Oculocerebrorenal Syndrome
Renal Tubular Transport, Inborn Errors
Phosphoric Monoester Hydrolases
Fanconi Syndrome
Acidosis, Renal Tubular
Encyclopedias as Topic
Dent Disease
Lowe's syndrome: identification of carriers by lens examination. (1/61)
Lens examinations were performed on 7 obligate and 7 possible carriers of the X-linked gene for Lowe's syndrome, and on 117 controls. By quantitatively grading punctate cortical opacities, it was possible to discriminate between the obligate carriers and the controls with a fair degree of confidence. In the age group most important for genetic counselling, that of child bearing, the data are too limited for the derivation of precise estimates, but may, nevertheless, be useful. More such data are needed. (+info)Characterization of a germline mosaicism in families with Lowe syndrome, and identification of seven novel mutations in the OCRL1 gene. (2/61)
The oculocerebrorenal syndrome of Lowe (OCRL) is an X-linked disorder characterized by major abnormalities of eyes, nervous system, and kidneys. Mutations in the OCRL1 gene have been associated with the disease. OCRL1 encodes a phosphatidylinositol 4, 5-biphosphate (PtdIns[4,5]P2) 5-phosphatase. We have examined the OCRL1 gene in eight unrelated patients with OCRL and have found seven new mutations and one recurrent in-frame deletion. Among the new mutations, two nonsense mutations (R317X and E558X) and three other frameshift mutations caused premature termination of the protein. A missense mutation, R483G, was located in the highly conserved PtdIns(4,5)P2 5-phosphatase domain. Finally, one frameshift mutation, 2799delC, modifies the C-terminal part of OCRL1, with an extension of six amino acids. Altogether, 70% of missense mutations are located in exon 15, and 52% of all mutations cluster in exons 11-15. We also identified two new microsatellite markers for the OCRL1 locus, and we detected a germline mosaicism in one family. This observation has direct implications for genetic counseling of Lowe syndrome families. (+info)Increased levels of plasma lysosomal enzymes in patients with Lowe syndrome. (3/61)
Lowe syndrome is an X-linked disorder that has a complex phenotype that includes progressive renal failure and blindness. The disease is caused by mutations in an inositol polyphosphate 5-phosphatase designated OCRL. It has been shown that the OCRL protein is found on the surface of lysosomes and that a renal tubular cell line deficient in OCRL accumulated substrate phosphatidylinositol 4, 5-bisphosphate. Because this lipid is required for vesicle trafficking from lysosomes, we postulate that there is a defect in lysosomal enzyme trafficking in patients with Lowe syndrome that leads to increased extracellular lysosomal enzymes and might lead to tissue damage and contribute to the pathogenesis of the disease. We have measured seven lysosomal enzymes in the plasma of 15 patients with Lowe syndrome and 15 age-matched male controls. We find a 1.6- to 2.0-fold increase in all of the enzymes measured. When the data was analyzed by quintiles of activity for all of the enzymes, we found that 95% of values in the lowest quintile come from normal subjects whereas in the highest quintile 85% of the values are from patients with Lowe syndrome. The increased enzyme levels are not attributable to renal insufficiency because there was no difference in enzyme activity in the four patients with the highest creatinine levels compared with the six patients with the lowest creatinine values. (+info)Urinary megalin deficiency implicates abnormal tubular endocytic function in Fanconi syndrome. (4/61)
Normal reabsorption of glomerular filtrate proteins probably requires recycling of the endocytic receptors megalin (gp330) and cubilin. Both receptors are located on the luminal surface of the renal proximal tubule epithelium. Whether abnormal amounts of receptor are present in the urine of patients with Dent's disease, Lowe's syndrome, or autosomal dominant idiopathic Fanconi syndrome was explored. They are all forms of the renal Fanconi syndrome and are associated with tubular proteinuria. Urine samples of equal creatinine contents were dialyzed, lyophilized, and subjected to electrophoresis on nonreducing sodium dodecyl sulfate-5% polyacrylamide gels. Proteins were blotted and probed with anti-megalin IgG, anti-cubilin IgG, or receptor-associated protein. Megalin and cubilin levels detected by immunochemiluminescence were measured as integrated pixels and expressed as percentages of the normal mean values. A striking deficiency of urinary megalin, compared with normal individuals (n = 42), was observed for eight of nine families with Dent's disease (n = 10) and for the two families with Lowe's syndrome (n = 3). The family with autosomal dominant idiopathic Fanconi syndrome (n = 2) exhibited megalin levels within the normal range. The measured levels of cubilin were normal for all patients. These results are consistent with defective recycling of megalin to the apical cell surface of the proximal tubules and thus decreased loss into urine in Dent's disease and Lowe's syndrome. This defect would interfere with the normal endocytic function of megalin, result in losses of potential ligands into the urine, and produce tubular proteinuria. (+info)The deficiency of PIP2 5-phosphatase in Lowe syndrome affects actin polymerization. (5/61)
Lowe syndrome is a rare X-linked disorder characterized by bilateral congenital cataracts, renal Fanconi syndrome, and mental retardation. Lowe syndrome results from mutations in the OCRL1 gene, which encodes a phosphatidylinositol 4,5 bisphosphate 5-phosphatase located in the trans-Golgi network. As a first step in identifying the link between ocrl1 deficiency and the clinical disorder, we have identified a reproducible cellular abnormality of the actin cytoskeleton in fibroblasts from patients with Lowe syndrome. The cellular abnormality is characterized by a decrease in long actin stress fibers, enhanced sensitivity to actin depolymerizing agents, and an increase in punctate F-actin staining in a distinctly anomalous distribution in the center of the cell. We also demonstrate an abnormal distribution of two actin-binding proteins, gelsolin and alpha-actinin, proteins regulated by both PIP(2) and Ca(+2) that would be expected to be altered in Lowe cells. Actin polymerization plays a key role in the formation, maintenance, and proper function of tight junctions and adherens junctions, which have been demonstrated to be critical in renal proximal tubule function, and in the differentiation of the lens. These findings point to a general mechanism to explain how this PIP(2) 5-phosphatase deficiency might produce the Lowe syndrome phenotype. (+info)Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. (6/61)
The oculocerebrorenal syndrome of Lowe (OCRL) is a rare X-linked disorder characterized by severe mental retardation, congenital cataracts and renal Fanconi syndrome. OCRL1 protein is a phosphatidylinositol 4,5-bisphosphate 5-phosphatase with a C-terminal RhoGAP domain. Considering the pleiotropic cellular functions of Rho GTPases (Rho, Rac and Cdc42) and their dysregulation in several forms of mental retardation, we have investigated the so far unexplored function of the RhoGAP domain of OCRL1. Activated Rac GTPase was found to stably associate with the OCRL1 RhoGAP domain in vitro and to co-immunoprecipitate with endogenous OCRL1. Contrasting with other GAPs, OCRL1 RhoGAP exhibited a significant interaction with GDP bound Rac in vitro. As compared to Rac, other Rho GTPases tested showed reduced (Cdc42) or no binding (RhoA, RhoG) to OCRL1 RhoGAP. Immunofluorescence studies in HEK and COS7 cells and Golgi perturbation assays with Brefeldin A demonstrated that a fraction of endogenous Rac co-localizes with OCRL1 and gamma-adaptin in the trans-Golgi network. The OCRL1 RhoGAP domain showed low Rac GAP activity in vitro, and when expressed in Swiss 3T3 cells induced specific inhibition of RacGTP dependent ruffles, consistent with OCRL1 being an active RacGAP. OCRL1 appears to be a bifunctional protein which, in addition to its PIP2 5-phosphatase activity, binds to Rac GTPase. This novel property may play a role in localizing OCRL1 to the trans-Golgi network. Moreover, loss of OCRL1 RhoGAP and the resulting alteration in Rho pathways may contribute to mental retardation in Lowe syndrome, as illustrated in other forms of X-linked mental retardation. (+info)Early proximal tubular dysfunction in Lowe's syndrome. (7/61)
The early diagnosis of Lowe's syndrome can be difficult. Urinary excretion of retinol binding protein (RBP) and the lysosomal enzyme N-acetyl-glucosaminidase (NAG) were significantly increased in boys with Lowe's syndrome. Measurement of these urine parameters is recommended in suspected cases. (+info)Lowe syndrome: proton mr spectroscopy, and diffusion mr imaging. (8/61)
A 3-year-old boy with Lowe syndrome had bilateral periatrial hyperintense lesions intermixed with small cyst-like changes in the periatial regions of the deep white matter at MR imaging. Proton MR spectroscopy revealed prominent myoinositol peaks suggesting the presence of gliosis. b = 1000 s/mm2 images of diffusion MR imaging were negative for the periatrial lesions. ADC maps, however, revealed high ADC values (1.76, and 1.66 x 10(-3) mm2/s), compared to the normal brain parenchyma (0.81 x 10(-3) mm2/s). These diffusion MR imaging findings likely represented gliosis. (+info)Oculocerebrorenal syndrome, also known as Lowe syndrome, is a rare genetic disorder that primarily affects the eyes, brain, and kidneys. It's characterized by congenital cataracts, intellectual disability, and progressive kidney disease. The condition is caused by mutations in the OCRL gene, which provides instructions for making an enzyme called phosphatidylinositol 4,5-bisphosphate 5-phosphatase. This enzyme plays a crucial role in cell signaling and trafficking within cells.
The symptoms of oculocerebrorenal syndrome can vary widely among affected individuals, but they typically include:
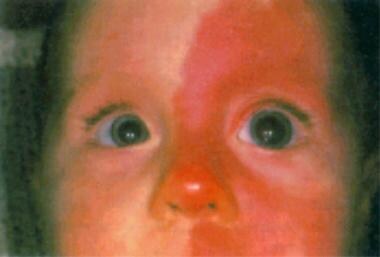
* Eye abnormalities: Most people with the condition are born with congenital cataracts that need to be removed soon after birth. Other eye problems may include glaucoma, strabismus (crossed eyes), and optic nerve damage, which can lead to vision loss.
* Brain abnormalities: Intellectual disability is a common feature of the condition, ranging from mild to severe. Affected individuals may also have delayed development, behavioral problems, and difficulty with coordination and movement.
* Kidney abnormalities: Progressive kidney disease is a hallmark of oculocerebrorenal syndrome. The kidneys may become enlarged and scarred, leading to kidney failure in some cases. Other kidney-related symptoms can include proteinuria (protein in the urine), hematuria (blood in the urine), and high blood pressure.
There is no cure for oculocerebrorenal syndrome, but treatments can help manage the symptoms. For example, cataract surgery can improve vision, while medications and dietary changes can help manage kidney disease. Early intervention and supportive care can also help improve outcomes for affected individuals.
Inborn errors of renal tubular transport refer to genetic disorders that affect the normal functioning of the kidney tubules. The kidney tubules are responsible for the reabsorption and secretion of various substances, including electrolytes and nutrients, as urine is formed. Inherited defects in the proteins that mediate these transport processes can lead to abnormal levels of these substances in the body and may result in a variety of clinical symptoms.
These disorders can affect different parts of the renal tubule, including the proximal tubule, loop of Henle, distal tubule, and collecting duct. Depending on the specific transporter affected, inborn errors of renal tubular transport can present with a range of clinical manifestations, such as electrolyte imbalances, acid-base disorders, growth retardation, kidney stones, nephrocalcinosis, or even kidney failure.
Examples of inborn errors of renal tubular transport include:
1. Distal renal tubular acidosis (dRTA): A genetic disorder that affects the ability of the distal tubule to acidify urine, leading to metabolic acidosis, hypokalemia, and nephrocalcinosis.
2. Bartter syndrome: A group of autosomal recessive disorders characterized by impaired sodium reabsorption in the loop of Henle, resulting in hypokalemia, metabolic alkalosis, and hyperreninemic hyperaldosteronism.
3. Gitelman syndrome: An autosomal recessive disorder caused by a defect in the thiazide-sensitive sodium chloride cotransporter in the distal tubule, leading to hypokalemia, metabolic alkalosis, and hypocalciuria.
4. Liddle syndrome: An autosomal dominant disorder characterized by increased sodium reabsorption in the collecting duct due to a gain-of-function mutation in the epithelial sodium channel (ENaC), resulting in hypertension, hypokalemia, and metabolic alkalosis.
5. Dent disease: An X-linked recessive disorder caused by mutations in the CLCN5 gene, which encodes a chloride channel in the proximal tubule, leading to low molecular weight proteinuria, hypercalciuria, and nephrolithiasis.
6. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC): An autosomal recessive disorder caused by mutations in the CLCN5 or CLDN16 genes, which encode chloride channels in the thick ascending limb of Henle's loop, resulting in hypomagnesemia, hypercalciuria, and nephrocalcinosis.
Phosphoric monoester hydrolases are a class of enzymes that catalyze the hydrolysis of phosphoric monoesters into alcohol and phosphate. This class of enzymes includes several specific enzymes, such as phosphatases and nucleotidases, which play important roles in various biological processes, including metabolism, signal transduction, and regulation of cellular processes.
Phosphoric monoester hydrolases are classified under the EC number 3.1.3 by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (IUBMB). The enzymes in this class share a common mechanism of action, which involves the nucleophilic attack on the phosphorus atom of the substrate by a serine or cysteine residue in the active site of the enzyme. This results in the formation of a covalent intermediate, which is then hydrolyzed to release the products.
Phosphoric monoester hydrolases are important therapeutic targets for the development of drugs that can modulate their activity. For example, inhibitors of phosphoric monoester hydrolases have been developed as potential treatments for various diseases, including cancer, neurodegenerative disorders, and infectious diseases.
Fanconi syndrome is a medical condition that affects the proximal tubules of the kidneys. These tubules are responsible for reabsorbing various substances, such as glucose, amino acids, and electrolytes, back into the bloodstream after they have been filtered through the kidneys.
In Fanconi syndrome, there is a defect in the reabsorption process, causing these substances to be lost in the urine instead. This can lead to a variety of symptoms, including:
* Polyuria (excessive urination)
* Polydipsia (excessive thirst)
* Dehydration
* Metabolic acidosis (an imbalance of acid and base in the body)
* Hypokalemia (low potassium levels)
* Hypophosphatemia (low phosphate levels)
* Vitamin D deficiency
* Rickets (softening and weakening of bones in children) or osteomalacia (softening of bones in adults)
Fanconi syndrome can be caused by a variety of underlying conditions, including genetic disorders, kidney diseases, drug toxicity, and heavy metal poisoning. Treatment typically involves addressing the underlying cause, as well as managing symptoms such as electrolyte imbalances and acid-base disturbances.
Renal tubular acidosis (RTA) is a medical condition that occurs when the kidneys are unable to properly excrete acid into the urine, leading to an accumulation of acid in the bloodstream. This results in a state of metabolic acidosis.
There are several types of RTA, but renal tubular acidosis type 1 (also known as distal RTA) is characterized by a defect in the ability of the distal tubules to acidify the urine, leading to an inability to lower the pH of the urine below 5.5, even in the face of metabolic acidosis. This results in a persistently alkaline urine, which can lead to calcium phosphate stones and bone demineralization.
Type 1 RTA is often caused by inherited genetic defects, but it can also be acquired due to various kidney diseases, drugs, or autoimmune disorders. Symptoms of type 1 RTA may include fatigue, weakness, muscle cramps, decreased appetite, and vomiting. Treatment typically involves alkali therapy to correct the acidosis and prevent complications.
An encyclopedia is a comprehensive reference work containing articles on various topics, usually arranged in alphabetical order. In the context of medicine, a medical encyclopedia is a collection of articles that provide information about a wide range of medical topics, including diseases and conditions, treatments, tests, procedures, and anatomy and physiology. Medical encyclopedias may be published in print or electronic formats and are often used as a starting point for researching medical topics. They can provide reliable and accurate information on medical subjects, making them useful resources for healthcare professionals, students, and patients alike. Some well-known examples of medical encyclopedias include the Merck Manual and the Stedman's Medical Dictionary.
Dent disease is a rare X-linked recessive genetic disorder that primarily affects the function of the proximal tubules in the kidneys. It is characterized by low molecular weight proteinuria, hypercalciuria, nephrolithiasis (kidney stones), and progressive kidney dysfunction leading to chronic kidney disease or end-stage renal failure in some cases. The disorder is caused by mutations in the CLCN5 gene, which provides instructions for making a chloride channel important for maintaining the proper function of proximal tubular cells in the kidneys. Dent disease primarily affects males, while females are typically asymptomatic carriers of the disorder.
 Oculocerebrorenal syndrome
Oculocerebrorenal syndrome
OCRL
David L. Nelson
Dent's disease
List of diseases (O)
List of MeSH codes (C10)
Charles Upton
List of MeSH codes (C16)
Charles Lowe
List of MeSH codes (C18)
List of MeSH codes (C12)
List of syndromes
Cataract
Leukoencephalopathy with vanishing white matter
List of congenital disorders
List of diseases (L)
Oculocerebrorenal syndrome - Wikipedia
 Oculocerebrorenal Dystrophy (Lowe Syndrome): Background, Pathophysiology, Epidemiology
Oculocerebrorenal Dystrophy (Lowe Syndrome): Background, Pathophysiology, Epidemiology
Lowe Syndrome (Oculocerebrorenal Syndrome): Background, Pathophysiology, Epidemiology
Oculocerebrorenal Syndrome: Background, Pathophysiology, Epidemiology
Oculocerebrorenal Syndrome: Background, Pathophysiology, Epidemiology
 The oculocerebrorenal syndrome of Lowe - diagnostic and therapeutic problems in Polish health care system
The oculocerebrorenal syndrome of Lowe - diagnostic and therapeutic problems in Polish health care system
 Lowe syndrome: MedlinePlus Genetics
Lowe syndrome: MedlinePlus Genetics
 Oculocerebrorenal Syndrome of Lowe (Lowe syndrome) and Dent Disease - 2 via the OCRL Gene Test - PreventionGenetics
Oculocerebrorenal Syndrome of Lowe (Lowe syndrome) and Dent Disease - 2 via the OCRL Gene Test - PreventionGenetics
Juvenile Glaucoma: Background, Pathophysiology, Epidemiology
 Phosphatidylinositol 5-Phosphatase Oculocerebrorenal Syndrome of Lowe Protein (OCRL) Controls Actin Dynamics during Early Steps...
Phosphatidylinositol 5-Phosphatase Oculocerebrorenal Syndrome of Lowe Protein (OCRL) Controls Actin Dynamics during Early Steps...
 Mitochondrial DNA deletion in a girl with manifestations of Kearns-Sayre and Lowe syndromes: an example of phenotypic mimicry?
Mitochondrial DNA deletion in a girl with manifestations of Kearns-Sayre and Lowe syndromes: an example of phenotypic mimicry?
 Research | Department of Ophthalmology / Hu Lab | Stanford Medicine
Research | Department of Ophthalmology / Hu Lab | Stanford Medicine
 urofacial syndrome - Ontology Browser - Rat Genome Database
urofacial syndrome - Ontology Browser - Rat Genome Database
 OCRL Proteins
OCRL Proteins
 Genetics of kidney stone disease | Nature Reviews Urology
Genetics of kidney stone disease | Nature Reviews Urology
 IPIP27 Coordinates PtdIns(4,5)P|sub|2|/sub| Homeostasis for Successful Cytokinesis. | IRIC - Institute for Research in...
IPIP27 Coordinates PtdIns(4,5)P|sub|2|/sub| Homeostasis for Successful Cytokinesis. | IRIC - Institute for Research in...
 Glycosuria Causes - Renal Glycosuria | Thyrotoxicosis
Glycosuria Causes - Renal Glycosuria | Thyrotoxicosis
 Mechanism of Human Tooth Eruption: Review Article Including a New Theory for Future Studies on the Eruption Process
Mechanism of Human Tooth Eruption: Review Article Including a New Theory for Future Studies on the Eruption Process
 Renal Glucosuria - Genitourinary Disorders - MSD Manual Professional Edition
Renal Glucosuria - Genitourinary Disorders - MSD Manual Professional Edition
 School of Psychology - Research Outputs
- Aston Research Explorer
School of Psychology - Research Outputs
- Aston Research Explorer
 Dhx40 Mouse Gene Details | DEAH-box helicase 40 | International Mouse Phenotyping Consortium
Dhx40 Mouse Gene Details | DEAH-box helicase 40 | International Mouse Phenotyping Consortium
 me Shared 'Renal' - 4 Picmonics
me Shared 'Renal' - 4 Picmonics
Specific PHGKB|Rare Diseases PHGKB|PHGKB
Picmonic Shared 'My Awesome Picmonic Playlist!' - 3 Picmonics
Long-term renal outcome in children with OCRL mutations: Retrospective analysis of a large international cohort - Fingerprint ...
March 2005
 Image Series List :: Spinal Cord
Image Series List :: Spinal CordTimm22 Mouse Gene Details | translocase of inner mitochondrial membrane 22 | International Mouse Phenotyping Consortium
herenciageneticayenfermedad: Health Conditions - Genetics Home Reference: O | Published: January 24, 2017
CIENCIASMEDICASNEWS: Health Conditions - Genetics Home Reference: O
OCRL16
- medical citation needed] This syndrome is caused by mutations in the OCRL gene which encodes an inositol polyphosphate-5-phosphatase. (wikipedia.org)
- In 1992, Nussbaum and colleagues reported that mutations of OCRL1 caused the rare X-linked disorder oculocerebrorenal syndrome of Lowe (OCRL), or Lowe syndrome, which includes the diagnostic triad of congenital cataracts, neonatal or infantile hypotonia with subsequent mental impairment, and renal tubular dysfunction. (medscape.com)
- Lowe syndrome is caused by an inherited mutation in the OCRL gene, mapped to chromosome Xq 26.1, which encodes the OCRL1 protein. (medscape.com)
- Membrane trafficking defects caused by mutation in OCRL may explain renal tubular defects observed in Lowe syndrome, including the inability of proximal tubular cells (PTC) to reabsorb low-molecular weight (LMW) proteins and other solutes such as phosphorus and bicarbonate from the glomerular filtrate. (medscape.com)
- Lowe syndrome, also called oculocerebrorenal syndrome (OCRS) and oculocerebrorenal syndrome of Lowe (OCRL), is an X-linked recessive metabolic disorder described by Lowe and coworkers in 1952. (medscape.com)
- Luo N, Kumar A, Conwell M, Weinreb RN, Anderson R, Sun Y. Compensatory Role of Inositol 5-Phosphatase INPP5B to OCRL in Primary Cilia Formation in Oculocerebrorenal Syndrome of Lowe. (medscape.com)
- The oculocerebrorenal syndrome described by C.U. Lowe in 1952 is a rare genetic defect (prevalence 1:500 000) caused by mutation of the OCRL gene which encodes phosphatidylinositol 4,5-bisphosphate 5-phosphatase. (edu.pl)
- Mutations in the OCRL gene cause Lowe syndrome. (medlineplus.gov)
- Researchers are working to determine how OCRL mutations cause the characteristic features of Lowe syndrome. (medlineplus.gov)
- In some cases of Lowe syndrome, an affected male inherits the mutation from a mother who carries one altered copy of the OCRL gene. (medlineplus.gov)
- Females who carry one mutated copy of the OCRL gene do not have the characteristic features of Lowe syndrome. (medlineplus.gov)
- The Oculocerebrorenal syndrome of Lowe (OCRL or Lowe syndrome) is a rare X-linked disorder, which is a characterized by a pleiotropic phenotype including congenital bilateral dense cataracts, infantile congenital muscular hypotonia, severe intellectual disability and Fanconi syndrome of the kidney proximal tubules (Lewis et al. (preventiongenetics.com)
- IPIP27 scaffolds the inositol phosphatase oculocerebrorenal syndrome of Lowe (OCRL) by coupling it to endocytic BAR domain proteins. (iric.ca)
- Currently, we focus on oculocerebrorenal syndrome of Lowe protein (OCRL), oligophrenin (OPHN) and fragile X mental retardation protein (FMRP). (neurosciences-duesseldorf.de)
- Lowe's syndrome is a result of mutations in a gene called the OCRL gene, which codes for an enzyme that helps modify fat molecules known as membrane phospholipids. (bhaskarhealth.com)
- For final confirmation of Lowe's syndrome, measurement of the activity of the inositol polyphosphate-5-phosphatase can be done in skin fibroblasts, although genetic testing for the OCRL gene is also an option. (bhaskarhealth.com)
Fanconi8
- Lowe syndrome can be considered a cause of Fanconi syndrome (bicarbonaturia, renal tubular acidosis, potassium loss and sodium loss). (wikipedia.org)
- When more patients were described, the phenotype was expanded to include the renal tubular defects that comprise Fanconi syndrome , and an X-linked inheritance pattern was noted. (medscape.com)
- It is characterized by congenital cataracts, infantile glaucoma, neonatal or infantile hypotonia, intellectual impairment, and renal tubular dysfunction (Fanconi syndrome). (medscape.com)
- A typical clinical triad characterizing the disease consists of congenital cataract, mental retardation and proximal tubulopathy (secondary Fanconi syndrome without glycosuria) with slow progression to end stage kidney disease in the 2nd-4th decade. (edu.pl)
- Kidney (renal) abnormalities, most commonly a condition known as renal Fanconi syndrome, frequently develop in individuals with Lowe syndrome. (medlineplus.gov)
- In individuals with renal Fanconi syndrome, the kidneys are unable to reabsorb important nutrients into the bloodstream. (medlineplus.gov)
- Fanconi Syndrome Fanconi syndrome consists of multiple defects in renal proximal tubular reabsorption, causing glucosuria, phosphaturia, generalized aminoaciduria, and bicarbonate wasting. (msdmanuals.com)
- The features of symptomatic Fanconi syndrome do not usually become manifest until after the first few months of life, except for LMW proteinuria. (nih.gov)
Phenotype3
- Böckenhauer D., Bökenkamp A., van't Hoff W., Levtchenko E., Kistvan Holthe J.E., Tasic V., Ludwig M. Renal phenotype in Lowe syndrome: a selective proximal tubular dysfunction. (edu.pl)
- Affected individuals are best described as having either a phenotype consistent with either severe (Hurler syndrome) or attenuated MPS I, a distinction that influences therapeutic options. (nih.gov)
- Bleeding Severity and Phenotype in 22q11.2 Deletion Syndrome-A Cross-Sectional Investigation. (harvard.edu)
Lowe Syndrome27
- Oculocerebrorenal syndrome (also called Lowe syndrome) is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. (wikipedia.org)
- glaucoma is present in about half of the individuals with Lowe syndrome, though usually not at birth. (wikipedia.org)
- The main substrates for Lowe syndrome are two phosphoinositides (Ptdln): PtdIn4,5P2 and PtdIn3,4,5P3. (medscape.com)
- [ 1 ] These studies provide strength to the classification of Lowe syndrome as part of the ciliopathy-associated diseases. (medscape.com)
- [ 2 ] and (2) Mehta ZB, Pietka G, Lowe M. The cellular and physiological functions of the Lowe syndrome protein OCRL1. (medscape.com)
- The low number of megalin at the PTC apical border explains the reduced endocytosis of low-molecular weight proteins that occur in Lowe syndrome. (medscape.com)
- [ 3 ] Proteomic analysis of urine from patients with Lowe syndrome typically show low levels of megalin and cubilin denoting a decrease in the number of these multiligand receptors in the PTC. (medscape.com)
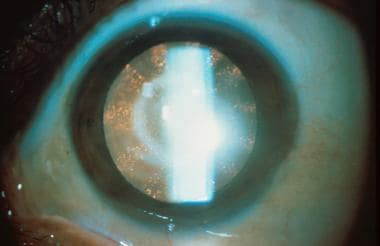
- Classic lenticular opacities in a female carrier for Lowe syndrome. (medscape.com)
- Lowe syndrome is caused by a mutation of the OCRL1 gene mapped to the chromosomal locus of Xq26.1. (medscape.com)
- Deficiency of the enzyme causes the protean manifestations of Lowe syndrome. (medscape.com)
- [ 4 , 5 ] Fibroblast activity was found to be decreased by 80%-90% in patients with Lowe syndrome compared to healthy controls. (medscape.com)
- The estimated prevalence of Lowe syndrome is 1 per 500,000 population. (medscape.com)
- The Lowe Syndrome Association (LSA) reported 190 males living with Lowe syndrome in 2000. (medscape.com)
- [ 7 ] As of 2005, the Italian Association of Lowe's Syndrome reported 34 individuals with Lowe syndrome living in Italy. (medscape.com)
- [ 6 ] No cases of Lowe syndrome have been reported in Africa, South America, and parts of Asia. (medscape.com)
- Lowe syndrome occurs almost exclusively in males. (medscape.com)
- Loi M. Lowe syndrome. (medscape.com)
- Here we reported a 2-year-old boy with the clinical picture of Lowe syndrome (congenital cataract, hypotonia, psychomotor development retardation and tubulopathy), examined and treated in many medical centers. (edu.pl)
- Nussbaum R.L., Orrison B.M., Janne P.A., Charnas L., Chinault A.C. Physical mapping and genomic structure of the Lowe syndrome gene OCRL1. (edu.pl)
- Lowe syndrome is a condition that primarily affects the eyes, brain, and kidneys. (medlineplus.gov)
- Many individuals with Lowe syndrome have delayed development, and intellectual ability ranges from normal to severely impaired. (medlineplus.gov)
- Progressive kidney problems in older children and adults with Lowe syndrome can lead to life-threatening renal failure and end-stage renal disease (ESRD). (medlineplus.gov)
- Lowe syndrome is an uncommon condition. (medlineplus.gov)
- Candidates for this test are patients with symptoms consistent with Lowe syndrome and Dent disease - 2 (negative for CLCN5 gene pathogenic variants ) , family members of patients who have known mutations and carrier testing for at-risk family members. (preventiongenetics.com)
- 2012). Female carriers of Lowe syndrome show no clinical symptoms, however, ~95% do show characteristic lenticular opacities that was absent in all proven noncarriers (Röschinger et al. (preventiongenetics.com)
- Although the biochemical defect in typical Lowe syndrome is not known, there is evidence suggesting that mitochondrial metabolism may be impaired. (nih.gov)
- Lowe syndrome (oculocerebrorenal syndrome) is characterized by involvement of the eyes, central nervous system, and kidneys. (nih.gov)
Cerebrooculorenal1
- Lowe's syndrome - also known as the cerebrooculorenal syndrome, the oculocerebrorenal syndrome of Lowe, and phosphatidylinositol-4,5-bisphosphate-5-phosphatase deficiency - represents a very rare multisystem disorder that affects the eye, the central nervous system, and the kidneys. (bhaskarhealth.com)
Inositol1
- Attre O., Olivos I.M., Okabe I., Bailey L.C., Nelson D.L., Lewis R.A., McInnes R.R., Nussbaum R.L. The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. (edu.pl)
Glaucoma2
- However, if there is an associated generalized defect in proximal tubular function, symptoms and signs may include hypophosphatemic rickets, volume depletion, short stature, muscle hypotonia, and ocular changes of cataracts or glaucoma (oculocerebrorenal syndrome) or Kayser-Fleischer rings (Wilson disease). (msdmanuals.com)
- Management of Lowe's syndrome includes early cataract surgery in order to avoid amblyopia, while ocular tone has to be tested repeatedly in order to diagnose glaucoma. (bhaskarhealth.com)
Autosomal Dominant1
- A group of syndromes caused by autosomal dominant mutation(s) in the WT1 gene, encoding Wilms tumor protein. (nih.gov)
Mutation1
- WT1 and NPHS2 gene mutation analysis and clinical management of steroid-resistant nephrotic syndrome. (nih.gov)
Dysfunction1
- This suggests that this syndrome is due to dysfunction of the cilia in these cells. (wikipedia.org)
Cataracts1
- In differential diagnosis, we have to bear in mind that bilateral cataracts of the eyes and hypotonia are also found in congenital infections (such as rubella), congenital myopathies, mitochondriopathies and some other syndromes. (bhaskarhealth.com)
Diagnosis1
- A triad of eye, nervous system, and kidney involvement is absolutely necessary for the diagnosis of Lowe's syndrome. (bhaskarhealth.com)
Disorder1
- FBN1-related Marfan syndrome (Marfan syndrome), a systemic disorder of connective tissue with a high degree of clinical variability, comprises a broad phenotypic continuum ranging from mild (features of Marfan syndrome in one or a few systems) to severe and rapidly progressive neonatal multiorgan disease. (nih.gov)
Kidney1
- Because of the three major organ systems involved (eyes, brain and kidney), it is known as oculocerebrorenal syndrome. (wikipedia.org)
Symptoms1
- We have studied a girl who presented with an oculocerebrorenal syndrome, but later developed symptoms and signs of mitochondrial encephalomyopathy. (nih.gov)
Phenotypic1
- Mitochondrial DNA deletion in a girl with manifestations of Kearns-Sayre and Lowe syndromes: an example of phenotypic mimicry? (nih.gov)
Proximal1
- The major morbidity and early mortality in Marfan syndrome relate to the cardiovascular system and include dilatation of the aorta at the level of the sinuses of Valsalva (predisposing to aortic tear and rupture), mitral valve prolapse with or without regurgitation, tricuspid valve prolapse, and enlargement of the proximal pulmonary artery. (nih.gov)
Lowe's Syndrome6
- What is Lowe's Syndrome? (bhaskarhealth.com)
- Home Bhaskar Health What is Lowe's Syndrome? (bhaskarhealth.com)
- The estimated prevalence of Lowe's syndrome is 1 in 500 thousand people. (bhaskarhealth.com)
- The gene resides on the X chromosome, hence Lowe's syndrome is inherited in an X-linked pattern. (bhaskarhealth.com)
- Renal involvement is another major clinical feature of Lowe's syndrome, usually occurring during the initial years of life with varying degrees of severity. (bhaskarhealth.com)
- Patients with Lowe's syndrome often exhibit typical facial determinants such as deep-set eyes, frontal bossing, chubby cheeks, as well as a fair complexion with a blonde hair. (bhaskarhealth.com)
Gene cause2
- Mutations in this gene cause oculocerebrorenal syndrome of Lowe and also Dent disease. (antibodies-online.com)
- A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. (nih.gov)
Disease1
- Lowe oculocerebrorenal syndrome is an X-linked recessive disease whose locus has been assigned to Xp25. (nih.gov)
Scheie1
- While affected individuals have traditionally been classified as having one of three MPS I syndromes (Hurler syndrome, Hurler-Scheie syndrome, or Scheie syndrome), no easily measurable biochemical differences have been identified and the clinical findings overlap. (nih.gov)
Deletion syndrome6
- 22q11 Deletion Syndrome" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (harvard.edu)
- This graph shows the total number of publications written about "22q11 Deletion Syndrome" by people in Harvard Catalyst Profiles by year, and whether "22q11 Deletion Syndrome" was a major or minor topic of these publication. (harvard.edu)
- Below are the most recent publications written about "22q11 Deletion Syndrome" by people in Profiles. (harvard.edu)
- Abnormalities in gray matter microstructure in young adults with 22q11.2 deletion syndrome. (harvard.edu)
- Failed Progenitor Specification Underlies the Cardiopharyngeal Phenotypes in a Zebrafish Model of 22q11.2 Deletion Syndrome. (harvard.edu)
- Frontal Hypoactivation During a Working Memory Task in Children With 22q11 Deletion Syndrome. (harvard.edu)
Abnormalities1
- It encompasses several syndromes with overlapping abnormalities including the DIGEORGE SYNDROME, VELOCARDIOFACIAL SYNDROME, and CONOTRUNCAL AMOMALY FACE SYNDROME. (harvard.edu)
Marfan1
- With proper management, the life expectancy of someone with Marfan syndrome approximates that of the general population. (nih.gov)
Noonan1
- Noonan Syndrome Turner Syndrome Normal Karyotype XO (60%) MC heart sisease 1. (mediconotebook.com)
Mutations cause1
- At least one mechanism by which these mutations cause this syndrome is by loss of its Rab-binding domain. (wikipedia.org)