Pyruvate Carboxylase Deficiency Disease
Pyruvate Carboxylase
Multiple Carboxylase Deficiency
Mannosidase Deficiency Diseases
Carbon-Carbon Ligases
Methylmalonyl-CoA Decarboxylase
Biotin
Carbamoyl-Phosphate Synthase I Deficiency Disease
Holocarboxylase Synthetase Deficiency
Carbon-Nitrogen Ligases
Immunologic Deficiency Syndromes
Biotinidase Deficiency
Multiple Sulfatase Deficiency Disease
Biotinidase
Amino Acid Metabolism, Inborn Errors
Carboxy-Lyases
Acetyl-CoA Carboxylase
Biotin-response organicaciduria. Multiple carboxylase defects and complementation studies with propionicacidemia in cultured fibroblasts. (1/12)
Fibroblast cultures from two individuals with biotin-responsive organicacidemia were found to have a pleiotropic deficiency of propionyl-CoA carboxylase, beta-methylcrotonyl-CoA carboxylase, and pyruvate carboxylase activities after growth in biotin limited culture medium, conditions which do not affect the carboxylase activities of normal cells. All three enzyme activities were restored to normal levels after transferring the mutant strains to biotin-rich medium. Both patients excreted abnormal levels of an array of metabolic intermediates, including beta-methylcrotonate, beta-hydroxyisovalerate, beta-hydroxypropionate, and lactate, which reflect metabolic blocks at all three carboxylase sites.14 mutants deficient in only propionyl-CoA carboxylase activity from patients with propionicacidemia and the two biotin-responsive strains were examined for complementation with seven previously mapped pcc mutants. No new pcc complementation groups were identified. Nine of the mutants were mapped to group pccA. The remaining 12 mutants mapped to pccBC or its B or C subgroups, confirming the complex nature of this group. The biotin-responsive mutants failed to complement each other but did complement mutants from all the pcc groups. Thus biotin-responsive organicacidemia is defined by a new complementation group, bio. The results obtained in this study suggest that the bio mutants have a defect of either biotin transport or a common holocarboxylase synthetase required for the biotin activation of all three mitochondrial carboxylases. (+info)The molecular basis of pyruvate carboxylase deficiency: mosaicism correlates with prolonged survival. (2/12)
(+info)The French and North American phenotypes of pyruvate carboxylase deficiency, correlation with biotin containing protein by 3H-biotin incorporation, 35S-streptavidin labeling, and Northern blotting with a cloned cDNA probe. (3/12)
Cultured skin fibroblasts from 16 patients with either French or American pyruvate carboxylase (PC) deficiency were examined for their ability to incorporate 3H-biotin into proteins. Cell extracts were also examined for the presence of biotin-containing proteins with 35S-streptavidin, immunoreactive protein with anti-PC antibody, and PC mRNA by Northern blotting with a PC cDNA probe. All the North American presentation patients showed a 3H-biotin protein, a streptavidin protein, and an anti-PC precipitable protein at 125 kilodaltons on sodium dodecyl sulfate-polyacrylamide gel electrophoresis of cellular proteins. They also showed a detectable mRNA species for PC on Northern blotting. Of the French presentation patients, five showed very low or absent 3H-biotin protein, streptavidin protein, and anti-PC precipitable protein at 125 kilodaltons. Three French presentation patients showed PC protein to be present on the basis of these techniques. Similarly, five showed either very low or absent mRNA for PC on Northern blotting whereas three gave evidence of the presence of PC-specific mRNA. Thus, whereas the North American presentation of PC deficiency is associated with the presence of a mature biotin containing protein of the correct molecular weight, the French presentation may, in some (but not in all) cases, have both absent PC protein and absent PC mRNA. (+info)Heterogeneity of holocarboxylase synthetase in patients with biotin-responsive multiple carboxylase deficiency. (4/12)
Holocarboxylase synthetase activity has been determined in fibroblasts of seven patients with the neonatal form of biotin-responsive multiple carboxylase deficiency. The normal Km for biotin was 15 +/- 3 nmol/l, while in the patients the values ranged from 48 to 1,062 nmol/l. The mean maximum velocity was 27% of normal. Differences among the values obtained for the Km for biotin and the heat stability of holocarboxylase synthetase suggested that the patients studied represented at least four distinct variants at the holocarboxylase synthetase locus. (+info)Inborn errors of metabolism. Vitamin-responsive genetic disease. (5/12)
The several ways in which vitamin administration may bring about a biochemical response in genetic abnormalities have been discussed. Two major interrelated lessons emerge from what we now know about vitamin-responsive genetic disease. First, it is possible to enhance metabolite flow through partially deficient reactions by suitable manipulation of the environment in which a fixed amount of enzyme functions or by changing the concentration of the enzyme itself. The latter approach may be the most versatile in the long run since there may be agents other than vitamins which increase enzyme concentrations. A striking example of such an effect in mammals is furnished by the work of Pitot and his collaborators, who by administration of casein hydrolysate to rats, increased threonine dehydratase activity several hundred-fold (Peraino and Pitot, 1964) by increasing the rate of enzyme synthesis (Jost, Khairallah, and Pitot, 1968). Other means of enhancing enzyme activities, ranging from tissue transplantation to transfer of genetic material, have been discussed elsewhere (for example, see Brady, 1973). These procedures will not be discussed here, other than to mention a recent report (Mukherjee and Krasner, 1973) who transferred several small plugs of liver tissue (approximately 5% of the liver) from normal rats to the livers of rats genetically deficient in bilirubin uridine diphosphate glucuronyltransferase activity. Twelve weeks later the specific activity of glucuronyltransferase had risen in the livers of the recipient rats to 6-23% of normal, and the serum bilirubin of these rats, which had initially been elevated, had fallen to close to, or within, the normal range. Thus liver grafts between suitably matched individuals, may in the near future, become a means of increasing hepatic activities of deficient enzymes to extents which are therapeutically meaningful. The second lesson to be learned from the review presented here is that enhancement of enzyme activity may be therapeutically beneficial even though the increase is small and the activity attained is still reduced relative to normal. It will be well to bear this in mind in any attempts to treat inborn errors of metabolism. (+info)Biotin in clinical medicine--a review. (6/12)
The recent developments in cofactor therapy of inherited biochemical disease has awakened interest in biotin as a therapeutic agent. This review briefly details the physiological and biochemical aspects of human biotin metabolism. The role of biotin in therapy of human disease is critically examined and the relevant literature extensively reviewed. It is our hope that this review will stimulate further clinical interest in the treatment of biotin-responsive human disorders. (+info)[3H]biotin-labeled proteins in cultured human skin fibroblasts from patients with pyruvate carboxylase deficiency. (7/12)
Biotin containing carboxylases in cultured human skin fibroblasts were radioactively labeled by addition of [8,9-3H]biotin to biotin-depleted cell cultures. Three major bands were visualized by fluorography after sodium dodecyl sulfate-polyacrylamide gel electrophoresis of the fibroblast proteins. These bands corresponded to pyruvate carboxylase (Mr = 125,000), the biotin-containing subunit of methyl crotonyl-CoA carboxylase (Mr = 75,000) and the biotin-containing subunit of propionyl-CoA carboxylase (Mr = 73,000) as judged by molecular weight markers, purified carboxylase protein standards, and interaction with monospecific antisera. Four out of 5 cell lines from patients with classical pyruvate carboxylase deficiency (less than 5% of normal activity) labeled with this technique displayed a normal band in the position of pyruvate carboxylase while one cell line showed complete absence of any labeled protein in this area. These results demonstrate heterogeneity in the etiology of pyruvate carboxylase deficiency. (+info)The molecular basis for the two different clinical presentations of classical pyruvate carboxylase deficiency. (8/12)
Eight cases of isolated human pyruvate carboxylase deficiency were examined from seven families. Although all patients presented with a chronic lacticacidemia, two particular patients presented with the added features of hyperammonemia, citrullinemia, and hyperlysinemia. When cultured skin fibroblasts from these patients were examined for their ability to synthesize [3H]biotin-containing proteins, it was found that the two patients who presented with hyperammonemia, citrullinemia, and hyperlysinemia did not synthesise a protein of the correct subunit molecular weight (Mr = 125 K daltons) corresponding to pyruvate carboxylase. In addition, when skin fibroblast proteins were labeled with [35S]methionine, cross-reacting material (CRM) corresponding to pyruvate carboxylase was immunoprecipitated by antipyruvate carboxylase antiserum in most patients, but again the two patients with the atypical presentation showed no CRM. We propose that the different clinical presentation of human pyruvate carboxylase deficiency is a manifestation of two different mutations in the pyruvate carboxylase gene, one that results in the synthesis of a relatively inactive pyruvate carboxylase protein CRM(+ve) and one that results in the lack of expression of the gene in the form of a recognizable protein CRM(-ve). (+info)Pyruvate carboxylase deficiency disease is a rare inherited metabolic disorder that affects the body's ability to break down proteins, fats, and carbohydrates for energy. It is caused by mutations in the Pyruvate Carboxylase (PC) gene, which provides instructions for making an enzyme called pyruvate carboxylase. This enzyme plays a critical role in gluconeogenesis, a process that takes place in the liver and kidneys to produce glucose, a simple sugar that is a primary source of energy for the body.
In pyruvate carboxylase deficiency disease, the enzyme's activity is significantly reduced or absent, leading to an accumulation of toxic levels of certain metabolic intermediates, such as lactic acid and pyruvic acid, in the blood and other tissues. This accumulation can cause a range of symptoms, including developmental delay, seizures, poor muscle tone, difficulty breathing, and feeding problems.
The severity of pyruvate carboxylase deficiency disease varies widely, depending on the degree of enzyme activity that is affected. Some individuals may have mild symptoms, while others may experience severe, life-threatening complications. Treatment typically involves managing symptoms and providing supportive care to help prevent complications. In some cases, a low-protein diet or supplementation with certain vitamins and minerals may be recommended to help reduce the accumulation of toxic metabolites.
Pyruvate carboxylase is a biotin-containing enzyme that plays a crucial role in gluconeogenesis, the process of generating new glucose molecules from non-carbohydrate sources. The enzyme catalyzes the conversion of pyruvate to oxaloacetate, an important intermediate in several metabolic pathways, particularly in the liver, kidneys, and brain.
The reaction catalyzed by pyruvate carboxylase is as follows:
Pyruvate + CO2 + ATP + H2O → Oxaloacetate + ADP + Pi + 2H+
In this reaction, pyruvate reacts with bicarbonate (HCO3-) to form oxaloacetate, consuming one molecule of ATP in the process. The generation of oxaloacetate provides a key entry point for non-carbohydrate precursors, such as lactate and certain amino acids, to enter the gluconeogenic pathway.
Pyruvate carboxylase deficiency is a rare but severe genetic disorder that can lead to neurological impairment and developmental delays due to the disruption of energy metabolism in the brain.
Multiple carboxylase deficiency (MCD) is a rare genetic disorder that affects the body's ability to metabolize certain amino acids, particularly those that contain sulfur. It is caused by mutations in the genes responsible for producing enzymes involved in the biotin-dependent carboxylation reactions, which are critical for various metabolic processes in the body.
There are two major types of MCD:
1. Profound multiple carboxylase deficiency (also known as Type II biotinidase deficiency): This form is more severe and is caused by a defect in the holocarboxylase synthetase enzyme, which is responsible for attaching biotin to several carboxylases.
2. Biotin-responsive multiple carboxylase deficiency (also known as Type I biotinidase deficiency): This form is milder and is caused by a defect in the biotinidase enzyme, which recycles biotin in the body. However, it can be treated with biotin supplementation.
Symptoms of MCD may include:
* Developmental delay
* Seizures
* Hypotonia (low muscle tone)
* Ataxia (lack of coordination)
* Rash
* Hair loss
* Acidosis (high levels of acid in the body)
* Coma and even death, if left untreated
Early diagnosis and treatment with biotin supplementation can significantly improve outcomes for individuals with MCD.
Mannosidosis is a rare inherited metabolic disorder caused by a deficiency of the enzyme alpha-mannosidase, which is responsible for breaking down complex sugar molecules called mannose-rich oligosaccharides. When the enzyme is not functioning properly, these sugar molecules accumulate in various tissues and organs, leading to progressive damage.
There are two main types of Mannosidosis: type I (also known as classic Mannosidosis) and type II (also known as mild or attenuated Mannosidosis). The symptoms and severity of the disease can vary widely between individuals and between the two types, but may include developmental delays, intellectual disability, coarse facial features, skeletal abnormalities, hearing loss, recurrent respiratory infections, and vision problems.
The diagnosis of Mannosidosis is typically made through enzyme assay and genetic testing. Treatment is primarily supportive and may include physical therapy, speech therapy, hearing aids, and management of respiratory and other medical issues as they arise. In some cases, bone marrow transplantation may be considered as a treatment option.
Carbon-carbon ligases are a type of enzyme that catalyze the formation of carbon-carbon bonds between two molecules. These enzymes play important roles in various biological processes, including the biosynthesis of natural products and the metabolism of carbohydrates and lipids.
Carbon-carbon ligases can be classified into several categories based on the type of reaction they catalyze. For example, aldolases catalyze the condensation of an aldehyde or ketone with another molecule to form a new carbon-carbon bond and a new carbonyl group. Other examples include the polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs), which are large multienzyme complexes that catalyze the sequential addition of activated carbon units to form complex natural products.
Carbon-carbon ligases are important targets for drug discovery and development, as they play critical roles in the biosynthesis of many disease-relevant molecules. Inhibitors of these enzymes have shown promise as potential therapeutic agents for a variety of diseases, including cancer, infectious diseases, and metabolic disorders.
Methylmalonyl-CoA decarboxylase is a mitochondrial enzyme that plays a crucial role in the metabolism of certain amino acids and fatty acids. Specifically, it catalyzes the conversion of methylmalonyl-CoA to propionyl-CoA through the decarboxylation of the thioester bond.
The reaction is as follows:
Methylmalonyl-CoA → Propionyl-CoA + CO2
This enzyme requires biotin as a cofactor, and its activity is reduced in individuals with methylmalonic acidemia, a rare inherited metabolic disorder caused by mutations in the MMAB or MCEE genes that encode subunits of the methylmalonyl-CoA decarboxylase enzyme complex.
Deficiency of this enzyme leads to an accumulation of methylmalonic acid and methylmalonyl-CoA, which can cause metabolic acidosis, hyperammonemia, and other symptoms associated with the disorder.
Biotin is a water-soluble vitamin, also known as Vitamin B7 or Vitamin H. It is a cofactor for several enzymes involved in metabolism, particularly in the synthesis and breakdown of fatty acids, amino acids, and carbohydrates. Biotin plays a crucial role in maintaining healthy skin, hair, nails, nerves, and liver function. It is found in various foods such as nuts, seeds, whole grains, milk, and vegetables. Biotin deficiency is rare but can occur in people with malnutrition, alcoholism, pregnancy, or certain genetic disorders.
Carbamoyl-phosphate synthase I (CPS1) deficiency disease is a rare inherited disorder of urea synthesis, which can lead to hyperammonemia (elevated blood ammonia levels) and life-threatening neurological symptoms. CPS1 is an enzyme that plays a crucial role in the first step of the urea cycle, where it catalyzes the conversion of ammonia and bicarbonate into carbamoyl phosphate.
In CPS1 deficiency disease, mutations in the CPS1 gene lead to reduced or absent enzyme activity, impairing the body's ability to detoxify ammonia. As a result, toxic levels of ammonia accumulate in the blood and can cause irreversible brain damage, intellectual disability, coma, or even death if not treated promptly and effectively.
Symptoms of CPS1 deficiency disease may include poor feeding, vomiting, lethargy, hypotonia (low muscle tone), seizures, and developmental delays. The severity of the disorder can vary widely, from a severe neonatal-onset form with early symptoms appearing within the first few days of life to a milder late-onset form that may not become apparent until later in infancy or childhood.
Treatment typically involves a combination of dietary restrictions, medications to lower ammonia levels and support liver function, and, in some cases, liver transplantation. Early diagnosis and intervention are critical for improving outcomes and minimizing the risk of long-term neurological complications.
Holocarboxylase Synthetase Deficiency (HCD) is a rare genetic disorder of biotin metabolism, characterized by the body's inability to properly utilize the vitamin biotin. Biotin plays a crucial role in various essential functions, such as the breakdown of fats, proteins, and carbohydrates, as well as the regulation of gene expression.
Holocarboxylase synthetase is an enzyme responsible for attaching biotin to four different carboxylases, which are necessary for these vital processes. In Holocarboxylase Synthetase Deficiency, this enzyme is either partially or completely nonfunctional due to mutations in the HLCS gene.
The symptoms of HCD can vary widely but often include:
1. Feeding difficulties and poor growth in infancy
2. Severe metabolic acidosis
3. Ketoacidosis
4. Delayed development
5. Hypotonia (low muscle tone)
6. Skin rashes
7. Hair loss
8. Neurological symptoms, such as seizures and ataxia (loss of coordination and balance)
If left untreated, Holocarboxylase Synthetase Deficiency can lead to severe complications, including developmental delays, neurological damage, and even death. However, with early diagnosis and proper treatment involving biotin supplementation, many of these symptoms can be managed, and the progression of the disorder can be slowed or stopped.
Carbon-Nitrogen (C-N) ligases are a class of enzymes that catalyze the joining of a carbon atom from a donor molecule to a nitrogen atom in an acceptor molecule through a process called ligase reaction. This type of enzyme plays a crucial role in various biological processes, including the biosynthesis of amino acids, nucleotides, and other biomolecules that contain both carbon and nitrogen atoms.
C-N ligases typically require ATP or another energy source to drive the reaction forward, as well as cofactors such as metal ions or vitamins to facilitate the chemical bond formation between the carbon and nitrogen atoms. The specificity of C-N ligases varies depending on the enzyme, with some acting only on specific donor and acceptor molecules while others have broader substrate ranges.
Examples of C-N ligases include glutamine synthetase, which catalyzes the formation of glutamine from glutamate and ammonia, and asparagine synthetase, which catalyzes the formation of asparagine from aspartate and ammonia. Understanding the function and regulation of C-N ligases is important for understanding various biological processes and developing strategies to modulate them in disease states.
Immunologic deficiency syndromes refer to a group of disorders characterized by defective functioning of the immune system, leading to increased susceptibility to infections and malignancies. These deficiencies can be primary (genetic or congenital) or secondary (acquired due to environmental factors, medications, or diseases).
Primary immunodeficiency syndromes (PIDS) are caused by inherited genetic mutations that affect the development and function of immune cells, such as T cells, B cells, and phagocytes. Examples include severe combined immunodeficiency (SCID), common variable immunodeficiency (CVID), Wiskott-Aldrich syndrome, and X-linked agammaglobulinemia.
Secondary immunodeficiency syndromes can result from various factors, including:
1. HIV/AIDS: Human Immunodeficiency Virus infection leads to the depletion of CD4+ T cells, causing profound immune dysfunction and increased vulnerability to opportunistic infections and malignancies.
2. Medications: Certain medications, such as chemotherapy, immunosuppressive drugs, and long-term corticosteroid use, can impair immune function and increase infection risk.
3. Malnutrition: Deficiencies in essential nutrients like protein, vitamins, and minerals can weaken the immune system and make individuals more susceptible to infections.
4. Aging: The immune system naturally declines with age, leading to an increased incidence of infections and poorer vaccine responses in older adults.
5. Other medical conditions: Chronic diseases such as diabetes, cancer, and chronic kidney or liver disease can also compromise the immune system and contribute to immunodeficiency syndromes.
Immunologic deficiency syndromes require appropriate diagnosis and management strategies, which may include antimicrobial therapy, immunoglobulin replacement, hematopoietic stem cell transplantation, or targeted treatments for the underlying cause.
Biotinidase deficiency is a genetic disorder that affects the body's ability to recycle and reuse biotin, a type of B vitamin. Biotinidase is an enzyme that helps release biotin from proteins in the food we eat and recycle it for use by the body.
In people with biotinidase deficiency, the biotinidase enzyme is either partially or completely missing, leading to a decrease in available biotin. This can result in a variety of symptoms, including seizures, developmental delays, hearing and vision loss, skin rashes, hair loss, and muscle weakness.
There are two main types of biotinidase deficiency: partial deficiency and profound deficiency. Partial deficiency means that some biotinidase activity is present, but not enough to prevent symptoms. Profound deficiency means that there is little or no biotinidase activity, resulting in more severe symptoms.
Biotinidase deficiency can be diagnosed through a blood test that measures the level of biotinidase enzyme activity. Treatment typically involves taking biotin supplements to replace the missing biotin and prevent symptoms from developing or worsening. With early diagnosis and treatment, people with biotinidase deficiency can often lead normal lives.
Multiple sulfatase deficiency (MSD) is a rare inherited metabolic disorder that affects multiple organ systems in the body. It is caused by mutations in the SUMF1 gene, which provides instructions for making an enzyme called formylglycine-generating enzyme (FGE). FGE is essential for the function of several sulfatase enzymes, which are responsible for removing sulfate groups from certain sugar molecules attached to proteins and lipids.
In MSD, the activity of all or most of these sulfatase enzymes is reduced or absent, leading to the accumulation of sulfated molecules in various tissues and organs. This can result in a wide range of symptoms that typically appear in infancy or early childhood, including developmental delay, intellectual disability, coarse facial features, skeletal abnormalities, vision and hearing loss, and problems with mobility and coordination.
MSD is an autosomal recessive disorder, which means that an individual must inherit two copies of the mutated gene (one from each parent) in order to develop the disease. The incidence of MSD is estimated to be less than 1 in 1 million people worldwide. Currently, there is no cure for MSD and treatment is focused on managing symptoms and improving quality of life.
Biotinidase is an enzyme that is responsible for the release of biotin, a vital nutrient, from proteins in the body. Biotin is essential for various metabolic processes, including the synthesis of fatty acids and glucose. Biotinidase deficiency can lead to serious health problems, such as seizures, developmental delays, and hearing and vision loss. Therefore, biotinidase levels are often measured in newborn screening tests to identify babies who may be at risk for this rare but treatable condition.
Inborn errors of amino acid metabolism refer to genetic disorders that affect the body's ability to properly break down and process individual amino acids, which are the building blocks of proteins. These disorders can result in an accumulation of toxic levels of certain amino acids or their byproducts in the body, leading to a variety of symptoms and health complications.
There are many different types of inborn errors of amino acid metabolism, each affecting a specific amino acid or group of amino acids. Some examples include:
* Phenylketonuria (PKU): This disorder affects the breakdown of the amino acid phenylalanine, leading to its accumulation in the body and causing brain damage if left untreated.
* Maple syrup urine disease: This disorder affects the breakdown of the branched-chain amino acids leucine, isoleucine, and valine, leading to their accumulation in the body and causing neurological problems.
* Homocystinuria: This disorder affects the breakdown of the amino acid methionine, leading to its accumulation in the body and causing a range of symptoms including developmental delay, intellectual disability, and cardiovascular problems.
Treatment for inborn errors of amino acid metabolism typically involves dietary restrictions or supplementation to manage the levels of affected amino acids in the body. In some cases, medication or other therapies may also be necessary. Early diagnosis and treatment can help prevent or minimize the severity of symptoms and health complications associated with these disorders.
Carboxy-lyases are a class of enzymes that catalyze the removal of a carboxyl group from a substrate, often releasing carbon dioxide in the process. These enzymes play important roles in various metabolic pathways, such as the biosynthesis and degradation of amino acids, sugars, and other organic compounds.
Carboxy-lyases are classified under EC number 4.2 in the Enzyme Commission (EC) system. They can be further divided into several subclasses based on their specific mechanisms and substrates. For example, some carboxy-lyases require a cofactor such as biotin or thiamine pyrophosphate to facilitate the decarboxylation reaction, while others do not.
Examples of carboxy-lyases include:
1. Pyruvate decarboxylase: This enzyme catalyzes the conversion of pyruvate to acetaldehyde and carbon dioxide during fermentation in yeast and other organisms.
2. Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO): This enzyme is essential for photosynthesis in plants and some bacteria, as it catalyzes the fixation of carbon dioxide into an organic molecule during the Calvin cycle.
3. Phosphoenolpyruvate carboxylase: Found in plants, algae, and some bacteria, this enzyme plays a role in anaplerotic reactions that replenish intermediates in the citric acid cycle. It catalyzes the conversion of phosphoenolpyruvate to oxaloacetate and inorganic phosphate.
4. Aspartate transcarbamylase: This enzyme is involved in the biosynthesis of pyrimidines, a class of nucleotides. It catalyzes the transfer of a carboxyl group from carbamoyl aspartate to carbamoyl phosphate, forming cytidine triphosphate (CTP) and fumarate.
5. Urocanase: Found in animals, this enzyme is involved in histidine catabolism. It catalyzes the conversion of urocanate to formiminoglutamate and ammonia.
Acetyl-CoA carboxylase (ACCA) is a biotin-dependent enzyme that plays a crucial role in fatty acid synthesis. It catalyzes the conversion of acetyl-CoA to malonyl-CoA, which is the first and rate-limiting step in the synthesis of long-chain fatty acids. The reaction catalyzed by ACCA is as follows:
acetyl-CoA + HCO3- + ATP + 2H+ --> malonyl-CoA + CoA + ADP + Pi + 2H2O
ACCA exists in two isoforms, a cytosolic form (ACC1) and a mitochondrial form (ACC2). ACC1 is primarily involved in fatty acid synthesis, while ACC2 is responsible for the regulation of fatty acid oxidation. The activity of ACCA is regulated by several factors, including phosphorylation/dephosphorylation, allosteric regulation, and transcriptional regulation. Dysregulation of ACCA has been implicated in various metabolic disorders, such as obesity, insulin resistance, and non-alcoholic fatty liver disease.
Ligases are a group of enzymes that catalyze the formation of a covalent bond between two molecules, usually involving the joining of two nucleotides in a DNA or RNA strand. They play a crucial role in various biological processes such as DNA replication, repair, and recombination. In DNA ligases, the enzyme seals nicks or breaks in the phosphodiester backbone of the DNA molecule by catalyzing the formation of an ester bond between the 3'-hydroxyl group and the 5'-phosphate group of adjacent nucleotides. This process is essential for maintaining genomic integrity and stability.
 Merton F. Utter
Merton F. Utter
Triheptanoin
Ketogenic diet
Pyruvate carboxylase deficiency
List of MeSH codes (C18)
List of MeSH codes (C10)
List of MeSH codes (C16)
Hypokinesia
Lactic acidosis
Hypotonia
List of diseases (P)
Biotin deficiency
Ketosis
Methylcrotonyl-CoA carboxylase
Propionyl-CoA
6-Phosphogluconate dehydrogenase
Glycolysis
Biotin
Citrate-malate shuttle
Fatty acid
Enzyme
Biosynthesis
List of OMIM disorder codes
Combined malonic and methylmalonic aciduria
Amino acid
Mitochondrion
Morpheein
Insulin
pyruvate carboxylase deficiency disease Disease Ontology Browser - DOID:3651
 Pyruvate carboxylase deficiency: MedlinePlus Genetics
Pyruvate carboxylase deficiency: MedlinePlus Genetics
Merton F. Utter - Wikipedia
 Pyruvate Carboxylase Deficiency: Overview, Pathophysiology, Etiology
Pyruvate Carboxylase Deficiency: Overview, Pathophysiology, Etiology
 Wolman Disease - Supra-Regional Assay Service
Wolman Disease - Supra-Regional Assay Service
Treatment of congenital lactic acidosis with dichloroacetate | Archives of Disease in Childhood
Bio2Vec
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
 DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
DeCS - Changed terms
Newborn Screening In India - MERD INDIA FOUNDATION: Support Group For Inborn Errors Of Metabolism In India,Genetic Disorders In...
 Familial Hyperlysinemia | Syndromes: Rapid Recognition and Perioperative Implications | AccessAnesthesiology | McGraw Hill...
Familial Hyperlysinemia | Syndromes: Rapid Recognition and Perioperative Implications | AccessAnesthesiology | McGraw Hill...
Specific PHGKB|Rare Diseases PHGKB|PHGKB
Vitamin B1 (Thiamine): Reference Range, Interpretation, Collection and Panels
 Childhood ataxia with central nervous system hypomyelination | MedLink Neurology
Childhood ataxia with central nervous system hypomyelination | MedLink Neurology
 Mitochondrial Diseases (medical concept explorer)
Mitochondrial Diseases (medical concept explorer)
 Búsqueda | Portal Regional de la BVS
Búsqueda | Portal Regional de la BVS
 Pyruvate Metabolism Disorders - Children's Health Issues - MSD Manual Consumer Version
Pyruvate Metabolism Disorders - Children's Health Issues - MSD Manual Consumer Version
Hartnup Disease | Profiles RNS
Metabolism17
- Later in his career, his lab became a leading center in the study of inborn errors of metabolism of pyruvate. (wikipedia.org)
- Pyruvate carboxylase (PC) deficiency is a rare inborn error of metabolism that can cause developmental delay and failure to thrive starting in the neonatal or early infantile period. (medscape.com)
- Pyruvate carboxylase (PC) deficiency affects metabolism in several major ways. (medscape.com)
- Pathways of pyruvate metabolism and oxidative phosphorylation. (bmj.com)
- The vision was to have a forum of like-minded individuals who can promote & help spreading awarenesses about significance of early detection of Inborn Errors of Metabolism and Rare Diseases, and thereby save and support thousands of lives. (merdindia.com)
- This website and support group MERD INDIA FOUNDATION is only to provide basic information and moral support to the parents of children with inborn errors of metabolism and rare genetic diseases. (merdindia.com)
- Deficiency leads to decreased transketolase activity in red blood cells (erythrocytes) and increases pyruvic acid in the blood, which, in turn, is not converted to acetyl-coA and is unable to enter the Krebs cycle (for aerobic oxidative metabolism). (medscape.com)
- A deficiency in any one of the enzymes involved in pyruvate metabolism leads to one of many disorders. (msdmanuals.com)
- In many pyruvate metabolism disorders, both parents of the affected child carry a copy of the abnormal gene. (msdmanuals.com)
- Many enzymes are involved in pyruvate metabolism. (msdmanuals.com)
- Symptoms of pyruvate metabolism disorders may develop any time between early infancy and late adulthood. (msdmanuals.com)
- Untangling the Spirals of Metabolic Disease: Primary Diagnoses and Secondary Effects: Implications for Treatment David A. H. Whiteman MD 1909 Archibald Garrod In his paper, Inborn Errors of Metabolism, the disease Alkaptonuria (Ochronosis: Homogentisic Acid Oxidase Deficiency) is described as being caused by a gene. (abcdocz.com)
- Some inherited metabolic disorders may alter pyruvate metabolism indirectly. (nih.gov)
- Disorders in pyruvate metabolism appear to lead to deficiencies in neurotransmitter synthesis and, consequently, to nervous system disorders. (nih.gov)
- Glucose metabolism and pyruvate carboxylase enhance glutathione synthesis and restrict oxidative stress in pancreatic islets. (scienceopen.com)
- Magnesium is required for three critical enzymatic reactions in glucose metabolism: pyruvate carboxylase, phosphoenol-pyruvate carboxykinase and fructose 1,6 biphosphatase. (metabolichealing.com)
- Inherited abnormalities of fructose metabolism, which include three known autosomal recessive types: hepatic fructokinase deficiency (essential fructosuria), hereditary fructose intolerance, and hereditary fructose-1,6-diphosphatase deficiency. (sdsu.edu)
Gluconeogenesis5
- He and his coworkers discovered the enzymes pyruvate carboxylase and phosphoenolpyruvate carboxykinase and their role in converting pyruvate to phosphoenolpyruvate via oxaloacetate in gluconeogenesis, a pathway not the reverse of that catalyzed in glycolysis by pyruvate kinase. (wikipedia.org)
- PC deficiency results in malfunction of the citric acid cycle and of gluconeogenesis, thereby depriving the body of energy. (medscape.com)
- 2 4 A few cases have been reported that involve deficiencies in enzymes of the tricarboxylic acid cycle, such as fumarase, or of gluconeogenesis, such as pyruvate carboxylase (PC) or phosphoenolpyruvate carboxykinase (PEPCK). (bmj.com)
- Pyruvate may be reduced to lactate in the cytoplasm or may be transported into the mitochondria for anabolic reactions, such as gluconeogenesis and lipogenesis, or for oxidation to acetyl CoA by the pyruvate dehydrogenase (PDH) complex (PDC). (bmj.com)
- They are difficult to diagnose and describe because pyruvate is a key intermediate in glycolysis, gluconeogenesis, and the tricarboxylic acid cycle. (nih.gov)
OMIM2
- The analysis uses data from IMPC, along with published data on other mouse mutants, in comparison to human disease reports in OMIM, Orphanet, and DECIPHER. (mousephenotype.org)
- HCP is an autosomal disease, meaning it is carried in one of the non-sex chromosomes(OMIM). (bionity.com)
Disorders3
- Metabolic Disorders and Rare Diseases Organization of India was conceived by two parents - Vikas Bhatia and Poonam Bhatia ,who wanted to set up a parent support and advocacy organization in India. (merdindia.com)
- Some specific pyruvate disorders are helped by changes in diet. (msdmanuals.com)
- A hereditary deficiency in any one of these enzymes results in one of a variety of disorders, depending on which enzyme is deficient. (msdmanuals.com)
Genetic6
- Evaluation of the patient by an expert in metabolic and genetic disease is necessary to confirm the diagnosis, guide appropriate treatment, and determine prognosis. (medscape.com)
- For example, Oklahoma screens for Pompe disease , a genetic condition that impacts the heart and other muscles. (kctv5.com)
- South Carolina screens for a genetic disorder that leads to heart disease, intellectual disabilities and skeletal problems. (kctv5.com)
- Eventually, doctors discovered in 2014 that Kiana had pyruvate carboxylase deficiency , or PCD, a genetic condition that now as a pre-teen has left her unable to walk, talk or sit. (kctv5.com)
- When your patients are looking to understand if they are a carrier for specific genetic conditions like cystic fibrosis, spinal muscular atrophy, fragile X syndrome, or Tay-Sachs disease, appropriate genetic screening and actionable results are essential . (questwomenshealth.com)
- Hereditary coproporphyria (HCP) is a hereditary genetic disease that causes purple urine, photo sensitivity , and attacks of abdominal pain. (bionity.com)
Abnormalities4
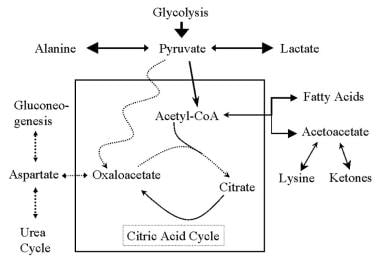
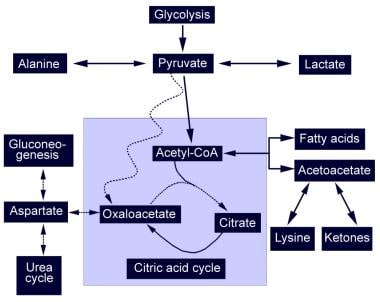
- Diagrammatic representation of the citric acid cycle and the abnormalities found in pyruvate carboxylase deficiency (PCD). (medscape.com)
- Patients may develop a wide spectrum of neurologic abnormalities, from prenatal-onset white matter disease to juvenile- or adult-onset ataxia and dementia, sometimes with ovarian insufficiency. (medlink.com)
- Childhood ataxia with CNS hypomyelination (or vanishing white matter disease) is a relatively common leukodystrophy in which most of the patients have a pathognomonic pattern of MRI and diffusion tensor imaging abnormalities. (medlink.com)
- Chitkara DK, Nurko S, Shoffner JM, Buie T, Flores A. Abnormalities in gastrointestinal motility are associated with diseases of oxidative phosphorylation in children. (springer.com)
Glycogen3
- Glycogen storage diseases presenting as hypertrophic cardiomyopathy. (springer.com)
- Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. (springer.com)
- Glycogen synthase deficiency (Glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. (springer.com)
Inborn1
- Guanidinoacetate methyltransferase (GAMT) deficiency is an inborn error of creatine synthesis resulting in creatine deficiency and an accumulation of the toxic metabolite (GAA) guanidinoacetate in tissues and body fluids, including the brain. (medicalhomeportal.org)
Glucose2
- Under aerobic conditions, the activity of PDC determines the rate at which all cells oxidise glucose, pyruvate, and lactate. (bmj.com)
- In the presence of oxygen, the process begins with glycolysis, which breaks down glucose into two molecules of pyruvate. (microbiologynote.com)
Conversion of pyruvate2
- PC catalyzes the conversion of pyruvate to oxaloacetate with biotin as a cofactor. (medscape.com)
- However, instead of proceeding through the remaining aerobic pathways, anaerobic respiration ends with the conversion of pyruvate into other compounds, such as lactate or ethanol. (microbiologynote.com)
Inherited disorder1
- Pyruvate carboxylase deficiency is an inherited disorder that causes lactic acid and other potentially toxic compounds to accumulate in the blood. (medlineplus.gov)
Enzymes3
- Most identifiable cases involve inherited or spontaneous mutations in the pyruvate dehydrogenase complex (PDC) or in one or more enzymes of the respiratory chain. (bmj.com)
- This disorder is caused by a lack of a group of enzymes needed to process pyruvate. (msdmanuals.com)
- Diseases caused by the loss of one or more enzymes involved in the hydrolysis of mannoside linkages (MANNOSIDASES). (ouhsc.edu)
Clinical7
- Most often, the disease is diagnosed by a general screening for metabolic disease initiated by clinical signs. (mhmedical.com)
- An early onset form of phytanic acid storage disease with clinical and biochemical signs different from those of REFSUM DISEASE . (nih.gov)
- Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. (springer.com)
- The major clinical findings of holocarboxylase synthetase deficiency include severe ketoacidosis, exfoliative dermatitis, and hypoglycemia . (medscape.com)
- Tammachote R, Janklat S, Tongkobpetch S, Suphapeetiporn K, Shotelersuk V. Holocarboxylase synthetase deficiency: novel clinical and molecular findings. (medscape.com)
- Clinical Presentation and Positive Outcome of Two Siblings with Holocarboxylase Synthetase Deficiency Caused by a Homozygous L216R Mutation. (medscape.com)
- Jonathon Wright, MD has noted after several decades of clinical practice that when hypochlorhydria is present, vitamin, mineral and amino acid deficiencies are very common. (metabolichealing.com)
Severe5
- In the most severe form, pyruvate carboxylase deficiency results in progressive neurologic symptoms, starting in the neonatal or early infantile period, including developmental delay, poor muscle tone, abnormal eye movements, and seizures. (medscape.com)
- A stimulation exceeding 20%-25% after the addition of TTP indicates severe thiamine deficiency (an activity coefficient of 1.25). (medscape.com)
- This deficiency results in a variety of symptoms, ranging from mild to severe. (msdmanuals.com)
- MCAD Deficiency A Child with Multiple Problems The Pregnancy Complicated by abdominal pain, severe nausea and vomiting, "black out spells" Emergency C-section because of maternal hemorrhage (unknown cause) A Child with Multiple Problems The Child Multiple hospital admissions for vomiting and dehydration in first year of life Nissen fundoplication at 18 months of age. (abcdocz.com)
- Reduced half-life of holocarboxylase synthetase from patients with severe multiple carboxylase deficiency. (medscape.com)
Deficient2
- Gordon N. Classic diseases revisited: carbohydrate-deficient glycoprotein syndromes. (springer.com)
- Deficient biotinidase activity in late-onset multiple carboxylase deficiency. (medscape.com)
Nervous system7
- Researchers suggest that the loss of pyruvate carboxylase function in the nervous system, particularly the role of the enzyme in myelin formation and neurotransmitter production, also contributes to the neurologic features of pyruvate carboxylase deficiency. (medlineplus.gov)
- Mutations affecting the eukaryotic initiation factor 2B (eIF2B) cause one of the most common leukodystrophies, the autosomal recessive childhood ataxia with central nervous system hypomyelination (CACH), or vanishing white matter disease (VWM). (medlink.com)
- Molecules like ISRIB (integrated stress response inhibitor) correct the eIF2B deficiency in most mutants and are likely to be tried as therapy for central nervous system hypomyelination/vanishing white matter disease. (medlink.com)
- Lysosomal Storage Diseases, Nervous System" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (childrensmercy.org)
- This graph shows the total number of publications written about "Lysosomal Storage Diseases, Nervous System" by people in this website by year, and whether "Lysosomal Storage Diseases, Nervous System" was a major or minor topic of these publications. (childrensmercy.org)
- Below are the most recent publications written about "Lysosomal Storage Diseases, Nervous System" by people in Profiles. (childrensmercy.org)
- Measurements of creatine phosphokinase are used in the diagnosis and treatment of myocardial infarction, skeletal muscle diseases, and diseases of the central nervous system. (cdc.gov)
Biotinidase Deficiency1
- Valproic acid can cause biotinidase deficiency, which may be helped by biotin supplements. (elispot.biz)
Enzyme deficiencies2
- 5 This concept is most readily appreciated by considering mitochondrial enzyme deficiencies. (bmj.com)
- enzyme deficiencies of α-aminoadipic semialdehyde dehydrogenase and the saccharopine dehydrogenases have been associated with increased serum levels of l -lysine. (mhmedical.com)
Mitochondrial2
- MELAS is a rare disease entity and occasionally comorbid with mitochondrial diabetes in childhood. (bvsalud.org)
- Aside from her brother, the family genogram (Figure 3) revealed no significant illness that would indicate a mitochondrial or hereditary neurodegenerative disease, and all other half-siblings from both parents' first marriages showed no signs of neurologic disorder. (jmust.org)
Oxidative phosphorylation1
- This process involves four main metabolic pathways: glycolysis, pyruvate oxidation, the Krebs cycle (also known as the citric acid cycle or the tricarboxylic acid cycle), and oxidative phosphorylation. (microbiologynote.com)
Inheritance1
- It is unclear at this time if the disease inheritance is dominant or recessive. (bionity.com)
Mutations3
- Mutations in the PC gene cause pyruvate carboxylase deficiency. (medlineplus.gov)
- Mutations in the PC gene reduce the amount of pyruvate carboxylase in cells or disrupt the enzyme's activity. (medlineplus.gov)
- Aside from varying intensity of symptoms there are no other known mutations, and it is not known at this time if mutations in other genes can trigger this same disease. (bionity.com)
Pathways2
- Pyruvate cannot produce oxaloacetate and is shunted to alternative pathways that produce lactic acid and alanine. (medscape.com)
- Hartnup Disorder, also known as hartnup disease, is related to aminoaciduria and cystinuria, and has symptoms including seizures An important gene associated with Hartnup Disorder is SLC6A19 (Solute Carrier Family 6 Member 19), and among its related pathways/superpathways are Disease and Transport of inorganic cations/anions and amino acids/oligopeptides. (silexon.tech)
Body's1
- However, a number of micronutrients should be suitable for the body's needs, so that it does not result in deficiency or of overdoses. (freedissertation.com)
Biochemical2
- Metabolic disease may present in a fulminate fashion to the pediatric intensivist with profound biochemical disturbances, encephalopathy and even cardiac failure. (springer.com)
- The diagnosis of citrin deficiency is established in an individual with characteristic biochemical findings (in general, increased blood or plasma concentration of ammonia, plasma or serum concentration of citrulline and arginine, plasma or serum threonine-to-serine ratio, and serum concentration of pancreatic secretory trypsin inhibitor) and identification of biallelic pathogenic variants in SLC25A13 . (nih.gov)
Lactic acid2
- Additionally, a loss of pyruvate carboxylase allows compounds such as lactic acid and ammonia to build up and damage organs and tissues. (medlineplus.gov)
- Problems with the breaking down (metabolizing) of pyruvate can limit a cell's ability to produce energy and allow a buildup of a waste product called lactic acid (lactic acidosis). (msdmanuals.com)
Infantile1
- Mutant holocarboxylase synthetase: evidence for the enzyme defect in early infantile biotin-responsive multiple carboxylase deficiency. (medscape.com)
Congenital1
- A disease that results from a congenital defect in ELECTRON TRANSPORT COMPLEX IV. (uams.edu)
Genetics1
- The benefits of integrating cross-species systems genetics platforms to advance knowledge in the underlying mechanisms that drive cardiometabolic diseases have been investigated. (elifesciences.org)
Symptoms4
- Researchers have identified at least three types of pyruvate carboxylase deficiency, which are distinguished by the severity of their signs and symptoms. (medlineplus.gov)
- Pyruvate carboxylase deficiency type B has life-threatening signs and symptoms that become apparent shortly after birth. (medlineplus.gov)
- This chapter serves as a guide to the recognition of metabolic disease based on presenting signs, symptoms and screening laboratory tests. (springer.com)
- Citrin deficiency can manifest in newborns or infants as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD), in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD), and in adults as recurrent hyperammonemia with neuropsychiatric symptoms in citrullinemia type II (CTLN2). (nih.gov)
Oxidation1
- These pyruvate molecules then enter the mitochondria , where they undergo oxidation, releasing carbon dioxide as a byproduct. (microbiologynote.com)
Leigh1
- For example, he showed that contrary to contemporary belief, Leigh disease is not associated with deficiency in pyruvate carboxylase activity. (wikipedia.org)
Syndrome2
- The expanding phenotype of GLUT1-deficiency syndrome. (scienceopen.com)
- Magnesium deficiency is strongly linked to diabetes and metabolic syndrome. (metabolichealing.com)
Lactate1
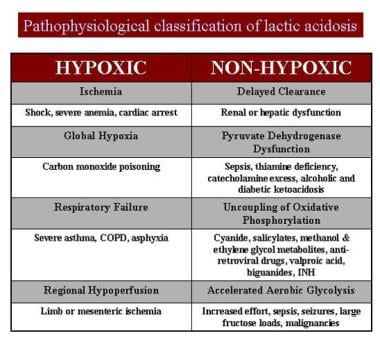
- Laboratory changes are dyslipidemia, increased lactate-to-pyruvate ratio, higher levels of urinary oxidative stress markers, and considerable deviation in tricarboxylic acid (TCA) cycle metabolites. (nih.gov)
Hepatic3
- Elevated levels of the transaminases can indicate myocardial infarction, hepatic disease, muscular dystrophy, or organ damage. (cdc.gov)
- It is currently the most sensitive enzymatic indicator of liver disease, with normal values rarely found in the presence of hepatic disease. (cdc.gov)
- Hereditary coproporphyria (HCP) is a form of hepatic porphyria associated with a deficiency of the enzyme coproporphyrinogen III oxidase. (bionity.com)
Biotin5
- Some cases of partial enzyme deficiency respond to the administration of excess biotin. (medscape.com)
- Biotin has been used in alternative medicine as a likely effective aid in treating or preventing biotin deficiency. (elispot.biz)
- Biotin deficiency can be caused by malnutrition, rapid weight loss, long-term tube feeding, and other medical conditions. (elispot.biz)
- Roth KS, Yang W, Foremann JW, Rothman R, Segal S. Holocarboxylase synthetase deficiency: a biotin-responsive organic acidemia. (medscape.com)
- A novel molecular mechanism to explain biotin-unresponsive holocarboxylase synthetase deficiency. (medscape.com)
Molecules1
- Note: If you'd like to get a target analysis report for Hartnup Disease , or if you are interested to learn how our AI-powered BDE-Chem can design therapeutic molecules to interact with the target(s) above against the disease of Hartnup Disease at a cost 90% lower than traditional approaches, please feel free to contact us at [email protected] . (silexon.tech)
Phenotypes3
- A knowledge graph of biological entities such as genes, gene functions, diseases, phenotypes and chemicals. (edu.sa)
- Phenotype comparisons summarize the similarity of mouse phenotypes with human disease phenotypes. (mousephenotype.org)
- Periodic measurement of plasma concentration of ammonia and citrulline, and serum concentration of PSTI for all phenotypes associated with citrin deficiency. (nih.gov)
Metabolic disorder1
- In humans, acetoacetyl-CoA is involved in the metabolic disorder called the short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (HADH) pathway. (hmdb.ca)