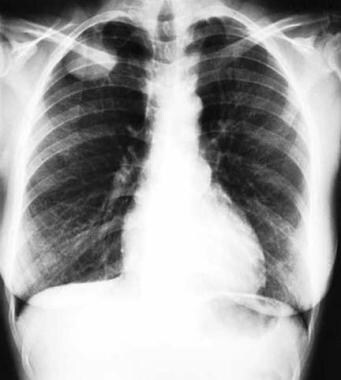
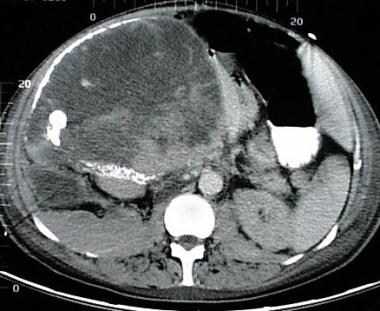
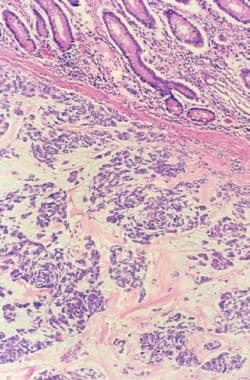
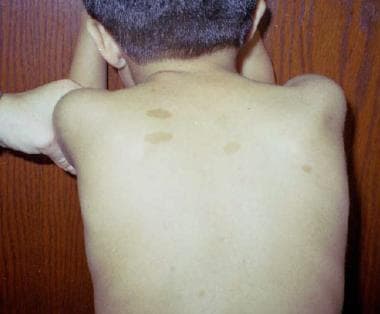
Neurofibrosarcoma
Neurofibroma
Neurofibromatosis 1
Nerve Sheath Neoplasms
Peripheral Nervous System Neoplasms
Neurilemmoma
Neurofibroma, Plexiform
Cardiac tumor biopsy under the guidance of intracardiac echocardiography. (1/17)
Transthoracic echocardiography or transesophageal echocardiography is sometimes useful in intracardiac tumor biopsy. Intracardiac echocardiography was used as an alternative to either of these for performing a biopsy of a right cardiac tumor in a 79-year-old woman. The procedure was well tolerated and no complications occurred. Histopathological findings and immunohistological staining were compatible with the diagnosis of neurogenic sarcoma. (+info)Case report of schwannoma of the rectum--clinical and pathological contribution. (2/17)
The case of benign schwannoma of the rectum primarily misdiagnosed as myogenic (neurogenic?) sarcoma is presented. A large tumor of 8 cm in diameter of the anterior rectal wall was removed with wide margins and an artificial anus was constructed. During 12 years of follow-up neither local recurrence nor distant metastases were observed. The patient is still alive and free of the disease. For that reason a surgery specimen of the tumor was pathologically reanalyzed and it showed features of type Antoni A and B tissues. These findings, together with strong reactivity for S-100 protein, Vimentin and negative for Actin supported the diagnosis of benign schwannoma. Because the localization of the tumor in the rectum is extremely rare, clinical and pathological features are presented and discussed. (+info)Malignant schwannoma of the sciatic nerve originating in a spinal plexiform neurofibroma associated with neurofibromatosis type 1--case report. (3/17)
A 26-year-old man with neurofibromatosis type 1 (NF1) presented with a giant malignant schwannoma of the sciatic nerve. The differential diagnosis of malignant peripheral nerve sheath tumor (MPNST) was based on clinical, radiological, and histological evidence. The tumor apparently originated in a spinal plexiform neurofibroma. The lesion was resected totally without neural damage to the sciatic nerve. However, the tumor recurred within 2 months. The patient died of unknown factors probably associated with the spinal involvement. MPNST associated with NF1 has a poor prognosis due to recurrence or metastasis despite complete macroscopic removal. (+info)Metastatic disease of the proximal femur. (4/17)
Since January 1999, ten patients had undergone surgical treatment for metastatic bony lesions of proximal femur at this centre. Seven of these patients were treated for complete pathological fractures, one for impending fracture and one for revision of internal fixation and loosening of hemiarthroplasty. Primary malignancies were located in breast in four cases, prostate in three and one in lung, thyroid and neurofibrosarcoma. Two patients had died within six months after surgery, four after 1 year while the remaining four were still alive. The mean duration of survival was eleven months. Nine patients had been ambulating pain free and there were no failure of reconstruction. (+info)A study of the influence of newly synthesized acyclonucleosides and 1,2,3,4-tetrahydroisoquinoline derivatives on deoxythymidine and deoxycytidine kinase activities in human neurofibrosarcoma and ovarian cancer. (5/17)
The influence of nine newly synthesized uracil acyclonucleosides, and 36 derivatives of 1,2,3,4-tetrahydroisoquinoline on the activity of enzymes catalysing dTMP and dGMP synthesis, on the content of dTTP and dGTP in acid soluble fraction and on the incorporation of [14C]dThd and [14C ]dGuo into DNA in tumour homogenates was studied. The influence of the compounds was studied in the cytosol from intraoperatively excised human tumours - neurofibrosarcoma and ovarian cancer. It was shown that dTMP and dGMP synthesis is inhibited competitively by 34.1+/-4.0% in both types of tumours by 0.2 mM 1-N-(3'-hydroxypropyl)-6-methyluracil (1) and 0.2 mM 1-N-(3'-hydroxypropyl)- 5,6- tetramethyleneuracil (2). The mentioned acyclonucleosides reduced the content of dTTP and dGTP in the acid soluble fraction of tumours (59.7+/-3.1% of control). 1-(4-chlorophenyl)-6,7-dihydroxy- 1,2,3,4-tetrahydroisoquinoline (3), 1-(2,3-dichlorophenyl)-6,7-dihydroxy 1,2,3,4-tetrahydroisoquinoline (4) and 1-(3-methoxyphenyl)-6,7-dihydroxy 1,2,3,4-tetrahydroisoquinoline (5) at 0.2 mM concentration caused a mixed type inhibition of the synthesis of dTMP and dGMP by, on average, 33.2+/-4.4%, and reduced the content of dTTP and dGTP in the acid soluble fraction (52.6+/-3.7% of control) but were active only in the cytosol of neurofibrosarcoma. While acyclonucleosides undergo phosphorylation in the cytosol by cellular kinases, with their triphosphates being active acyclonucleoside metabolites, active 1,3,4,5-tetrahydroisoquinoline derivatives (compounds not containing a deoxyribose moiety), cannot be phosphorylated. ACN and THI derivatives which inhibit dThd and dCyd kinase activities, inhibit also the incorporation of [14C]dThd and [14C]dGuo (ACN - 50.2+/-2.7%, THI - 53.4+/-3.9% of incorporation inhibition) into tumour DNA. The obtained results point to the mechanism of uracil acyclonucleosides and 1,2,3,4-tetrahydroisoquinoline biological activity consisting in inhibiting the synthesis of DNA components. (+info)Diagnosis, classification, and management of soft tissue sarcomas. (6/17)
BACKGROUND: Soft tissue sarcomas are challenging to oncologists due to their unique character, the infrequency of their occurrence, and the difficulties in predicting outcomes. Advances in imaging, as well as improvements in surgical techniques and adjunctive treatment methods, have improved care for patients with these unusual disorders. METHODS: The various types of soft tissue tumors are defined, and the statistics for the Orthopaedic Oncology Group in relation to them are reviewed and compared with literature references. RESULTS: The overall survival rate for 1,220 tumors treated at our institute from June 1972 to June of 2001 was 72%, with a wide range. Patients with leiomyosarcomas, clear cell sarcomas, and malignant fibrous histiocytomas had a poorer survival rate, while those with fibrosarcomas, liposarcomas, and neurofibrosarcomas fared better. Outcome was affected by patient age, tumor anatomic site, tumor stage, and a history of recurrence. CONCLUSIONS: Competent imaging, predictive immunological and genetic studies, improved surgery, and newer methods of adjunctive and neoadjunctive treatment should result in improvements in outcomes for patients with these tumors. (+info)Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. (7/17)
BACKGROUND: Patients affected by neurofibromatosis type 1 (NF1) are at higher risk of developing soft-tissue sarcomas (STS) than the general population. The clinical findings and outcome in 43 children and adolescents with NF1 treated for STS in the Italian protocols between 1988 and 2004 are reported. METHODS: The study included 37 patients with neurogenic sarcomas (36 malignant peripheral nerve sheath tumors [MPNST], 1 triton tumor) and 6 cases of rhabdomyosarcoma (RMS). The prevalence of NF1 observed during the study period was 43% in the MPNST population and 1% in the RMS group. RESULTS: Most patients with neurogenic sarcomas had large, invasive tumors. Five-year event-free and overall survival rates were 19% and 28%, respectively. Two of 16 patients with evaluable disease responded to chemotherapy. All 6 RMS patients were +info)Could an osteoinductor result in degeneration of a neurofibroma in NF1? (8/17)
(+info)Neurofibrosarcoma is a rare type of soft tissue sarcoma, which is a cancer that develops in the soft tissues of the body such as fat, muscle, tendons, blood vessels, and nerves. Neurofibrosarcoma specifically arises from the nerve sheath cells, also known as the Schwann cells, that cover and protect the peripheral nerves.
This type of cancer typically forms a painless mass or tumor in the affected area, which can grow and invade nearby tissues and organs over time. Neurofibrosarcoma can occur anywhere in the body but is most commonly found in the arms, legs, trunk, or head and neck region.
Neurofibrosarcoma can be classified into two main types: conventional and malignant peripheral nerve sheath tumor (MPNST). Conventional neurofibrosarcoma is more common and tends to occur in older adults, while MPNST is a more aggressive form that is associated with genetic disorders such as neurofibromatosis type 1.
Treatment for neurofibrosarcoma typically involves surgical removal of the tumor, along with radiation therapy and/or chemotherapy to help prevent recurrence and spread of the cancer. The prognosis for neurofibrosarcoma varies depending on several factors, including the size and location of the tumor, the patient's age and overall health, and the stage of the disease at diagnosis.
A neurofibroma is a benign (non-cancerous) tumor that develops from the nerve sheath, which is the protective covering around nerves. These tumors can grow anywhere on the body and can be found under the skin or deep inside the body. Neurofibromas can vary in size, and they may cause symptoms such as pain, numbness, or tingling if they press on nearby nerves.
Neurofibromas are a common feature of neurofibromatosis type 1 (NF1), a genetic disorder that affects approximately 1 in every 3,000 people worldwide. NF1 is characterized by the development of multiple neurofibromas and other tumors, as well as skin changes such as café-au-lait spots and freckling.
It's important to note that while most neurofibromas are benign, they can rarely undergo malignant transformation and become cancerous. If you have a neurofibroma or are concerned about your risk of developing one, it's important to seek medical advice from a healthcare professional who is familiar with this condition.
Neurofibromatosis 1 (NF1) is a genetic disorder that affects the development and growth of nerve tissue. It's also known as von Recklinghausen disease. NF1 is characterized by the growth of non-cancerous tumors on the nerves, as well as skin and bone abnormalities.
The symptoms of Neurofibromatosis 1 can vary widely, even among members of the same family. Some common features include:
* Multiple café au lait spots (flat, light brown patches on the skin)
* Freckles in the underarms and groin area
* Benign growths on or under the skin called neurofibromas
* Larger, more complex tumors called plexiform neurofibromas
* Optic gliomas (tumors that form on the optic nerve)
* Distinctive bone abnormalities, such as a curved spine (scoliosis) or an enlarged head (macrocephaly)
* Learning disabilities and behavioral problems
Neurofibromatosis 1 is caused by mutations in the NF1 gene, which provides instructions for making a protein called neurofibromin. This protein helps regulate cell growth and division. When the NF1 gene is mutated, the production of neurofibromin is reduced or absent, leading to uncontrolled cell growth and the development of tumors.
NF1 is an autosomal dominant disorder, which means that a person has a 50% chance of inheriting the mutated gene from an affected parent. However, about half of all cases are the result of new mutations in the NF1 gene, and occur in people with no family history of the disorder.
There is currently no cure for Neurofibromatosis 1, but treatments are available to manage the symptoms and complications of the disease. These may include medications to control pain or reduce the size of tumors, surgery to remove tumors or correct bone abnormalities, and physical therapy to improve mobility and strength. Regular monitoring by a healthcare team experienced in treating Neurofibromatosis 1 is also important to detect any changes in the condition and provide appropriate care.
Nerve sheath neoplasms are a group of tumors that arise from the cells surrounding and supporting the nerves. These tumors can be benign or malignant and include schwannomas, neurofibromas, and malignant peripheral nerve sheath tumors (MPNSTs). Schwannomas develop from the Schwann cells that produce the myelin sheath of the nerve, while neurofibromas arise from the nerve's supporting cells called fibroblasts. MPNSTs are cancerous tumors that can grow rapidly and invade surrounding tissues. Nerve sheath neoplasms can cause various symptoms depending on their location and size, including pain, numbness, weakness, or paralysis in the affected area.
Peripheral nervous system (PNS) neoplasms refer to tumors that originate in the peripheral nerves, which are the nerves outside the brain and spinal cord. These tumors can be benign or malignant (cancerous). Benign tumors, such as schwannomas and neurofibromas, grow slowly and do not spread to other parts of the body. Malignant tumors, such as malignant peripheral nerve sheath tumors (MPNSTs), can invade nearby tissues and may metastasize (spread) to other organs.
PNS neoplasms can cause various symptoms depending on their location and size. Common symptoms include pain, weakness, numbness, or tingling in the affected area. In some cases, PNS neoplasms may not cause any symptoms until they become quite large. Treatment options for PNS neoplasms depend on several factors, including the type, size, and location of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
A neurilemmoma, also known as schwannoma or peripheral nerve sheath tumor, is a benign, slow-growing tumor that arises from the Schwann cells, which produce the myelin sheath that surrounds and insulates peripheral nerves. These tumors can occur anywhere along the course of a peripheral nerve, but they most commonly affect the acoustic nerve (vestibulocochlear nerve), leading to a type of tumor called vestibular schwannoma or acoustic neuroma. Neurilemmomas are typically encapsulated and do not invade the surrounding tissue, although larger ones may cause pressure-related symptoms due to compression of nearby structures. Rarely, these tumors can undergo malignant transformation, leading to a condition called malignant peripheral nerve sheath tumor or neurofibrosarcoma.
A plexiform neurofibroma is a type of neurofibroma, which is a benign tumor that develops from the nerve sheath. In a plexiform neurofibroma, the tumor grows along the nerves and can involve multiple fascicles, leading to a large, diffuse mass. These tumors can occur anywhere in the body but are most commonly found in the head, neck, and trunk.
Plexiform neurofibromas can be associated with neurofibromatosis type 1 (NF1), a genetic disorder that affects approximately 1 in every 3,000 people worldwide. In individuals with NF1, plexiform neurofibromas can cause significant morbidity, including disfigurement, pain, and functional impairment. Additionally, there is a small risk of malignant transformation into a type of cancer called malignant peripheral nerve sheath tumor (MPNST).
The diagnosis of plexiform neurofibromas is typically made based on clinical examination, medical history, and imaging studies such as MRI. A biopsy may be necessary to confirm the diagnosis. Treatment options for plexiform neurofibromas include surgery, radiation therapy, and medication. The choice of treatment depends on several factors, including the size and location of the tumor, the presence of symptoms, and the risk of malignant transformation.
 Malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumor
MXI1
Fibrosarcoma
Schwannoma
List of skin conditions
List of MeSH codes (C04)
List of diseases (N)
List of MeSH codes (C10)
Nervous tissue
 Neurofibrosarcoma- Symptoms, Treatment & Support - Without a Ribbon
Neurofibrosarcoma- Symptoms, Treatment & Support - Without a Ribbon
Malignant peripheral nerve sheath tumor - Wikipedia
 Adrenal Carcinoma: Practice Essentials, Background, Pathophysiology
Adrenal Carcinoma: Practice Essentials, Background, Pathophysiology
 Sarcoma and Bone Cancer Types | Dana-Farber Cancer Institute
Sarcoma and Bone Cancer Types | Dana-Farber Cancer Institute
 Ralph E. Vatner, MD,PhD
Ralph E. Vatner, MD,PhD
 Edward C. McCarron, MD| Surgical Oncology | MedStar Health
Edward C. McCarron, MD| Surgical Oncology | MedStar Health
Rare diseases in the arts
 Occurrence and prognosis of lymph node metastases in patients selected for isolated limb perfusion with soft tissue sarcoma
Occurrence and prognosis of lymph node metastases in patients selected for isolated limb perfusion with soft tissue sarcoma
 Canine Sarcoma | The National Canine Cancer Foundation
Canine Sarcoma | The National Canine Cancer Foundation
Search Results | AVMA
 Tumors of the Skin in Dogs - Dog Owners - Merck Veterinary Manual
Tumors of the Skin in Dogs - Dog Owners - Merck Veterinary Manual
 Isophorone (EHC 174, 1995)
Isophorone (EHC 174, 1995)
Search Index - Alpha: n
00249517 | PEIR Digital Library
MR Imaging of the Muscular Component of Myocutaneous Flaps in the Head and Neck | American Journal of Neuroradiology
 Update on Feline Fibrosarcoma - WSAVA2002 - VIN
Update on Feline Fibrosarcoma - WSAVA2002 - VIN
 Cat is howling, can't stand, not eating, not drinking. | Ask A Vet
Cat is howling, can't stand, not eating, not drinking. | Ask A Vet
Tumors of the Skin in Dogs - Dog Owners - MSD Veterinary Manual
 Michigan Mesothelioma Doctors, Cancer Centers & Treatment
Michigan Mesothelioma Doctors, Cancer Centers & Treatment
Enablement | Patently-O
Neurogenic Tumors of the Mediastinum Treatment & Management: Medical Therapy, Surgical Therapy, Preoperative Details
 Academic Institute - Research output
- Houston Methodist Scholars
Academic Institute - Research output
- Houston Methodist Scholars
 Generic cialis drugs great britain - Top One Canadian Pharmacy Store
Generic cialis drugs great britain - Top One Canadian Pharmacy Store
 Namespace
Namespace
 Loss of neurofibromatosis type I (NFI) gene expression in pheochromocytomas from patients without NFI - Fingerprint
-...
Loss of neurofibromatosis type I (NFI) gene expression in pheochromocytomas from patients without NFI - Fingerprint
-...
 Browsing by Subject "Computer assisted tomography"
Browsing by Subject "Computer assisted tomography"
Epigenomic reordering induced by polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant...
Specific PHGKB|Rare Diseases PHGKB|PHGKB
Malignant1
- The most common soft-tissue primary malignant tumors are fibrosarcomas (desmoids, neurofibrosarcomas) and malignant fibrous histiocytomas. (msdmanuals.com)
Tumor5
- The p53 (a tumor suppressor gene in the normal population) genome on 17p in neurofibrosarcoma patients is mutated, increasing the probability of cancer. (wikipedia.org)
- The most conclusive test for a patient with a potential neurofibrosarcoma is a tumor biopsy (taking a sample of cells directly from the tumor itself). (wikipedia.org)
- For patients who have neurofibrosarcomas in an extremity, if the tumor is vascularized (has its own blood supply) and has many nerves going through it and/or around it, amputation of the extremity may be necessary. (wikipedia.org)
- Histologically, the mass was identified as a peripheral nerve sheath tumor (neurofibrosarcoma). (avma.org)
- Peripheral Nerve Sheath Tumor Two years ago, Sheena was diagnosed with neurofibrosarcoma, or peripheral nerve. (askavetquestion.com)
Schwannoma1
- In 50/50 Anna Kenrick plays Katherine McKay, a young, inexperienced therapist who is assigned to help Joseph Gordon-Levitt deal the fact he has schwannoma neurofibrosarcoma and presumably the fact that no matter how many great Christopher Nolan or Rian Johnson films he appears in, we'll all always think of him as the kid in Third Rock From The Sun. (prairiesmokepress.com)
Genetic2
- There is no well-known cause of neurofibrosarcoma, however, certain genetic mutations are held responsible for the development of these tumours. (withoutaribbon.org)
- Soft tissue sarcomas have been linked within families, so it is hypothesized that neurofibrosarcoma may be genetic, although researchers still do not know the exact cause of the disease. (wikipedia.org)
Cancer3
- A neurofibrosarcoma is a type of connective tissue (tissues that bind, separate, or support other tissues or organs) cancer that surrounds the nerves, these tissue cells receive the signals from the brain and stimulate the movements. (withoutaribbon.org)
- Neurofibrosarcoma is rare cancer, meaning it is not as well known as other forms of cancer. (withoutaribbon.org)
- If you suffer from rare cancer such as Neurofibrosarcoma, we can help and support you through your journey thanks to the generous donations we receive. (withoutaribbon.org)
Treatment1
- Treatment for neurofibrosarcoma is similar to that of other cancers. (wikipedia.org)
Surgery1
- In some instances, the oncologist may choose chemotherapy drugs when treating a patient with neurofibrosarcoma, usually in conjunction with surgery. (wikipedia.org)
Soft1
- The neurofibrosarcoma showed low AgNOR count as compared to other soft tissues sarcomas. (aleijten.com)
Tumors2
- since p53 is inactivated in neurofibrosarcoma patients, they are much more susceptible to developing tumors. (wikipedia.org)
- Although I will focus on how the genetic abnormalities and the formation of NF1 neurofibromas are interrelated- with a less intense discussion of neurofibrosarcomas- I also wish to emphasize that the NF1 phenotype entails much more than tumors of the central and peripheral nervous systems and, conversely, that NF1 neurofibromas cannot be explained solely in terms of NF1 mutations and abnormal function of neurofibromin. (medscape.com)
Neurofibromas1
- These findings suggest that methylation of some tumour-related genes may play a significant role in the tumourigenesis of neurofibromas/neurofibrosarcomas. (nih.gov)
Extremity1
- For patients who have neurofibrosarcomas in an extremity, if the tumor is vascularized (has its own blood supply) and has many nerves going through it and/or around it, amputation of the extremity may be necessary. (wikipedia.org)
Genome1
- The p53 (a tumor suppressor gene in the normal population) genome on 17p in neurofibrosarcoma patients is mutated, increasing the probability of cancer. (wikipedia.org)
Neoplasms1
- Peripheral and central nervous system neoplasms occur frequently, especially OPTIC NERVE GLIOMA and NEUROFIBROSARCOMA. (edu.au)
Sarcomas1
- Soft tissue sarcomas have been linked within families, so it is hypothesized that neurofibrosarcoma may be genetic, although researchers still do not know the exact cause of the disease. (wikipedia.org)
Similar1
- Treatment for neurofibrosarcoma is similar to that of other cancers. (wikipedia.org)