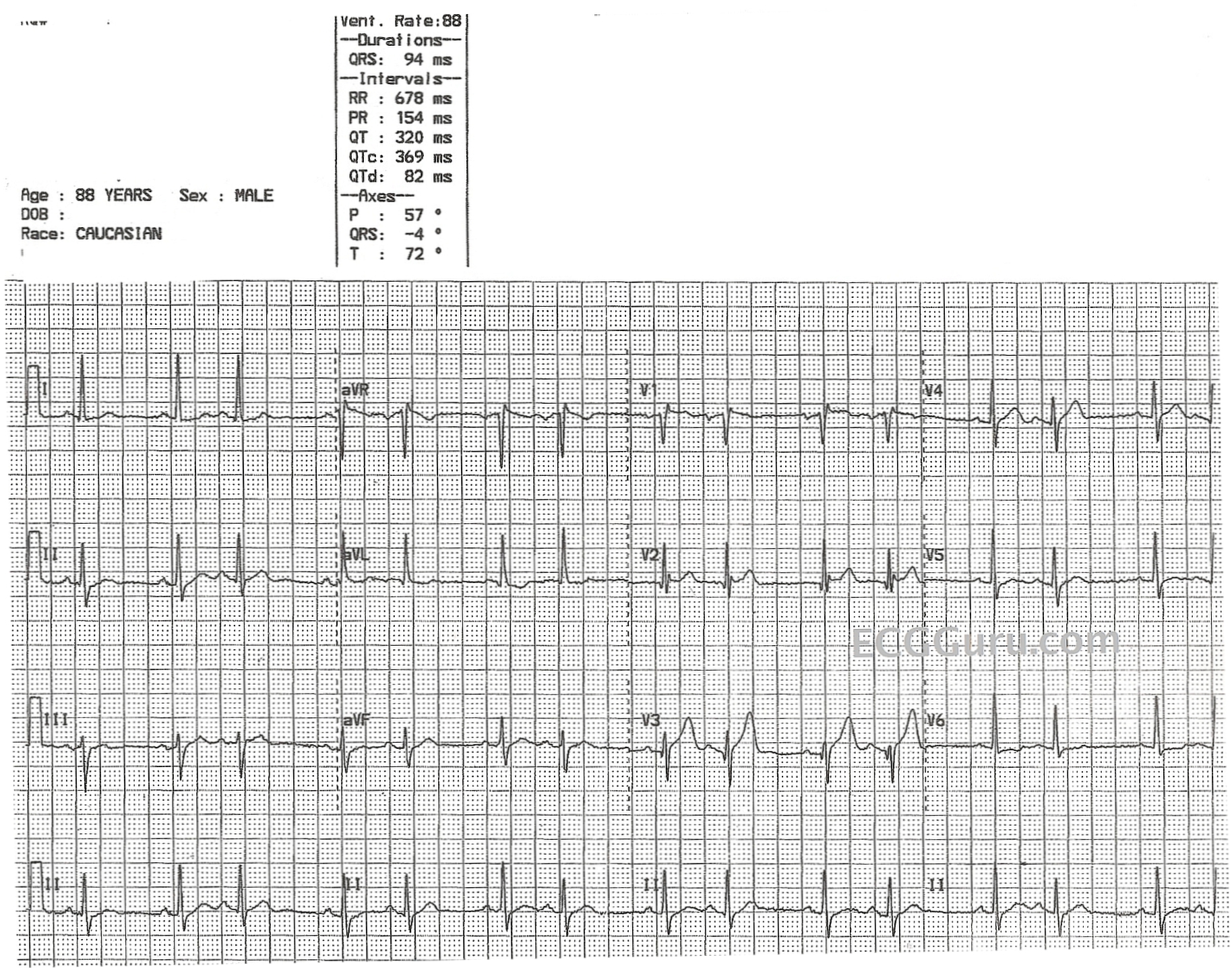
Situs Inversus
Dextrocardia
Kartagener Syndrome
Ciliary Motility Disorders
Cilia
Blepharophimosis
Heterotaxy Syndrome
Abnormalities, Multiple
Biliary Atresia
Heart Septal Defects
Kidney Diseases, Cystic
Cholecystectomy, Laparoscopic
Blepharoptosis
Observations on some additional abnormalities in situs inversus viscerum. (1/163)
The abnormal findings in a case of Situs inversus totalis are described. The duodenum was placed abnormally and retained its primitive mesentery. The proximal 22 in of jejunum were retroperitoneal. The attachment of the root of the mesentery to the posterior abdominal wall had a 7-shaped appearance, and there was a partial failure of the primitive mesocolon to adhere to the posterior abdominal wall. The common hepatic artery arose from the superior meseneric artery, which also provided a branch to the proximal jejunal loop. The right vagus nerve was found anterior to the oesophagus at the oesophageal hiatus in the diaphragm, and the left vagus was posterior. A double ureter was present on the right side. The findings are discussed in relation to mid-gut development. (+info)Liver transplantation in patients with situs inversus. (2/163)
Two patients with situs inversus and biliary atresia were treated with hepatic transplantation, one with an auxiliary liver and the other with an orthotopic graft which was placed using a piggy-back technique. Both transplants functioned well initially. The auxiliary liver was rejected after 1 1/2 months, and the patient died after an attempt at retransplantation many months later. The recipient of the orthotopic liver has perfect liver function 10 months postoperatively. (+info)Asymmetry of cilia and of mice and men. (3/163)
Evidence is given for the opinion that cilia in the early embryo, by their work, determine the laterality of the body; without ciliary work body laterality would be randomized. More exactly, monocilia in the primitive node are responsible for this determination. They have been described as being of the 9+0 type, but with dynein arms and with a gyrating movement. The orientation of the monocilia on the epithelium is of no importance but the direction of their gyration is, as may also be the shape of the node. The chirality of the cilia is thus reflected directly in the asymmetry of the body. The dynein arms go clockwise as seen from the base to tip and the ciliary rotation is in the same direction. The resulting waterflow is towards the left and so is the movement of the forming heart. In most subgroups of the immotile-cilia syndrome this mechanism does not work and equally many individuals will be born with situs inversus as with situs solitus. An exception is the immotile-cilia subgroup, named 'microtubule transposition', which is characterized by all cilia having a 9+0 structure throughout most of their length. (+info)Abnormal nodal flow precedes situs inversus in iv and inv mice. (4/163)
We examined the nodal flow of well-characterized mouse mutants, inversus viscerum (iv) and inversion of embryonic turning (inv), and found that their laterality defects are always accompanied by an abnormality in nodal flow. In a randomized laterality mutant, iv, the nodal cilia were immotile and the nodal flow was absent. In a situs inversus mutant, inv, the nodal cilia was motile but could only produce very weak leftward nodal flow. These results consistently support our hypothesis that the nodal flow produces the gradient of putative morphogen and triggers the first L-R determination event. (+info)Situs inversus with cholelithiasis. (5/163)
Situs inversus totalis is a form of heterotaxia which is usually detected accidentally while investigating for any associated condition. If undetected, this condition can create a diagnostic puzzle. We report one such case in which situs inversus was associated with cholelithiasis. (+info)A locus for primary ciliary dyskinesia maps to chromosome 19q. (6/163)
Primary ciliary dyskinesia is an autosomal recessive condition characterised by chronic sinusitis, bronchiectasis, and subfertility. Situs inversus occurs in 50% of cases (Kartagener syndrome). It has an estimated incidence of 1 in 20 000 live births. The clinical phenotype is caused by defective ciliary function associated with a range of ultrastructural abnormalities including absent dynein arms, absent radial spokes, and disturbed ciliary orientation. The molecular genetic basis is unknown. A genome scan was performed in five Arabic families. Using GENEHUNTER, a maximal multipoint lod score (HLOD) of 4.4 was obtained on chromosome 19q13.3-qter at alpha (proportion of linked families) = 0.7. A 15 cM critical region is defined by recombinations at D19S572 and D19S218. These data provide significant evidence for a PCD locus on chromosome 19q and confirm locus heterogeneity. (+info)Neonatal arterial switch operation for transposition of the great arteries in a patient with mirror image dextrocardia and situs inversus totalis. (7/163)
The neonatal arterial switch operation has become the standard therapy for D-transposition of the great arteries in the absence of left ventricular outflow tract obstruction. We describe our experience of successful arterial switch operation after balloon atrial septostomy in a 5-day-old infant girl who had atrial and visceral situs inversus totalis, mirror image dextrocardia, and D-transposition of the great arteries. To our knowledge, ours is the first report of this operation in a patient with this anatomy. (+info)Laparoscopic cholecystectomy and appendectomy in situs inversus totalis. (8/163)
Situs inversus totalis is an uncommon anatomic anomaly that complicates diagnosis and management of acute abdominal pain. Expedient diagnosis of common intraperitoneal disease processes such as biliary colic, acute appendicitis and diverticulitis is often delayed as a result of seemingly incongruous physical findings. We present the case of a young woman with prior emergency room visits for complaints of a vague left upper quadrant abdominal pain. An ultrasound performed on her third presentation revealed visceral situs inversus with cholelithiasis and dilated intra- and extrahepatic biliary ducts. Standard laparoscopic cholecystectomy and cholangiography with a mirror-image surgical approach was performed successfully and without complication. (+info)Situs Inversus is a congenital condition in which the major visceral organs are situated in mirror-image positions to their normal locations. Instead of being on the left side, the heart and its large blood vessels are on the right side, while the liver is on the left side and the lungs are reversed. The stomach, spleen, and pancreas may also be affected. It's important to note that this condition is generally asymptomatic and often goes unnoticed unless there are complications or associated abnormalities.
There are two types of Situs Inversus: total (complete reversal of all organs) and partial (reversal of only some organs). Total Situs Inversus is also sometimes referred to as "mirror-image dextrocardia" because the heart, which is usually on the left side, is located on the right side in a mirrored position.
While Situs Inversus itself does not typically cause health problems, people with this condition may have an increased risk for certain medical conditions, such as congenital heart defects or primary ciliary dyskinesia (PCD), which can lead to chronic respiratory infections and infertility.
Dextrocardia is a medical condition in which the heart is positioned on the right side of the chest instead of the left side. This is a congenital condition, meaning it is present at birth. In people with dextrocardia, the heart's structure and function are usually normal, but the orientation of the heart within the chest is reversed.
There are two main types of dextrocardia:
1. Dextrocardia without visceral situs inversus: In this type, the heart is on the right side of the chest, but the other organs in the chest and abdomen are in their normal positions. This is a rare condition and can be associated with other congenital heart defects.
2. Dextrocardia with visceral situs inversus: In this type, the heart is on the right side of the chest, and the other organs in the chest and abdomen are mirrored or reversed from their normal positions. This is a less common form of dextrocardia and is often referred to as "situs inversus totalis."
It's important to note that while dextrocardia itself is not a life-threatening condition, people with this condition may have other heart defects or medical issues that require treatment. If you or someone you know has been diagnosed with dextrocardia, it's essential to consult with a healthcare professional for proper evaluation and management.
Kartagener Syndrome is a rare genetic disorder that primarily affects the respiratory system. It is characterized by the triad of chronic sinusitis, bronchiectasis (damage and widening of the airways in the lungs), and situs inversus totalis - a condition where the major visceral organs are mirrored or reversed from their normal positions.
In Kartagener Syndrome, the cilia (tiny hair-like structures) lining the respiratory tract are abnormal or dysfunctional, which impairs their ability to clear mucus and other particles. This leads to recurrent respiratory infections, bronchiectasis, and ultimately, progressive lung damage.
The condition is inherited as an autosomal recessive trait, meaning that an individual must inherit two copies of the defective gene - one from each parent - to develop the syndrome. Kartagener Syndrome is a subtype of primary ciliary dyskinesia (PCD), a group of disorders affecting ciliary structure and function.
Ciliary motility disorders are a group of rare genetic conditions that affect the function of cilia, which are tiny hair-like structures on the surface of cells in the body. Cilia play an important role in moving fluids and particles across the cell surface, including the movement of mucus and other substances in the respiratory system, the movement of eggs and sperm in the reproductive system, and the movement of fluid in the inner ear.
Ciliary motility disorders are caused by mutations in genes that are responsible for the proper functioning of cilia. These mutations can lead to abnormalities in the structure or function of cilia, which can result in a range of symptoms depending on the specific disorder and the parts of the body that are affected.
Some common symptoms of ciliary motility disorders include recurrent respiratory infections, chronic sinusitis, hearing loss, infertility, and situs inversus, a condition in which the major organs are reversed or mirrored from their normal positions. There are several different types of ciliary motility disorders, including primary ciliary dyskinesia, Kartagener syndrome, and immotile cilia syndrome.
Treatment for ciliary motility disorders typically involves addressing the specific symptoms and underlying causes of the disorder. This may include antibiotics to treat respiratory infections, surgery to correct structural abnormalities, or assisted reproductive technologies to help with infertility.
Axonemal dyneins are motor proteins that are located in the axoneme of eukaryotic cilia and flagella. The axoneme is the internal structure of these cellular appendages, and it is composed of nine microtubule doublets arranged in a ring around two central single microtubules.
Dyneins are large protein complexes that use the energy from ATP hydrolysis to move along microtubules, generating force and motion. Axonemal dyneins are responsible for the sliding of the microtubule doublets relative to each other, which leads to the bending and movement of cilia and flagella.
There are several types of axonemal dyneins, classified based on their structure and function. The outer dynein arms are larger complexes that generate the power stroke for ciliary beating, while the inner dynein arms are smaller complexes involved in regulating the beat pattern and frequency.
Defects in axonemal dyneins can lead to a variety of genetic disorders known as ciliopathies, which affect the structure and function of cilia and flagella. These disorders can cause a range of symptoms, including respiratory problems, infertility, and developmental abnormalities.
Cilia are tiny, hair-like structures that protrude from the surface of many types of cells in the body. They are composed of a core bundle of microtubules surrounded by a protein matrix and are covered with a membrane. Cilia are involved in various cellular functions, including movement of fluid or mucus across the cell surface, detection of external stimuli, and regulation of signaling pathways.
There are two types of cilia: motile and non-motile. Motile cilia are able to move in a coordinated manner to propel fluids or particles across a surface, such as those found in the respiratory tract and reproductive organs. Non-motile cilia, also known as primary cilia, are present on most cells in the body and serve as sensory organelles that detect chemical and mechanical signals from the environment.
Defects in cilia structure or function can lead to a variety of diseases, collectively known as ciliopathies. These conditions can affect multiple organs and systems in the body, including the brain, kidneys, liver, and eyes. Examples of ciliopathies include polycystic kidney disease, Bardet-Biedl syndrome, and Meckel-Gruber syndrome.
Blepharophimosis is a medical term that refers to the abnormal drooping or narrowing of the eyelid openings (palpebral fissures) due to tightening or shortening of the muscles or tendons that control eyelid movement. This condition can affect one or both eyes and may be present at birth (congenital) or acquired later in life due to various causes such as aging, nerve damage, or certain medical conditions like thyroid eye disease.
Blepharophimosis can cause various symptoms, including difficulty in fully opening the eyes, blurred vision, and irritation of the eyes due to exposure to air, light, or foreign particles. In severe cases, it may also lead to corneal damage or visual impairment if left untreated. Treatment options for blepharophimosis depend on the underlying cause and severity of the condition and may include medications, surgery, or a combination of both.
Heterotaxy syndrome is a rare and complex congenital disorder characterized by the abnormal lateralization or arrangement of internal organs in the chest and abdomen. In this condition, the normal left-right (LR) asymmetry of the thoracic and abdominal organs is disrupted, resulting in either complete or partial reversal of the usual LR orientation. The term "heterotaxy" literally means "different arrangement."
Heterotaxy syndrome can be further classified into two main types:
1. **Ivemark's syndrome** (or left atrial isomerism): In this type, there is a mirror-image reversal of the normal LR organization of the thoracic and abdominal organs. This results in both sides of the body having structures that are typically found on the left side (left atrial isomerism). Common features include:
* Complete heart block or complex congenital heart defects, such as transposition of the great arteries, double outlet right ventricle, and total anomalous pulmonary venous return.
* Bilateral bilobed lungs with a central location of the liver (situs ambiguus).
* Bronchial malformations, including bilateral eparterial bronchi.
* Gastrointestinal tract abnormalities, such as intestinal malrotation and biliary atresia.
* Increased incidence of situs inversus totalis (complete mirror-image reversal of the normal LR arrangement).
2. **Right atrial isomerism** (or asplenia syndrome): In this type, there is a lack of normal LR organization, and both sides of the body have structures that are typically found on the right side (right atrial isomerism). Common features include:
* Complex congenital heart defects, such as single ventricle, double outlet right ventricle, pulmonary stenosis or atresia, and total anomalous pulmonary venous return.
* Absent or multiple spleens (polysplenia) with varying degrees of functional asplenia.
* Bilateral trilobed lungs with a right-sided location of the liver (situs ambiguus).
* Bronchial malformations, including bilateral hyperarterial bronchi.
* Gastrointestinal tract abnormalities, such as intestinal malrotation and biliary atresia.
* Increased incidence of congenital diaphragmatic hernia.
Both situs ambiguus and heterotaxy syndrome are associated with increased morbidity and mortality due to the complex congenital heart defects, gastrointestinal tract abnormalities, and immunological dysfunction in cases of asplenia or hyposplenia. Early diagnosis and management by a multidisciplinary team are crucial for improving outcomes in these patients.
The mesocolon is a peritoneal fold that attaches the colon to the posterior abdominal wall. It contains blood vessels, lymphatics, and nerves that supply the colon. The mesocolon allows for the mobility and flexibility of the colon within the abdominal cavity. There are several parts of the mesocolon, including the mesentery of the ascending colon (right mesocolon), the transverse mesocolon, and the mesentery of the descending and sigmoid colon (left mesocolon).
'Abnormalities, Multiple' is a broad term that refers to the presence of two or more structural or functional anomalies in an individual. These abnormalities can be present at birth (congenital) or can develop later in life (acquired). They can affect various organs and systems of the body and can vary greatly in severity and impact on a person's health and well-being.
Multiple abnormalities can occur due to genetic factors, environmental influences, or a combination of both. Chromosomal abnormalities, gene mutations, exposure to teratogens (substances that cause birth defects), and maternal infections during pregnancy are some of the common causes of multiple congenital abnormalities.
Examples of multiple congenital abnormalities include Down syndrome, Turner syndrome, and VATER/VACTERL association. Acquired multiple abnormalities can result from conditions such as trauma, infection, degenerative diseases, or cancer.
The medical evaluation and management of individuals with multiple abnormalities depend on the specific abnormalities present and their impact on the individual's health and functioning. A multidisciplinary team of healthcare professionals is often involved in the care of these individuals to address their complex needs.
Biliary atresia is a rare, progressive liver disease in infants and children, characterized by the inflammation, fibrosis, and obstruction of the bile ducts. This results in the impaired flow of bile from the liver to the intestine, leading to cholestasis (accumulation of bile in the liver), jaundice (yellowing of the skin and eyes), and eventually liver cirrhosis and failure if left untreated.
The exact cause of biliary atresia is not known, but it is believed to be a combination of genetic and environmental factors. It can occur as an isolated condition or in association with other congenital anomalies. The diagnosis of biliary atresia is typically made through imaging studies, such as ultrasound and cholangiography, and confirmed by liver biopsy.
The standard treatment for biliary atresia is a surgical procedure called the Kasai portoenterostomy, which aims to restore bile flow from the liver to the intestine. In this procedure, the damaged bile ducts are removed and replaced with a loop of intestine that is connected directly to the liver. The success of the Kasai procedure depends on several factors, including the age at diagnosis and surgery, the extent of liver damage, and the skill and experience of the surgeon.
Despite successful Kasai surgery, many children with biliary atresia will eventually develop cirrhosis and require liver transplantation. The prognosis for children with biliary atresia has improved significantly over the past few decades due to earlier diagnosis, advances in surgical techniques, and better postoperative care. However, it remains a challenging condition that requires close monitoring and multidisciplinary management by pediatric hepatologists, surgeons, and other healthcare professionals.
A heart septal defect is a type of congenital heart defect, which means it is present at birth. It involves an abnormal opening in the septum, the wall that separates the two sides of the heart. This opening allows oxygen-rich blood to leak into the oxygen-poor blood chambers in the heart.
There are several types of heart septal defects, including:
1. Atrial Septal Defect (ASD): A hole in the atrial septum, the wall between the two upper chambers of the heart (the right and left atria).
2. Ventricular Septal Defect (VSD): A hole in the ventricular septum, the wall between the two lower chambers of the heart (the right and left ventricles).
3. Atrioventricular Septal Defect (AVSD): A combination of an ASD and a VSD, often accompanied by malformation of the mitral and/or tricuspid valves.
The severity of a heart septal defect depends on the size of the opening and its location in the septum. Small defects may cause no symptoms and may close on their own over time. Larger defects can lead to complications, such as heart failure, pulmonary hypertension, or infective endocarditis, and may require medical or surgical intervention.
A "torsion abnormality" is not a standard medical term, but I believe you are asking about torsional deformities or abnormalities related to torsion. Torsion refers to a twisting force or movement that can cause structures to rotate around their long axis. In the context of medical definitions:
Torsional abnormality could refer to a congenital or acquired condition where anatomical structures, such as blood vessels, muscles, tendons, or bones, are twisted or rotated in an abnormal way. This can lead to various complications depending on the structure involved and the degree of torsion.
For instance, in congenital torsional deformities of long bones (like tibia or femur), the rotation of the bone axis can cause issues with gait, posture, and joint function. In some cases, this may require surgical intervention to correct the abnormality.
In the context of vascular torsion abnormalities, such as mesenteric torsion, it could lead to bowel ischemia due to the twisting of blood vessels that supply the intestines. This can be a surgical emergency and requires immediate intervention to restore blood flow and prevent further damage.
It's essential to consult with a medical professional for a precise diagnosis and treatment options if you or someone else experiences symptoms related to torsional abnormalities.
Cystic kidney diseases are a group of genetic disorders that cause fluid-filled sacs called cysts to form in the kidneys. These cysts can vary in size and can grow over time, which can lead to damage in the kidneys and affect their function. There are two main types of cystic kidney diseases: autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD).
ADPKD is the most common type and is characterized by the presence of numerous cysts in both kidneys. It is usually diagnosed in adulthood, but it can also occur in children. The cysts can cause high blood pressure, kidney stones, urinary tract infections, and eventually kidney failure.
ARPKD is a rare, inherited disorder that affects both the kidneys and liver. It is characterized by the presence of numerous cysts in the kidneys and abnormalities in the bile ducts of the liver. ARPKD is usually diagnosed in infancy or early childhood and can cause serious complications such as respiratory distress, kidney failure, and liver fibrosis.
Other types of cystic kidney diseases include nephronophthisis, medullary cystic kidney disease, and glomerulocystic kidney disease. These conditions are also inherited and can cause kidney damage and kidney failure.
Treatment for cystic kidney diseases typically involves managing symptoms such as high blood pressure, pain, and infections. In some cases, surgery may be necessary to remove large cysts or to treat complications such as kidney stones. For individuals with advanced kidney disease, dialysis or a kidney transplant may be necessary.
Laparoscopic cholecystectomy is a surgical procedure to remove the gallbladder using a laparoscope, a thin tube with a camera, which allows the surgeon to view the internal structures on a video monitor. The surgery is performed through several small incisions in the abdomen, rather than a single large incision used in open cholecystectomy. This approach results in less postoperative pain, fewer complications, and shorter recovery time compared to open cholecystectomy.
The procedure is typically indicated for symptomatic gallstones or chronic inflammation of the gallbladder (cholecystitis), which can cause severe abdominal pain, nausea, vomiting, and fever. Laparoscopic cholecystectomy has become the standard of care for gallbladder removal due to its minimally invasive nature and excellent outcomes.
Blepharoptosis is a medical term that refers to the drooping or falling of the upper eyelid. It is usually caused by weakness or paralysis of the muscle that raises the eyelid, known as the levator palpebrae superioris. This condition can be present at birth or acquired later in life due to various factors such as aging, nerve damage, eye surgery complications, or certain medical conditions like myasthenia gravis or brain tumors. Blepharoptosis may obstruct vision and cause difficulty with daily activities, and treatment options include eyedrops, eye patches, or surgical correction.
Dyneins are a type of motor protein that play an essential role in the movement of cellular components and structures within eukaryotic cells. They are responsible for generating force and motion along microtubules, which are critical components of the cell's cytoskeleton. Dyneins are involved in various cellular processes, including intracellular transport, organelle positioning, and cell division.
There are several types of dyneins, but the two main categories are cytoplasmic dyneins and axonemal dyneins. Cytoplasmic dyneins are responsible for moving various cargoes, such as vesicles, organelles, and mRNA complexes, toward the minus-end of microtubules, which is usually located near the cell center. Axonemal dyneins, on the other hand, are found in cilia and flagella and are responsible for their movement by sliding adjacent microtubules past each other.
Dyneins consist of multiple subunits, including heavy chains, intermediate chains, light-intermediate chains, and light chains. The heavy chains contain the motor domain that binds to microtubules and hydrolyzes ATP to generate force. Dysfunction in dynein proteins has been linked to various human diseases, such as neurodevelopmental disorders, ciliopathies, and cancer.
 Situs inversus
Situs inversus Situs Inversus Imaging: Practice Essentials, Radiography, Computed Tomography
Situs Inversus Imaging: Practice Essentials, Radiography, Computed Tomography 001045 - situs inversus viscerum Strain Details
001045 - situs inversus viscerum Strain Details Roux-en-Y Gastric Bypass Surgery in Situs Inversus Patient - SAGES Abstract Archives
Roux-en-Y Gastric Bypass Surgery in Situs Inversus Patient - SAGES Abstract Archives Inverse problems and Bayesian tracking: An application to neurophysiological data | Department of Computer Science [pre 2018...
Inverse problems and Bayesian tracking: An application to neurophysiological data | Department of Computer Science [pre 2018... Agnathia-holoprosencephaly-situs inversus syndrome - About the Disease - Genetic and Rare Diseases Information Center
Agnathia-holoprosencephaly-situs inversus syndrome - About the Disease - Genetic and Rare Diseases Information Center Nephronophthisis and Situs Inversus via the ANKS6 Gene Test - PreventionGenetics
Nephronophthisis and Situs Inversus via the ANKS6 Gene Test - PreventionGenetics Situs Inversus | Profiles RNS
Situs Inversus | Profiles RNS Research Articles about Situs Inversus - ijSciences Publications
Research Articles about Situs Inversus - ijSciences Publications situs inversus 6k 01 framegrabs 08 - Ian Gibbins
situs inversus 6k 01 framegrabs 08 - Ian Gibbins situs inversus with levocardia - Ontology Report - Rat Genome Database
situs inversus with levocardia - Ontology Report - Rat Genome Database Leukemia and Situs inversus totalis, related diseases and genetic alterations | MENDELIAN.CO
Leukemia and Situs inversus totalis, related diseases and genetic alterations | MENDELIAN.CO 20 Extremely Rare Diseases
20 Extremely Rare Diseases Fluorescence cholangiography during laparoscopic cholecystectomy in a patient with situs inversus totalis: a case report and...
Fluorescence cholangiography during laparoscopic cholecystectomy in a patient with situs inversus totalis: a case report and... Transjugular intrahepatic portosystemic shunt for pyrrolidine-alkaloid-induced hepatic sinusoidal obstruction syndrome in a...
Transjugular intrahepatic portosystemic shunt for pyrrolidine-alkaloid-induced hepatic sinusoidal obstruction syndrome in a... Another 10 Bizarre Medical Tales - Listverse
Another 10 Bizarre Medical Tales - Listverse Recent Emails
Recent Emails Right Hand, Left Hand - Chris McManus | Harvard University Press
Right Hand, Left Hand - Chris McManus | Harvard University Press Primary ciliary dyskinesia: MedlinePlus Genetics
Primary ciliary dyskinesia: MedlinePlus Genetics Infections
Infections William O'Malley, M.D. | UR Medicine
William O'Malley, M.D. | UR Medicine





















![El general Failure, leo tu disco duro: [S29] Tres trailers de videojuegos que vería una y otra vez](data:image/jpg;base64,/9j/4AAQSkZJRgABAQAAAQABAAD/2wBDAAgGBgcGBQgHBwcJCQgKDBQNDAsLDBkSEw8UHRofHh0aHBwgJC4nICIsIxwcKDcpLDAxNDQ0Hyc5PTgyPC4zNDL/2wBDAQkJCQwLDBgNDRgyIRwhMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjIyMjL/wAARCAC7APoDASIAAhEBAxEB/8QAHwAAAQUBAQEBAQEAAAAAAAAAAAECAwQFBgcICQoL/8QAtRAAAgEDAwIEAwUFBAQAAAF9AQIDAAQRBRIhMUEGE1FhByJxFDKBkaEII0KxwRVS0fAkM2JyggkKFhcYGRolJicoKSo0NTY3ODk6Q0RFRkdISUpTVFVWV1hZWmNkZWZnaGlqc3R1dnd4eXqDhIWGh4iJipKTlJWWl5iZmqKjpKWmp6ipqrKztLW2t7i5usLDxMXGx8jJytLT1NXW19jZ2uHi4+Tl5ufo6erx8vP09fb3+Pn6/8QAHwEAAwEBAQEBAQEBAQAAAAAAAAECAwQFBgcICQoL/8QAtREAAgECBAQDBAcFBAQAAQJ3AAECAxEEBSExBhJBUQdhcRMiMoEIFEKRobHBCSMzUvAVYnLRChYkNOEl8RcYGRomJygpKjU2Nzg5OkNERUZHSElKU1RVVldYWVpjZGVmZ2hpanN0dXZ3eHl6goOEhYaHiImKkpOUlZaXmJmaoqOkpaanqKmqsrO0tba3uLm6wsPExcbHyMnK0tPU1dbX2Nna4uPk5ebn6Onq8vP09fb3+Pn6/9oADAMBAAIRAxEAPwD5/ooooAKKKKACiiigAooooAKKKKACiiigAooooAKKKKACiiigAooooAKKKKACiiigAooooAKKKKACiiigAooooAujR9SIyNPuiP8Ari3+FL/Y+pf9A+6/78t/hXrOlald3vhZxbzsl3YnDgYO5Px9v5VRGu6l3uWP4CtakacLXb1OrLcuxeYRlKlyrldmm3f8jzT+x9T/AOgfdf8Aflv8KP7H1P8A6B91/wB+W/wr04a5qGObk/lQ3iC+Qf8AHyajno92em+GcxSveH3v/I8x/sfUv+gfdf8Aflv8KP7H1L/oH3X/AH5b/CvRJPE+oD7tyfyFVz4o1Yn5bpvyFJzpLqznlkWOi7Xj97/yOD/sfU/+gddf9+W/wo/sbU/+gdd/9+W/wr3nw5PPd6Haz3Dl5X3bmI64dh/ICtpUrsp4WM4qSe58vicdUw9aVGcdYu2582f2Nqf/AEDrv/vy3+FH9jap/wBA67/78t/hX0p5dL5dX9SXcw/taX8n4nzV/Y2p/wDQOu/+/Lf4Uf2Nqn/QOu/+/Lf4V9K+X7Uvl+1H1Jdw/taX8v4nzT/Y2qf9A27/AO/Lf4Uv9jap/wBA67/78t/hX0r5dKI6Pqce4/7Vl/J+J80/2Nqn/QOu/wDvy3+FJ/Y2qf8AQOu/+/Lf4V9L+X7UeXml9TX8wf2rL+T8T5o/sXVP+gdd/wDflv8ACj+xtU/6Bt3/AN+W/wAK+mPLpfLo+pr+YP7Ul/J+J8z/ANjap/0Drv8A78t/hSf2Lqn/AEDrv/vy3+FfTPl0vlij6mu4/wC1Jfy/ifM39i6p/wBA27/78t/hR/Yuqf8AQNu/+/Lf4V9M+WKXy/aj6mu4f2pL+X8T5k/sXVP+gbd/9+W/wpf7F1T/AKBt3/35b/Cvpry6Xy6Pqa7h/akv5fxPmP8AsXVP+gbd/wDfhv8ACj+xdU/6Bt3/AN+G/wAK+nPLpfLo+pruH9qS/l/E+Y/7F1T/AKBt3/34b/Cj+xdU/wCgbd/9+G/wr6c8ul8uj6mu4/7Tf8v4nzF/Y2qf9A27/wC/Df4Uf2Lqn/QNvP8Avw3+FfTvl0vl0vqi7h/acv5fxPmH+xdU/wCgbd/9+G/wqE2F2Dg2kwI/6ZmvpTW7sadpks2fnI2oPUmuBFg7je2Szck+9YVacKb1Z2YWvUxCbUdjK0S+/srWkZjiCf8AdyDtz0r1my0jw0+l/ap7C2URjEhI4FeN3MPmRH1HSux8Pa3JPpflmRg/Eb4GTkHrj3/rXmZrg5YzC8tOTUou+nbr/mdntp4LEqpFtRno/Xo/0Nqafw+dRe0i0KL5DtLAg49zz0qFtN0i5VXhsIGRxlSo6ismLR9Rj1hTBDLJDJLuZihJTk/Nn6E16No+ir9njkkySRn5l2n8j0r5HGyhhacZwqNv1Z7U69Zv4nb1OXtfBdjdEE2EYH0Nb1r4D0KIZk06E/XNdYkMUCcADAqpcXHp0rwZ5jiajtGTS9RKtNfaZzV7a2Om3EdpaJHDEqZCA8DJOaEZSOCD9DWT4luf+JuvPWIfzNZguvev2jJqcpYCi29eVfkfnOZ4r/a6l+7Os4pa5UXrL0Zh9DThqMw6Sv8A99GvS9gziWLj2Oo4peK5karOP+WrUo1e4H/LT9BS9jIf1uB6Jd2drL4Zt7y3hRJFIEjKOT2OfxxV2DRrRtCCmJDePAZA38XqP6CsjwRef2zpWoaZcPyCGBA5wf8A64/Wnt4i2fENLMMv2dQLb8cf44FeZKFTmdNfZ1PfhVouEaz+3ZfMl020tl0K8vriFHYHbHuHQ4/xP6VXuWk/sK3BsEjj3cXGRluv4/8A6qf40vV0PSbTT7cjMsrSHP8Adzn+ZH5VX1a+ki+HWmXA27nlGePZ6qMXLln3ZFScYc1JfZjqTasztDaB7BLVQnylSPn6elWLSztn8MXVy0KGZWIVyOR0qj44vpLS20koR80Rzkey1N4ZuX1nwff2kEifbQ5wp47Aj+RFOz9jGfS5KnH61Onu7foS+G7S3vL+VJ4lkUREgN65FZUwVZ5FHADEAfjWv4Vs9S0yS8vdYQW8CREDcR65J47cVwM+tTyzyOshCs5I4HTNbUoOdWXK7rQ5sTUVKhDnVm7+p0mRS8Vyp1W4P/LZvwNMOoynrK5/4Ea6vYM8/wCuQ7HXZFNMka/eYD6muRN4zdWJ+ppv2qn9XfcX1xdjrTdW46yp+BqM6hbD/lpn6A1y32qj7VT9gT9cZ0x1SAdFY1G2qj+GMfUmud+1Uv2qn7BEvFyN1tTkPTaPwqNr6Vv+Wh/Disb7V71FcX4hgeQnoKbppK4lXlJ2RDqty1/qUdqGLJH8zc55rSW3UKB7Vl6LAX3XEn3nOc1t5PpXy+Jr+0qtrY/SMuwPsMPGL36nne3NMsZjY6iVJIjmG04OMHsa0RaGoLrTmkT5eo6GtoVFGVysThvbUnAcb6+t3KC+uRg4/wBa3+NOGt6kOmo3f/f5v8abDdaxDGEikZV9FJFSi/13tPJ/30a55YPDOTaa+49ahxDiKdKMJ4ZNpau6189iJ9b1LvqV3/3+b/Gq0ut6hj/j/uv+/wA3+NXTqGuj/ltJ/wB9GmHU9b7zSf8AfRqlhMMuq+4U+IK8tsL+K/yLlpdyT6faSSyNI5Rss7ZJ+dqsif3rFkvdUlYNId5AxlsmmfadQH8C/lXs0cXSpQUL7HwGOyrFYrETrKFuZ3tc3fP96PPPrWH9p1H+6n5Vi3Hiq/tp3hktkDKcHn/61a/X6Xc4pZFio7x/E7bzz60ef71x9t4mvLhCwjQEHpVy31e+nm8sKgan9fpdyVk2Jbsl+J1lpqt3YSNJaXMsDsMFo2IJFRm/nNz9oMr+du3+Zn5t2c5z61hifUv7qf8AfNSQyalLJtGwDudtT9eoXv8AoavJMala2i8zdu9WvL91e7uZZ2UYBkYnAok1a8ls0tJLmVreM5WIsdoP0/E1i30tzYR7pZlLAZICjissa9OWHof9nJ/KhY2j0/Ih5Ti7t9/M6661a8vRGLq5lmEYwgds7R7U211O6sZhNa3EkMg/iRiDWboxl1n5Y7lUk7K0fX8adqNpqunXJikKY6htvWl9eoW5f0GsnxkpcyWvqbV94k1XUovKvL+aWP8AuFsA/gKzvPPrWYG1Ij+D/vmqOp6pf6bGjMqNvJGMYojjqC0j+RVTJcbL3pq/zOh88+tHnn1riz4rvQP9UlRf8JneZx5Cfn/9ar+vUu5ismxD6L7zufPPrR5/vWVavqs9tHK6IhcZ246VOE1I90/75qf7Ro9zX+wMY/s/iXvPPrR559apeTqXqv8A3zR9n1H1X/vmj+0qPcf+r+M/l/Eu+f70eefWqf2XUT3X/vmj7HqPqv5Uv7So9w/1fxn8v4lzzz61Vmka6uY7deRnc1N+xah/eX/vmtLSNMkimMsxy56nFc2LzGEqTjB6s78tyGtDERnWWi1Nm1iEMCqPSpqBRXgH26VjL+xY7Uw2fPSrVrqCzHDYH1rTgWGU5yMVs20ZGRFYg9VqRrdI/wCHNb6RwKMsyj61XeGGaQqhDY9KnmYjH8iN1yUAqCS0Q8bRXRLYHZjHepk0tJh05o5wukciLDJ4FDadx0rso9FP92rA0RWX7pzR7QOeJwX2L/ZrlPF+ilVS+jT/AGX4/I17M2gBecGsXxB4ckvNLmt4wA7DK59RTVTUibjOLR4VZb4rlcj5WODT7y7mtb7dDKyN1yDVqeL7JcbXGCrdKzb9S1wxzn0+ldd00eY9GaEPiPU/MEYuNw91HSui0y5axs1k3Eyy/Oc9uw/qfxrjbKIgtIRyeBXS3B/dbVP3VAFFkDqSe7J7y8W7aNXcAn5yCfy/SlW2j25+XmsUwXU8jmNYmUDOCMHpnH17Vt2WiXV3pk21/wB5GMhd36VElY6aWuiRb0y8/s673qwPovoa2/F7/wBreF49QDlZrWXbwfvAnBH8j+FcZplvcpqEf2iCBI4zvcMcMQOcD3OK6XVUZ/A1uEzl7wL9DjP9DUv3WmS9djjpPEGp2sUcUVxtjAwDtBP5kVlXmp3l+yi4uHkx03HpWprtqi3txDEOFY7R7jj+h/OufzzWiS3RlKcvhbLk2dnA61f8MaUdS1iIOuYYvnf3x0H51ntKm0Jn5u9d54K05rezku3IHnYCj2GaKjsi8NDmmdQoAAGKeoGelNp61ynsWHYHoKcAPQUgp6ikwHADHSlwPSlHSlCk9qkEgwOKeKAh708KAOTSAKMCnDA9aXj0oA5NdyrmrMF1JkAMQKqKxPFXLS1kmkCoMk10syRo28T3TgbjW9aaW0C+YTkmnaNos0bBpU4611J07ICuuPTFc05djOdVIx4kwAKuK9ragG4miiB7yMBn86kn0uRFZ0OAoySa8a8R63calrLYkykZ+X6Dp/j+NEI8zMpzVrnu8Nssih0IZWGQQcg1bjtOMba8i8JeOrjTFENwC8IbBRjnH0r1zRdcsNbthLaSDdjlCeRVODickpsmNiCv3RVabTlIxtrYximHaTg0mkSqjPmHxbo/k67fQquNkpxx71zSaTPcyCJIyz9AMda+ptY8N6LfXK3N5aI8397JGfrjrUcfhzRPPW5i063WRejKuP0q1VsU7PVny39kdJ4o1HG4Zq/KfvHtux+RrrPE2gf2V4murcL+6Ql0P+yen6H9K5eaPch2js5rpjLmVzOUbMbDKFUAY4qayvHjvW2XgCP8pjz1/wAnFY6ybzgng1atoXLffTYD9wipaOmE2dGjo8oUgcnHSt+xvLK28HTSXwBCuTCAgY7txAIB4yMZrjLeYxXUSjOFO7HXpzUmsXkkmlBI2/dWsQU7T1Y8HPuOf1qHG6saVZ2aZm+IphceIrp4QPJ3gAgYzgAZ/TNYVxbMknmBMK/K/wCfrWsIpmdUkQiV1VhnqcgEfnmvSIPC1omm2sVzbpI8SdSO55P61o5KCSMqdJ1ZNnjSRENlu5r1vRIfJ0e1QjB8sHFJN4X0r7QJvsihhjABIXj26VoBdowOlZylzI7aFD2bbFAp4xTQM0T3FvZQma4lVEHr1P0rM6G0tyUYogmjnkeOJ1kdPvqpyV+tcTq3iuS5zHZhooe7fxN/hVXwxrkmi+IYb8nERHlzKRkFDwSR7dfwrT2Ttc5J4uKlZHow607J9aQFnZiwUOCQwXpn29qeErB6HWpJq4gzmrMMe9TkVEEqzEQtJg2RiE5p/kt6Vr2SROMOAD2q39ltv+eorNyM3Us7HmEMOWHFdLpH2a3Id8E1h3s0VlavO7BY0GSRXI3HjKd38u0jKA8Ak5Y12NORnOcY7nuyazC0QVAAa0LHV5GkCnYR74rwK0t/Emp/NDY3UvvtY1eOjeKbZBI+mTAHvtNZOml1OeTi9LHsHxA8T22neHpII2AuLkbeDyF7/n0/E14UuDvkJ/fPyc9Klvhq2wtd6fNtUcllOAKyoLuzaXbM727HjcRlfxx/hW1NJHLOLWiNOOQjheorr9L0nxJo+nrq0UTCE/vCqt8wHriub0uykN5byMEmti4JkibcuM+1dLf/ABAuYPEpSIqbKGXaqjjKjj8qwrzqcyUEcspTi7I7nw98Q474x2t6VWVuA/rXWxXkcsuVfIHvXg2qlNJ8RsbYgQMVniA7KwDAfrivQY9SJVXjf5GUMOaTXMlJHdRgqkbnYatNEWB8z/61JaXUaxhQciuQN/JMwUsWzXQWVuBbgk5YjgZxWbRtKkoxszkviOlvMwuYyA6KI2P948nH4ZryoEDA9yK7zx/dGK4S0DAlMs2P7x6/59q86nk8twd3X+HNdlH4TlqpLYyLq2dJ28vOM/L9KbCt0Gyucn3rWguG1FyPIACcblxgD0PrV2KwdWDlPlJ61bKjbTUi0W3eO7S4mO7bnGfXFdhaxxpaWkroDGzr29v/ANdchf3l3YzpAsYVZDhctw4rbOrGe2igjI2wE5C+v/6j+tQ07FykuY7S28Bx3+r2msTToAiLuiVfvsucHP0x+VdRLpcLcZrJ0LV5ptEi2c4G0+2KfLqE6ksWP0rmlds6aadroS70ZRyMVjXFkYieOKvvrEpPPNU7i9afgrgU1dHRG5zWtawmkhY1QPO4yAegHqawLbSta8UNJLCjSherE4A9hUXiKb7Z4gkQdFIiH9f1zWpqmvT6Jcwadp8piSzAQ7f4n6sT+P8AKrnKUUlHdnkY3ETcuSJy13YzWUjQToVkQ4INVx83BA4rrPGk0dzcW17tCtcW6SMB6kc1xrOM/eC+9b0JuUE2ctOTkrno/gnUBqNk9tNcKLu1UKUc8yIOFI9wMA+wFdQFFeJWFxDYX6XYmV5EO4BuRn8K6hPiDdB+XhI9PKP+NZ1Kbbuj1qFdRjaR6bbwCUkE4oeLynI9K4nT/iHGrjzzBtPXCsv+NdXaa/pWqhfIvoTI3/LPeM/lWMoSR0xqxlsy8krZFTfaB6/rVVhjpUJJzUWLsc74z8Paja+HvOeF1i3jcSK8qJMEwYcYPWveviP4xXUdIi0qAqGlO+UAfdUdMn9a8J1FkZjs+6DgH1rshqjy6spOV2djbfETV7S2WKG62gjBIRc/nimr4/1qSdGkv5SAQcbuD+FcErErkngVYR1RA27J9DR7NEqqz6I03xd4Y1HSEfULhIZnXEkRjJwfwHSvFPFFvp9vrNwthOJrQsTG+COD9ay2nEaIu87m9D0qU6Te3fmCJonKAEjfjr9amFPldxyk5qxVs9UutIn8/T7x4nzyB3HuOhH1rtNM8UaF4hYQeJLZbS4bhb62GBn/AGl//WPpXnwsrqSR0WIlkOCB6/XpUbLJBIUkUq46g1pKKZhqj0/x4V026sFhdbiNrSNY50OVlAGNwP6VhWXi3VYXjia62KibQrKCOBxWFpXnai32Hz3WMBpEXPCvjr7Zxg02PTruWQhA/BwWY4H61EYKMeUuk5R+E9l8I+ONOuYXTVUhjmUja4+UMKua18QbZGltdMARx/y23A9PQV5D/ZYtbbzbq98tc4yORVBpBHI5E5aPcEDqD39vpmp9km7nTKbS1O6vrptQKz3LF5WUFiTjnrWBdPBKxjtrUTzDODkkLUB1Eyx5n3mBBjaXxu+p6k1Ja6o1wvlR5tc/dEMW84+nWtFFohzjLREUD3OkMvnRIschyVyOfr7VoS6lpMcYljLs+OIW6A+vp/Sr8Phm2fbcXEstyXGQZRgfl1/Oq02k2kd5nyl+mOPy6U/aImWEctTNittS1phcW6RlIydilh8vfHrXR2s1rCEg1CxW0lOQWIIRz6gjjmqcmmWEAM8Lz27KM/6Ocn8B/hWXJ4jv7dzHvee36NHdICT9eKT97Y0UVSXvHeyNPHprWtqxjib5gUYjn61PoGqT3dhLFOu6aB9h3nnPpn8DXLaRqM72ZvdPiV7cH99aqfu+4ByR+HHtUGja/J9t1CRwwhmlbyyRg5U8A/gQKycWbKrG6sdNruuWem2spWWM3gA2wZyQT64rgLnxjqrOrCcJtOdqqMGut1WwsfEsCsZPJu0XCSeo9CO4rzrVtLvNIuRFeQlRn5WHKuPY1dNKxNedRbbF3Tb/AM3WIp7nkNMHcn65NWPEN1byaxdTxybo3lZlPqCTiufkkaJUdTywIHtVZ5WYkscn3puHvXPNlG8uY0Zb+e6+UEkAYyaq+WXuFjLgsxxkngVWG8gYBx69qsQQyyf6vgdyatRsVFWZ1uow6FZaXDBaCCedEzPPnO5vQVjW+qWaTKZNMgdAemMZFZ5hdThpME9COQang09riQwRp5kzDKMpwP14xRy2WprKXM9DrJdf8LT2Lj/hHUiuNpCskpwG/DFcY022XMZwM8VDcJNbTPBMhSVDhlPY0yMguA1JRsW6j6nunhO8bU/DdtNKSZAu0k98cZ/StUxjNcf4K1EparZJEwg8sSI/UKSTlc/UE/Q11hmGa5Jq0menSfNFM8dv9dkmN0z4Ms8mGYdl64H44/KsOWaN4m3Bt/G3B4/Gunj8HXH9oMJHU2hbOc/NisPXdHl0y+8leYn+aM57V1prY86pRmlzMzS/yHB9KaXxt5OaiYyIcEml+YsN2MVoc2xYST9/GD7Z5pJ7tpHJ3Nz7ioTvL7lOMdPakK/3jlsUguXra7iaCRLgSNhcR7WCgHI5PHI61FcRK0pWCQuo5G5cE1AoZkxHwvdSe9XbJfKyXfnsvXH40DWpL4fuEh1WLe23dlefcUl0Dc3MkzTMu49B0ouFs8bwv7wnPHFU2kJ47UhpPYdJI8alRJvT0NWrUF9MMmOPtGCfT5cj+tQWthdahMIreFpHPp2+tb2iWKmHUtLu1IIKscdVYZGamTSRapyZiyTmSTYWyi/zq3aXjWsyzQttdeQRTZtDnhYiO5gkOTxuKn9Rj9ary6dfW8wjmjMRIyCTkH6Y601JMhRkmdxY+MomVY7sNhiFJ7Cm6lqVmzCSGeM84xvBNctDoksigvcY9gtT/wBjY63Dk+oFTyxudanVa1RPNri4YR5JHAJ6VlSXPmMWY7ieTViTQXCFo52OOxWqiaTeSylI9rY5YngAe9UmkYVFUe4QalPp9wJbSRo265BrftZWbSXvQNqzXapjPBO0k/yFZEWiwk/6Re4I/hijJ/U4/lW/rMHlaXoWn2iAb3eQID1zgAn9aznJNpDpQlqyWC8KEEHBrRu9TS60a6gnhSf90xRXGfmA4rMu9IvLJFkdMqRyV5Aqok7Ie9FkzpblFWZzN5gQxAdRnj8qYsLQiNpYypflQw6jsR/jW0uhQ3N35jzOQXzsx79M1Y8VW8832WWCIMsEezKdQM8DHoP61pzanJ7F8rZzz/MuS6j/AGe9Sw3QVdqlR9WP+FUWZv8AVspBHrmrEWm3jwPMsRKou7BOCR7DvVNmav0Ottkt7zQJ4TGm4wmRXA5Drz1/AirNmtsmnSCOJFeMKyuByeR1PfrXPaTeNDpzMfuoHyM9sdP1NWYNQW308NJnb5Kg49eMVm0zqhJaM2tR8nU7ExMiF5YxhyOQ68Dn6ACuEEY2lhIowfumtKTWjFLGIPnRV5zx8xP/AOqqen2kmoXsVqnBlbGcdKaVkRN87VjrfBPiIafc/Y7gsbe4IUY52SdAfoeh/D0r0vOefWvJNL8N6rFrsET2siLFKGZyp2nB7Hv0r1oIAoBPOKwq2bud+EjJRakYxXmsbxFox1O1DxHE8WWTPQ+1bxUUbRTTNZJSVmeQ3EAbKyIVdeCD1BqjLDIjEgZHqO1evXej2N7kzQKWP8QGD+dZv/CNWlja3sse5i0LKA3O0YrVVDilhHfQ8yh3uxQYBxnmpDbu0iqWXLHAycD86WNlhug+OATVgupUsVDL1q7nE1Zlaa2mtbh4JojHKjbXHXBHbNaVhoGqX2GigZUP8cnAq3pGtDTgwe1WcSkMQ55BHofxrVm1bVdZPlWcDQxHjbGOT9TUuTOinTg1dsLTwHuIa8us+0Y/qa6Cz8NaZZ4KWysw/if5jWnY2rW9hBC4+ZIwp+uKuJbSSZ2qTWLmz0IUoRWiKcdvFF9xFX6DFcDqM5tfF84DFVlIQ/iK9Jkt3iBLgjFeXahIL69nnRCpLl1DdRThqRiHaKNyPS4yu8uzM3O4npUmtzfZ9EtGkRZJBL14BI5/pirOmTebZrnBckKQPWq+vRre3KwA4WIcH3pfaSMZpKPulS3mjnQNE2R6dx9amMdYMltc2EwkGVPZh0Naun6gl1iOQhZuw7N9K0aIp1b6S0ZYClGrRt4I5rO4QKN7Dt16U0QCQccH0qWzRre5Un7pODms57G9jPj0CKUq53YJ6d6zTO0vjOBVyY7ZhGM9Bjr+tdZdE6f58knCxgkZ/SuHieS1kabh5d28+/NFO8tSqvLGyR6n94YIyCOhrF1Dw3b3RL2/7hz2H3T+HatywIu9Nt7kjY0kYYr6EipTGV7is+az0N2lJanCSaNe6e+9o96D+JOaz5rhgTXpJGe1Zl1oNjdOXaPax67TitFU7mMqPY82eJUvvtgTcSPmGM4P94VFeXTpatIrHJ4ywKgfTPJNdD4kjsdEjMUM5e7bBVMZCj1NcLcTy3EheWRnb1NbRd9ThqpQdiIOQCNxIPUdBUm6aSIIWYxg8DPApqqOtejeGdAsta8M+XNlR5u8MnUHGDTlLl1Io03UdkeepGB2r0DwT4acSLqd0CuB+6Q/zrpNP8JaVYYZbcSOP4n5NbqIqKFUAAdhWE6t1ZHdRw3K+aQoFJin0lYHWeTf8LIuP+gdF/38P+FH/CyJ/wDoHR/9/D/hXD0V28kTxfrNXudx/wALIn/6B0f/AH8P+FD/ABHneN4zp0eGUqf3h/wrh6KOSIfWavcsi6Afd5YI3ZwTUbShv4cVFRVGLbZq6XrTaZMHWFJFB3YJwQfY11EHxLkg5/smAt3IkIz+lcFRScU9y4VZw+FnpDfFlyB/xI4M+vnn/CiD4tzwSbk0eDB6qZTj+Veb0VHso9ivrFTuem3HxgmuVw+iQD1xMf8ACuMn14S3Tyx2ccSsxIRW4XPasWiqjCMdhOtNqzZ0mm+LZdOdmFqkmWLAFyMZpW8XytK8htE3Mcn5zXNUUcqvcXtZ9zpT4udlKvZRsp6gsazpdXDSFkgVQe24nFZdFOyFKcpbnTWnjO6tk2vAk2OhZjn8+9W28fSsuDp8f18w/wCFcdRS5UUq00rJnY6r4+m1Sz8hrCOMlApdXJzgg+ntWMuvOrhvIXjHeseihRUVZA603uzvYviddRxLH/Z0R2jGfMPP6Uv/AAtC5/6BsX/f0/4VwNFL2cexX1mr3O//AOFoXH/QMi/7+n/Cj/haFx/0DIv+/p/wrgKKPZx7B9Zq9zQvtVkv7qa4lX55XLHnOM1T832qOirMnJvVlmO62K42Ahl28/XP9K6fRPHkuiaetpHYRyAEnc0hH9K4+ik0nuOFSUHeLPQv+Fq3P/QLh/7+n/Cl/wCFq3P/AEC4f+/p/wAK88oqfZx7Gn1mr3PQ/wDha1z/ANAuH/v6f8KP+Fq3P/QKh/7+n/CvPKKPZw7B9Zq9woooqzAKKKKACiiigAooooAKKKKACiiigAooooAKKKKACiiigAooooAKKKKACiiigAooooAKKKKACiiigAooooA//9k=)





