by defects in certain genes, with the type determined by the abnormal gene. (healthnbeautytips.co)
- Other symptoms depend on the type of muscular dystrophy a person has. (cvs.com)