Spinocerebellar Ataxias
Spinocerebellar Degenerations
Machado-Joseph Disease
Cerebellar Ataxia
Spinocerebellar Tracts
Ataxia
Trinucleotide Repeat Expansion
Trinucleotide Repeats
Nerve Tissue Proteins
Friedreich Ataxia
Cerebellum
DNA Repeat Expansion
Gait Ataxia
Intranuclear Inclusion Bodies
Age of Onset
Nuclear Proteins
Pedigree
Heredodegenerative Disorders, Nervous System
Shaw Potassium Channels
Peptides
Inclusion Bodies
Anticipation, Genetic
Regression (Psychology)
Genes, Dominant
Mutation
Nerve Degeneration
Neurodegenerative Diseases
Ataxia Telangiectasia
Myoclonic Cerebellar Dyssynergia
Chromosomes, Human, Pair 6
Chromosomes, Human, Pair 20
Repressor Proteins
Ataxia Telangiectasia Mutated Proteins
Multiple System Atrophy
Atrophy
DNA Probes, HLA
Founder Effect
Magnetic Resonance Imaging
Chromosomes, Human, Pair 19
Family Health
Genetic Linkage
Myoclonic Epilepsies, Progressive
Phenotype
Mice, Transgenic
Calcium Channels, P-Type
Brain
Cerebellar Diseases
Rotarod Performance Test
Alleles
Mutation, Missense
Disease Models, Animal
Ocular Motility Disorders
Repetitive Sequences, Nucleic Acid
Neurons
Huntington Disease
Brain Stem
Licensure
Haplotypes
Molecular Sequence Data
Inositol 1,4,5-Trisphosphate Receptors
Calcium Channels
Animals, Genetically Modified
Spinal Cord
Genetic Testing
Chromosome Mapping
DNA Breaks, Single-Stranded
TATA-Box Binding Protein
Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. (1/377)
Spinocerebellar ataxia 6 (SCA6) is caused by expansion of a polyglutamine stretch, encoded by a CAG trinucleotide repeat, in the human P/Q-type Ca(2+) channel alpha(1A) subunit. Although SCA6 shares common features with other neurodegenerative glutamine repeat disorders, the polyglutamine repeats in SCA6 are exceptionally small, ranging from 21 to 33. Because this size is too small to form insoluble aggregates that have been blamed for the cause of neurodegeneration, SCA6 is the disorder suitable for exploring the pathogenic mechanisms other than aggregate formation, whose universal role has been questioned. To characterize the pathogenic process of SCA6, we studied the effects of polyglutamine expansion on channel properties by analyzing currents flowing through the P/Q-type Ca(2+) channels with an expanded stretch of 24, 30, or 40 polyglutamines, recombinantly expressed in baby hamster kidney cells. Whereas the Ca(2+) channels with +info)Nuclear localization of the spinocerebellar ataxia type 7 protein, ataxin-7. (2/377)
Spinocerebellar ataxia type 7 (SCA7) belongs to a group of neurological disorders caused by a CAG repeat expansion in the coding region of the associated gene. To gain insight into the pathogenesis of SCA7 and possible functions of ataxin-7, we examined the subcellular localization of ataxin-7 in transfected COS-1 cells using SCA7 cDNA clones with different CAG repeat tract lengths. In addition to a diffuse distribution throughout the nucleus, ataxin-7 associated with the nuclear matrix and the nucleolus. The location of the putative SCA7 nuclear localization sequence (NLS) was confirmed by fusing an ataxin-7 fragment with the normally cytoplasmic protein chicken muscle pyruvate kinase. Mutation of this NLS prevented protein from entering the nucleus. Thus, expanded ataxin-7 may carry out its pathogenic effects in the nucleus by altering a matrix-associated nuclear structure and/or by disrupting nucleolar function. (+info)Very large (CAG)(n) DNA repeat expansions in the sperm of two spinocerebellar ataxia type 7 males. (3/377)
Genetic anticipation, i.e. increasing disease severity and decreasing age of onset from one generation to the next, is observed in a number of diseases, including myotonic dystrophy type 1, Huntington's disease and several of the spinocerebellar ataxias. All of these disorders are associated with the expansion of a trinucleotide repeat and array length is positively correlated with disease severity and inversely correlated with the age of onset. The expanded repeat is highly unstable and continues to expand from one generation to the next, providing a molecular explanation for anticipation. Spinocerebellar ataxia type 7 (SCA7) is one of the latest additions to the list of triplet repeat diseases and is distinct from the other SCAs in that it is accompanied by retinal degeneration. Pedigree analyses have previously revealed that the SCA7 repeat is highly unstable and liable to expand, in particular when transmitted by a male. Surprisingly, though, an under-representation of male transmission has also been reported. We now demonstrate directly by single molecule analyses that the expanded repeat is extraordinarily unstable in the male germline and biased toward massive increases. Nearly all of the mutant sperm of two SCA7 males contain alleles that are so large that most of the affected offspring would at best have a severe infantile form of the disease. Indeed, the gross under-representation of such very large expanded alleles in patients suggests that a significant proportion of such alleles might be associated with embryonic lethality or dysfunctional sperm. (+info)A common disease haplotype segregating in spinocerebellar ataxia 2 (SCA2) pedigrees of diverse ethnic origin. (4/377)
The identification of a CAG trinucleotide repeat expansion, located within the coding sequence of the ataxin-2 gene, as the mutation underlying spinocerebellar ataxia 2 (SCA2) has facilitated direct investigation of pedigrees previously excluded from linkage analysis due to insufficient size or pedigree structure. We have previously described the identification of the ancestral disease haplotype segregating in the Cuban founder population used to assign the disease locus to chromosome 12q23-24.1. We now report evidence for the segregation of the identical core haplotype in pedigrees of diverse ethnic origin from India, Japan and England, established by the analysis of the loci D12S1672 and D12S1333 located 20kb proximal and 200 kb distal to the triplet repeat motif respectively. Interpretation of this data is suggestive that for these pedigrees at least, the mutation has arisen on a single ancestral or predisposing chromosome. (+info)Multiple origins of the spinocerebellar ataxia 7 (SCA7) mutation revealed by linkage disequilibrium studies with closely flanking markers, including an intragenic polymorphism (G3145TG/A3145TG). (5/377)
Spinocerebellar ataxia 7 (SCA7) is a neurodegenerative disease characterised by the association of cerebellar ataxia and, in most patients, progressive macular degeneration leading to loss of autonomy and blindness. The patients die after 5-30 years of evolution. The cause of the disease has been identified as a (CAG)n repeat expansion in the coding sequence of the SCA7 gene on chromosome 3p. De novo mutations occur on intermediate-sized alleles carrying from 28 to 35 CAG repeats. Neomutations explain the persistence of the disease in spite of the great instability of the repeat sequence which results in the appearance of juvenile onset patients and the extinction of the disease within families. This rare disorder has been reported in a wide variety of countries and ethnic groups. In a large number of SCA7 families (n = 41) of different origins, we have determined the haplotypes segregating with the mutation of several microsatellite markers close to the SCA7 gene and of a new intragenic polymorphism (G3145TG/A3145TG). Four different haplotypes were found for centromeric markers (G3145TG/A3145TG-D3S1287-D3S3635) in the majority of the kindreds from four different geographic regions: A-2-4 in Korea; A-3-6 in North Africa, B-3-6 in continental Europe and A-4-6 in the UK and USA. The haplotypes in the Jamaican, Filipino, Brazilian and German families were different, suggesting that independent regional founders are at the origin of the SCA7 mutation in each population. Two different haplotypes were observed, however, in two families from the same rural area in central Italy in which de novo SCA7 mutations on intermediate alleles have been observed, suggesting the existence of different pools of at-risk chromosomes in this population. (+info)Spinocerebellar ataxia type 2 in seven Korean families: CAG trinucleotide expansion and clinical characteristics. (6/377)
Studies on spinocerebellar ataxias (SCA) have been hampered by a lack of disease markers. Clinical and pathological heterogeneity also made the classification unreliable. Linkage studies established that there are multiple subtypes of SCA. Five types are found to have unstable CAG expansion; the diagnosis can be established by molecular genetic study. Therefore, we systemically screened degenerative ataxia patients for these five SCA types, and identified eight patients with SCA2 (seven from six families and one sporadic case). This paper presents the clinical information on the seven patients, whose clinical information was available in detail. CAG repeat expansion in the patients ranged from 38 to 47 (normal control, 19 to 27). The onset ages ranged from 16 to 41 with 27.1 years as the mean, which correlated inversely with repeat lengths. All patients presented dysarthria and gait ataxia. Upper limb dysmetria or dysdiadochokinesia appeared later but progressed, causing severe disability. Slow saccade (4 patients in 7) and decreased DTR (4 in 7) were common. MRIs showed severe atrophy of the brainstem and cerebellum in all patients. We conclude that SCA2 is the most frequent type in Korea and carries rather pure cerebellar syndrome, slow saccade, and hyporeflexia. (+info)An unstable trinucleotide-repeat region on chromosome 13 implicated in spinocerebellar ataxia: a common expansion locus. (7/377)
Larger CAG/CTG trinucleotide-repeat tracts in individuals affected with schizophrenia (SCZ) and bipolar affective disorder (BPAD) in comparison with control individuals have previously been reported, implying a possible etiological role for trinucleotide repeats in these diseases. Two unstable CAG/CTG repeats, SEF2-1B and ERDA1, have recently been cloned, and studies indicate that the majority of individuals with large repeats as detected by repeat-expansion detection (RED) have large repeat alleles at these loci. These repeats do not show association of large alleles with either BPAD or SCZ. Using RED, we have identified a BPAD individual with a very large CAG/CTG repeat that is not due to expansion at SEF2-1B or ERDA1. From this individual's DNA, we have cloned a highly polymorphic trinucleotide repeat consisting of (CTA)n (CTG)n, which is very long ( approximately 1,800 bp) in this patient. The repeat region localizes to chromosome 13q21, within 1.2 cM of fragile site FRA13C. Repeat alleles in our sample were unstable in 13 (5.6%) of 231 meioses. Large alleles (>100 repeats) were observed in 14 (1. 25%) of 1,120 patients with psychosis, borderline personality disorder, or juvenile-onset depression and in 5 (.7%) of 710 healthy controls. Very large alleles were also detected for Centre d'Etude Polymorphisme Humaine (CEPH) reference family 1334. This triplet expansion has recently been reported to be the cause of spinocerebellar ataxia type 8 (SCA8); however, none of our large alleles above the disease threshold occurred in individuals either affected by SCA or with known family history of SCA. The high frequency of large alleles at this locus is inconsistent with the much rarer occurrence of SCA8. Thus, it seems unlikely that expansion alone causes SCA8; other genetic mechanisms may be necessary to explain SCA8 etiology. (+info)High germinal instability of the (CTG)n at the SCA8 locus of both expanded and normal alleles. (8/377)
The autosomal dominant spinocerebellar ataxias (SCAs) are a group of late-onset, neurodegenerative disorders for which 10 loci have been mapped (SCA1, SCA2, SCA4-SCA8, SCA10, MJD, and DRPLA). The mutant proteins have shown an expanded polyglutamine tract in SCA1, SCA2, MJD/SCA3, SCA6, SCA7, and DRPLA; a glycine-to-arginine substitution was found in SCA6 as well. Recently, an untranslated (CTG)n expansion on chromosome 13q was described as being the cause of SCA8. We have now (1) assessed the repeat size in a group of patients with ataxia and a large number of controls, (2) examined the intergenerational transmission of the repeat, and (3) estimated the instability of repeat size in the sperm of one patient and two healthy controls. Normal SCA8 chromosomes showed an apparently trimodal distribution, with classes of small (15-21 CTGs), intermediate (22-37 CTGs), and large (40-91 CTGs) alleles; large alleles accounted for only0.7% of all normal-size alleles. No expanded alleles (>/=100 CTGs) were found in controls. Expansion of the CTG tract was found in five families with ataxia; expanded alleles (all paternally transmitted) were characterized mostly by repeat-size contraction. There was a high germinal instability of both expanded and normal alleles: in one patient, the expanded allele (152 CTGs) had mostly contraction in size (often into the normal range); in the sperm of two normal controls, contractions were also more frequent, but occasional expansions into the upper limit of the normal size range were also seen. In conclusion, our results show (1) no overlapping between control (15-91) and pathogenic (100-152) alleles and (2) a high instability in spermatogenesis (both for expanded and normal alleles), suggesting a high mutational rate at the SCA8 locus. (+info)Spinocerebellar ataxias (SCAs) are a group of genetic disorders that affect the cerebellum, which is the part of the brain responsible for coordinating muscle movements. SCAs are characterized by progressive problems with balance, speech, and coordination. They are caused by mutations in various genes that result in the production of abnormal proteins that accumulate in neurons, leading to their degeneration.
There are over 40 different types of SCAs, each caused by a different genetic mutation. Some of the more common types include SCA1, SCA2, SCA3, SCA6, and SCA7. The symptoms and age of onset can vary widely depending on the type of SCA.
In addition to problems with coordination and balance, people with SCAs may also experience muscle weakness, stiffness, tremors, spasticity, and difficulty swallowing or speaking. Some types of SCAs can also cause visual disturbances, hearing loss, and cognitive impairment. Currently, there is no cure for SCAs, but treatments such as physical therapy, speech therapy, and medications can help manage the symptoms.
Spinocerebellar degenerations (SCDs) are a group of genetic disorders that primarily affect the cerebellum, the part of the brain responsible for coordinating muscle movements, and the spinal cord. These conditions are characterized by progressive degeneration or loss of nerve cells in the cerebellum and/or spinal cord, leading to various neurological symptoms.
SCDs are often inherited in an autosomal dominant manner, meaning that only one copy of the altered gene from either parent is enough to cause the disorder. The most common type of SCD is spinocerebellar ataxia (SCA), which includes several subtypes (SCA1, SCA2, SCA3, etc.) differentiated by their genetic causes and specific clinical features.
Symptoms of spinocerebellar degenerations may include:
1. Progressive ataxia (loss of coordination and balance)
2. Dysarthria (speech difficulty)
3. Nystagmus (involuntary eye movements)
4. Oculomotor abnormalities (problems with eye movement control)
5. Tremors or other involuntary muscle movements
6. Muscle weakness and spasticity
7. Sensory disturbances, such as numbness or tingling sensations
8. Dysphagia (difficulty swallowing)
9. Cognitive impairment in some cases
The age of onset, severity, and progression of symptoms can vary significantly among different SCD subtypes and individuals. Currently, there is no cure for spinocerebellar degenerations, but various supportive treatments and therapies can help manage symptoms and improve quality of life.
Machado-Joseph Disease (MJD) is a genetic disorder that affects the part of the brain that controls movement. It is also known as spinocerebellar ataxia type 3 (SCA3). MJD is characterized by progressive problems with coordination, speech, and swallowing, along with muscle stiffness, tremors, and in some cases, eye movement abnormalities.
MJD is caused by a mutation in the ATXN3 gene, which results in an expanded CAG repeat sequence. This genetic defect leads to the production of an abnormal protein that accumulates in nerve cells, causing them to die. The severity and age of onset of MJD can vary widely, even within families, but symptoms typically begin between the ages of 10 and 60.
MJD is inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the disease-causing mutation from an affected parent. Currently, there is no cure for MJD, but treatments can help manage symptoms and improve quality of life.
Cerebellar ataxia is a type of ataxia, which refers to a group of disorders that cause difficulties with coordination and movement. Cerebellar ataxia specifically involves the cerebellum, which is the part of the brain responsible for maintaining balance, coordinating muscle movements, and regulating speech and eye movements.
The symptoms of cerebellar ataxia may include:
* Unsteady gait or difficulty walking
* Poor coordination of limb movements
* Tremors or shakiness, especially in the hands
* Slurred or irregular speech
* Abnormal eye movements, such as nystagmus (rapid, involuntary movement of the eyes)
* Difficulty with fine motor tasks, such as writing or buttoning a shirt
Cerebellar ataxia can be caused by a variety of underlying conditions, including:
* Genetic disorders, such as spinocerebellar ataxia or Friedreich's ataxia
* Brain injury or trauma
* Stroke or brain hemorrhage
* Infections, such as meningitis or encephalitis
* Exposure to toxins, such as alcohol or certain medications
* Tumors or other growths in the brain
Treatment for cerebellar ataxia depends on the underlying cause. In some cases, there may be no cure, and treatment is focused on managing symptoms and improving quality of life. Physical therapy, occupational therapy, and speech therapy can help improve coordination, balance, and communication skills. Medications may also be used to treat specific symptoms, such as tremors or muscle spasticity. In some cases, surgery may be recommended to remove tumors or repair damage to the brain.
Spinocerebellar tracts are a type of white matter tract in the spinal cord that carry information related to proprioception, muscle tone, and movement coordination from the peripheral nervous system to the cerebellum. There are several different spinocerebellar tracts, including the dorsal (or posterior) spinocerebellar tract and the ventral (or anterior) spinocerebellar tract.
The dorsal spinocerebellar tract carries information about the position and movement of joints and muscles from receptors in the skin, muscles, and tendons to the cerebellum. This information is used by the cerebellum to help coordinate movements and maintain balance.
The ventral spinocerebellar tract carries information about muscle stretch and tension from receptors in the muscles to the cerebellum. This information is used by the cerebellum to regulate muscle tone and coordination.
Damage to the spinocerebellar tracts can result in a variety of neurological symptoms, including ataxia (loss of coordination), dysmetria (impaired ability to judge distance or speed of movement), and hypotonia (decreased muscle tone).
Ataxia is a medical term that refers to a group of disorders affecting coordination, balance, and speech. It is characterized by a lack of muscle control during voluntary movements, causing unsteady or awkward movements, and often accompanied by tremors. Ataxia can affect various parts of the body, such as the limbs, trunk, eyes, and speech muscles. The condition can be congenital or acquired, and it can result from damage to the cerebellum, spinal cord, or sensory nerves. There are several types of ataxia, including hereditary ataxias, degenerative ataxias, cerebellar ataxias, and acquired ataxias, each with its own specific causes, symptoms, and prognosis. Treatment for ataxia typically focuses on managing symptoms and improving quality of life, as there is no cure for most forms of the disorder.
Trinucleotide Repeat Expansion is a genetic mutation where a sequence of three DNA nucleotides is repeated more frequently than what is typically found in the general population. In this type of mutation, the number of repeats can expand or increase from one generation to the next, leading to an increased risk of developing certain genetic disorders.
These disorders are often neurological and include conditions such as Huntington's disease, myotonic dystrophy, fragile X syndrome, and Friedreich's ataxia. The severity of these diseases can be related to the number of repeats present in the affected gene, with a higher number of repeats leading to more severe symptoms or an earlier age of onset.
It is important to note that not all trinucleotide repeat expansions will result in disease, and some people may carry these mutations without ever developing any symptoms. However, if the number of repeats crosses a certain threshold, it can lead to genetic instability and an increased risk of disease development.
Trinucleotide repeats refer to a specific type of DNA sequence expansion where a particular trinucleotide (a sequence made up of three nucleotides) is repeated multiple times. In normal genomic DNA, these repeats are usually present in a relatively stable and consistent range. However, when the number of repeats exceeds a certain threshold, it can result in an unstable genetic variant known as a trinucleotide repeat expansion.
These expansions can occur in various genes and are associated with several neurogenetic disorders, such as Huntington's disease, myotonic dystrophy, fragile X syndrome, and Friedreich's ataxia. The length of the trinucleotide repeat tends to expand further in subsequent generations, which can lead to anticipation – an earlier age of onset and increased severity of symptoms in successive generations.
The most common trinucleotide repeats involve CAG (cytosine-adenine-guanine) or CTG (cytosine-thymine-guanine) repeats, although other combinations like CGG, GAA, and GCT can also be involved. These repeat expansions can result in altered gene function, protein misfolding, aggregation, and toxicity, ultimately leading to the development of neurodegenerative diseases and other clinical manifestations.
Nerve tissue proteins are specialized proteins found in the nervous system that provide structural and functional support to nerve cells, also known as neurons. These proteins include:
1. Neurofilaments: These are type IV intermediate filaments that provide structural support to neurons and help maintain their shape and size. They are composed of three subunits - NFL (light), NFM (medium), and NFH (heavy).
2. Neuronal Cytoskeletal Proteins: These include tubulins, actins, and spectrins that provide structural support to the neuronal cytoskeleton and help maintain its integrity.
3. Neurotransmitter Receptors: These are specialized proteins located on the postsynaptic membrane of neurons that bind neurotransmitters released by presynaptic neurons, triggering a response in the target cell.
4. Ion Channels: These are transmembrane proteins that regulate the flow of ions across the neuronal membrane and play a crucial role in generating and transmitting electrical signals in neurons.
5. Signaling Proteins: These include enzymes, receptors, and adaptor proteins that mediate intracellular signaling pathways involved in neuronal development, differentiation, survival, and death.
6. Adhesion Proteins: These are cell surface proteins that mediate cell-cell and cell-matrix interactions, playing a crucial role in the formation and maintenance of neural circuits.
7. Extracellular Matrix Proteins: These include proteoglycans, laminins, and collagens that provide structural support to nerve tissue and regulate neuronal migration, differentiation, and survival.
Friedreich Ataxia is a genetic disorder that affects the nervous system and causes issues with movement. It is characterized by progressive damage to the nerves (neurons) in the spinal cord and peripheral nerves, which can lead to problems with muscle coordination, gait, speech, and hearing. The condition is also associated with heart disorders, diabetes, and vision impairment.
Friedreich Ataxia is caused by a mutation in the FXN gene, which provides instructions for making a protein called frataxin. This protein plays a role in the production of energy within cells, particularly in the mitochondria. The mutation in the FXN gene leads to reduced levels of frataxin, which can cause nerve damage and other symptoms associated with Friedreich Ataxia.
The condition typically begins in childhood or early adulthood and progresses over time, often leading to significant disability. There is currently no cure for Friedreich Ataxia, but treatments are available to help manage the symptoms and improve quality of life.
The cerebellum is a part of the brain that lies behind the brainstem and is involved in the regulation of motor movements, balance, and coordination. It contains two hemispheres and a central portion called the vermis. The cerebellum receives input from sensory systems and other areas of the brain and spinal cord and sends output to motor areas of the brain. Damage to the cerebellum can result in problems with movement, balance, and coordination.
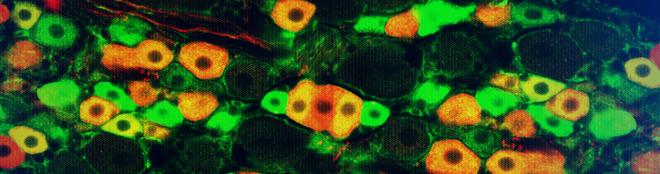
Purkinje cells are a type of neuron located in the cerebellar cortex, which is the outer layer of the cerebellum, a part of the brain that plays a crucial role in motor control and coordination. These cells have large branching dendrites and receive input from many other neurons, particularly granule cells. The axons of Purkinje cells form the principal output pathway of the cerebellar cortex, synapsing with deep cerebellar nuclei. They are named after Johannes Evangelista Purkinje, a Czech physiologist who first described them in 1837.
DNA repeat expansion is a genetic alteration in which a particular sequence of DNA base pairs is repeated multiple times. In normal genes, these repeats are relatively short and stable, but in certain diseases, the number of repeats can expand beyond a threshold, leading to changes in the structure or function of the gene. This type of mutation is often associated with neurological and neuromuscular disorders, such as Huntington's disease, myotonic dystrophy, and fragile X syndrome. The expanded repeats can also be unstable and may increase in size over generations, leading to more severe symptoms or earlier age of onset.
Gait ataxia is a type of ataxia, which refers to a lack of coordination or stability, specifically involving walking or gait. It is characterized by an unsteady, uncoordinated, and typically wide-based gait pattern. This occurs due to dysfunction in the cerebellum or its connecting pathways, responsible for maintaining balance and coordinating muscle movements.
In gait ataxia, individuals often have difficulty with controlling the rhythm and pace of their steps, tend to veer or stagger off course, and may display a reeling or stumbling motion while walking. They might also have trouble performing rapid alternating movements like quickly tapping their foot or heel. These symptoms are usually worse when the person is tired or attempting to walk in the dark.
Gait ataxia can be caused by various underlying conditions, including degenerative neurological disorders (e.g., cerebellar atrophy, multiple sclerosis), stroke, brain injury, infection (e.g., alcoholism, HIV), or exposure to certain toxins. Proper diagnosis and identification of the underlying cause are essential for effective treatment and management of gait ataxia.
Intranuclear inclusion bodies are abnormal, rounded structures found within the nucleus of a cell. They are composed of aggregated proteins or other cellular components and can be associated with various viral infections and certain genetic disorders. These inclusion bodies can interfere with normal nuclear functions, leading to cell damage and contributing to the pathogenesis of diseases such as cytomegalovirus infection, rabies, and some forms of neurodegenerative disorders like polyglutamine diseases. The presence of intranuclear inclusion bodies is often used in diagnostic pathology to help identify specific underlying conditions.
The "age of onset" is a medical term that refers to the age at which an individual first develops or displays symptoms of a particular disease, disorder, or condition. It can be used to describe various medical conditions, including both physical and mental health disorders. The age of onset can have implications for prognosis, treatment approaches, and potential causes of the condition. In some cases, early onset may indicate a more severe or progressive course of the disease, while late-onset symptoms might be associated with different underlying factors or etiologies. It is essential to provide accurate and precise information regarding the age of onset when discussing a patient's medical history and treatment plan.
Nuclear proteins are a category of proteins that are primarily found in the nucleus of a eukaryotic cell. They play crucial roles in various nuclear functions, such as DNA replication, transcription, repair, and RNA processing. This group includes structural proteins like lamins, which form the nuclear lamina, and regulatory proteins, such as histones and transcription factors, that are involved in gene expression. Nuclear localization signals (NLS) often help target these proteins to the nucleus by interacting with importin proteins during active transport across the nuclear membrane.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Heredodegenerative disorders of the nervous system are a group of inherited conditions that involve progressive degeneration of the nervous system over time. These disorders are caused by genetic mutations that affect the development and function of nerve cells in the brain and spinal cord. The symptoms and severity of these disorders can vary widely, depending on the specific condition and the location and extent of nerve cell damage.
Examples of heredodegenerative disorders of the nervous system include:
1. Huntington's disease: a genetic disorder that causes the progressive breakdown of nerve cells in the brain, leading to uncontrolled movements, emotional problems, and cognitive decline.
2. Friedreich's ataxia: an inherited disorder that affects the nerves and muscle coordination, causing symptoms such as difficulty walking, poor balance, and speech problems.
3. Spinal muscular atrophy: a genetic disorder that affects the motor neurons in the spinal cord, leading to muscle weakness and wasting.
4. Hereditary sensory and autonomic neuropathies: a group of inherited disorders that affect the nerves that control sensation and automatic functions such as heart rate and digestion.
5. Leukodystrophies: a group of genetic disorders that affect the white matter of the brain, leading to symptoms such as motor and cognitive decline, seizures, and vision loss.
Treatment for heredodegenerative disorders of the nervous system typically focuses on managing symptoms and improving quality of life. There is no cure for most of these conditions, but research is ongoing to develop new treatments and therapies that may help slow or stop the progression of nerve cell damage.
Shaw potassium channels, also known as KCNA4 channels, are a type of voltage-gated potassium channel that is encoded by the KCNA4 gene in humans. These channels play a crucial role in regulating the electrical excitability of cells, particularly in the heart and nervous system.
Shaw channels are named after James E. Shaw, who first identified them in 1996. They are composed of four subunits that arrange themselves to form a central pore through which potassium ions can flow. The channels are activated by depolarization of the cell membrane and help to repolarize the membrane during action potentials.
Mutations in the KCNA4 gene have been associated with various cardiac arrhythmias, including familial atrial fibrillation and long QT syndrome type 3. These conditions can cause irregular heart rhythms and may increase the risk of sudden cardiac death. Therefore, understanding the function and regulation of Shaw potassium channels is important for developing therapies to treat these disorders.
Peptides are short chains of amino acid residues linked by covalent bonds, known as peptide bonds. They are formed when two or more amino acids are joined together through a condensation reaction, which results in the elimination of a water molecule and the formation of an amide bond between the carboxyl group of one amino acid and the amino group of another.
Peptides can vary in length from two to about fifty amino acids, and they are often classified based on their size. For example, dipeptides contain two amino acids, tripeptides contain three, and so on. Oligopeptides typically contain up to ten amino acids, while polypeptides can contain dozens or even hundreds of amino acids.
Peptides play many important roles in the body, including serving as hormones, neurotransmitters, enzymes, and antibiotics. They are also used in medical research and therapeutic applications, such as drug delivery and tissue engineering.
Inclusion bodies are abnormal, intracellular accumulations or aggregations of various misfolded proteins, protein complexes, or other materials within the cells of an organism. They can be found in various tissues and cell types and are often associated with several pathological conditions, including infectious diseases, neurodegenerative disorders, and genetic diseases.
Inclusion bodies can vary in size, shape, and location depending on the specific disease or condition. Some inclusion bodies have a characteristic appearance under the microscope, such as eosinophilic (pink) staining with hematoxylin and eosin (H&E) histological stain, while others may require specialized stains or immunohistochemical techniques to identify the specific misfolded proteins involved.
Examples of diseases associated with inclusion bodies include:
1. Infectious diseases: Some viral infections, such as HIV, hepatitis B and C, and herpes simplex virus, can lead to the formation of inclusion bodies within infected cells.
2. Neurodegenerative disorders: Several neurodegenerative diseases are characterized by the presence of inclusion bodies, including Alzheimer's disease (amyloid-beta plaques and tau tangles), Parkinson's disease (Lewy bodies), Huntington's disease (Huntingtin aggregates), and amyotrophic lateral sclerosis (TDP-43 and SOD1 inclusions).
3. Genetic diseases: Certain genetic disorders, such as Danon disease, neuronal intranuclear inclusion disease, and some lysosomal storage disorders, can also present with inclusion bodies due to the accumulation of abnormal proteins or metabolic products within cells.
The exact role of inclusion bodies in disease pathogenesis remains unclear; however, they are often associated with cellular dysfunction, oxidative stress, and increased inflammation, which can contribute to disease progression and neurodegeneration.
Genetic anticipation is a phenomenon observed in certain genetic disorders where the severity and/or age of onset of the disease tend to worsen in successive generations. This occurs due to an expansion of triplet repeat sequences (sequences of three consecutive DNA base pairs) in the affected gene, which can lead to an increased production of abnormal proteins associated with the disorder. The expanded repeats are more likely to be inherited when the parent who carries them is a female. Examples of genetic disorders that exhibit anticipation include Huntington's disease, myotonic dystrophy, and fragile X syndrome.
Dominant genes refer to the alleles (versions of a gene) that are fully expressed in an individual's phenotype, even if only one copy of the gene is present. In dominant inheritance patterns, an individual needs only to receive one dominant allele from either parent to express the associated trait. This is in contrast to recessive genes, where both copies of the gene must be the recessive allele for the trait to be expressed. Dominant genes are represented by uppercase letters (e.g., 'A') and recessive genes by lowercase letters (e.g., 'a'). If an individual inherits one dominant allele (A) from either parent, they will express the dominant trait (A).
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Nerve degeneration, also known as neurodegeneration, is the progressive loss of structure and function of neurons, which can lead to cognitive decline, motor impairment, and various other symptoms. This process occurs due to a variety of factors, including genetics, environmental influences, and aging. It is a key feature in several neurological disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease, and multiple sclerosis. The degeneration can affect any part of the nervous system, leading to different symptoms depending on the location and extent of the damage.
Neurodegenerative diseases are a group of disorders characterized by progressive and persistent loss of neuronal structure and function, often leading to cognitive decline, functional impairment, and ultimately death. These conditions are associated with the accumulation of abnormal protein aggregates, mitochondrial dysfunction, oxidative stress, chronic inflammation, and genetic mutations in the brain. Examples of neurodegenerative diseases include Alzheimer's disease, Parkinson's disease, Huntington's disease, Amyotrophic Lateral Sclerosis (ALS), and Spinal Muscular Atrophy (SMA). The underlying causes and mechanisms of these diseases are not fully understood, and there is currently no cure for most neurodegenerative disorders. Treatment typically focuses on managing symptoms and slowing disease progression.
Ataxia telangiectasia is a rare, inherited genetic disorder that affects the nervous system, immune system, and overall development. The condition is characterized by progressive difficulty with coordination and balance (ataxia), as well as the development of small, dilated blood vessels (telangiectasias) on the skin and eyes.
The underlying cause of ataxia telangiectasia is a mutation in the ATM gene, which provides instructions for making a protein that plays a critical role in DNA repair and maintaining genetic stability. When this gene is mutated, cells are unable to properly repair damaged DNA, leading to an increased risk of cancer and other health problems.
Individuals with ataxia telangiectasia typically begin to show symptoms during early childhood, with progressive difficulties in coordination and balance, slurred speech, and recurrent respiratory infections due to weakened immune function. Over time, these symptoms can worsen, leading to significant disability and reduced life expectancy.
There is currently no cure for ataxia telangiectasia, and treatment is focused on managing the symptoms and complications of the condition. This may include physical therapy, speech therapy, and medications to help control infections and other health problems.
Myoclonic cerebellar dyssynergia is not a widely recognized or formally defined medical term. However, based on its individual components, it can be inferred to refer to a neurological condition characterized by:
1. Myoclonus: These are sudden, involuntary jerking movements of a muscle or group of muscles. They typically occur as a result of hyperexcitability of the neurons in the brain that control movement (motor neurons).
2. Cerebellar: The cerebellum is a part of the brain responsible for coordinating muscle movements, maintaining posture and balance, and fine-tuning motor skills. When a condition is described as "cerebellar," it implies that there is some dysfunction or abnormality in this region of the brain.
3. Dyssynergia: This term refers to a lack of coordination between muscles and muscle groups during voluntary movements. It can result from damage to the cerebellum or other parts of the nervous system involved in motor control.
Therefore, myoclonic cerebellar dyssynergia could be interpreted as a condition characterized by involuntary muscle jerks (myoclonus) and impaired coordination of voluntary movements (dyssynergia), likely due to cerebellar dysfunction. However, it is essential to consult with a medical professional for an accurate diagnosis and treatment plan if you or someone else experiences symptoms that may align with this description.
Human chromosome pair 6 consists of two rod-shaped structures present in the nucleus of each human cell. They are identical in size and shape and contain genetic material, made up of DNA and proteins, that is essential for the development and function of the human body.
Chromosome pair 6 is one of the 23 pairs of chromosomes found in humans, with one chromosome inherited from each parent. Each chromosome contains thousands of genes that provide instructions for the production of proteins and regulate various cellular processes.
Chromosome pair 6 contains several important genes, including those involved in the development and function of the immune system, such as the major histocompatibility complex (MHC) genes. It also contains genes associated with certain genetic disorders, such as hereditary neuropathy with liability to pressure palsies (HNPP), a condition that affects the nerves, and Waardenburg syndrome, a disorder that affects pigmentation and hearing.
Abnormalities in chromosome pair 6 can lead to various genetic disorders, including numerical abnormalities such as trisomy 6 (three copies of chromosome 6) or monosomy 6 (only one copy of chromosome 6), as well as structural abnormalities such as deletions, duplications, or translocations of parts of the chromosome.
Human chromosome pair 20 is one of the 23 pairs of human chromosomes present in every cell of the body, except for the sperm and egg cells which contain only 23 individual chromosomes. Chromosomes are thread-like structures that carry genetic information in the form of genes.
Human chromosome pair 20 is an acrocentric chromosome, meaning it has a short arm (p arm) and a long arm (q arm), with the centromere located near the junction of the two arms. The short arm of chromosome 20 is very small and contains few genes, while the long arm contains several hundred genes that play important roles in various biological processes.
Chromosome pair 20 is associated with several genetic disorders, including DiGeorge syndrome, which is caused by a deletion of a portion of the long arm of chromosome 20. This syndrome is characterized by birth defects affecting the heart, face, and immune system. Other conditions associated with abnormalities of chromosome pair 20 include some forms of intellectual disability, autism spectrum disorder, and cancer.
Repressor proteins are a type of regulatory protein in molecular biology that suppress the transcription of specific genes into messenger RNA (mRNA) by binding to DNA. They function as part of gene regulation processes, often working in conjunction with an operator region and a promoter region within the DNA molecule. Repressor proteins can be activated or deactivated by various signals, allowing for precise control over gene expression in response to changing cellular conditions.
There are two main types of repressor proteins:
1. DNA-binding repressors: These directly bind to specific DNA sequences (operator regions) near the target gene and prevent RNA polymerase from transcribing the gene into mRNA.
2. Allosteric repressors: These bind to effector molecules, which then cause a conformational change in the repressor protein, enabling it to bind to DNA and inhibit transcription.
Repressor proteins play crucial roles in various biological processes, such as development, metabolism, and stress response, by controlling gene expression patterns in cells.
Ataxia telangiectasia mutated (ATM) proteins are a type of protein that play a crucial role in the maintenance and repair of DNA in cells. The ATM gene produces these proteins, which are involved in several important cellular processes such as:
1. DNA damage response: When DNA is damaged, ATM proteins help to detect and respond to the damage by activating various signaling pathways that lead to DNA repair or apoptosis (programmed cell death) if the damage is too severe.
2. Cell cycle regulation: ATM proteins regulate the cell cycle by controlling checkpoints that ensure proper DNA replication and division. This helps prevent the propagation of cells with damaged DNA.
3. Telomere maintenance: ATM proteins help maintain telomeres, which are the protective caps at the ends of chromosomes. Telomeres shorten as cells divide, and when they become too short, cells can no longer divide and enter a state of senescence or die.
Mutations in the ATM gene can lead to Ataxia-telangiectasia (A-T), a rare inherited disorder characterized by neurological problems, immune system dysfunction, increased risk of cancer, and sensitivity to ionizing radiation. People with A-T have defective ATM proteins that cannot properly respond to DNA damage, leading to genomic instability and increased susceptibility to disease.
Multiple System Atrophy (MSA) is a rare, progressive neurodegenerative disorder that affects multiple systems in the body. It is characterized by a combination of symptoms including Parkinsonism (such as stiffness, slowness of movement, and tremors), cerebellar ataxia (lack of muscle coordination), autonomic dysfunction (problems with the autonomic nervous system which controls involuntary actions like heart rate, blood pressure, sweating, and digestion), and pyramidal signs (abnormalities in the corticospinal tracts that control voluntary movements).
The disorder is caused by the degeneration of nerve cells in various parts of the brain and spinal cord, leading to a loss of function in these areas. The exact cause of MSA is unknown, but it is thought to involve a combination of genetic and environmental factors. There is currently no cure for MSA, and treatment is focused on managing symptoms and improving quality of life.
Atrophy is a medical term that refers to the decrease in size and wasting of an organ or tissue due to the disappearance of cells, shrinkage of cells, or decreased number of cells. This process can be caused by various factors such as disuse, aging, degeneration, injury, or disease.
For example, if a muscle is immobilized for an extended period, it may undergo atrophy due to lack of use. Similarly, certain medical conditions like diabetes, cancer, and heart failure can lead to the wasting away of various tissues and organs in the body.
Atrophy can also occur as a result of natural aging processes, leading to decreased muscle mass and strength in older adults. In general, atrophy is characterized by a decrease in the volume or weight of an organ or tissue, which can have significant impacts on its function and overall health.
Calcium channels, Q-type, are a type of voltage-gated calcium channel found in various tissues, including the brain and heart. They are called "Q-type" because they exhibit a distinctive "q-wave" in their current trace during electrical activity. These channels play important roles in regulating physiological processes such as neurotransmitter release, hormone secretion, and cardiac muscle contraction.
The pore-forming subunit of Q-type calcium channels is the CaV2.1 (or α1A) subunit, which is encoded by the CACNA1A gene. These channels are activated by depolarization of the cell membrane and allow the influx of calcium ions into the cell. The resulting increase in intracellular calcium concentration triggers various downstream signaling pathways that mediate the physiological responses mentioned above.
Dysfunction of Q-type calcium channels has been implicated in several neurological and cardiovascular disorders, including migraine, epilepsy, cerebellar ataxia, and hypertension. Therefore, understanding the structure, function, and regulation of these channels is an important area of research for developing new therapeutic strategies to treat these conditions.
DNA probes for HLA (Human Leukocyte Antigen) are specific DNA sequences that are used in laboratory tests to detect and identify the presence or absence of particular HLA genes or alleles in an individual's genetic material. HLAs are proteins found on the surface of cells that play a critical role in the immune system's ability to distinguish between "self" and "non-self."
DNA probes for HLA are typically composed of short, single-stranded DNA molecules that are complementary to a specific region of the HLA gene. These probes are labeled with a detectable marker, such as a radioactive isotope or a fluorescent dye, allowing them to be visualized and detected during laboratory testing.
When a DNA probe for HLA is hybridized to a sample of an individual's genetic material, it will bind specifically to the complementary sequence of the target HLA gene, if present. The presence or absence of the probe-target hybrid can then be detected and used to identify the specific HLA allele.
DNA probes for HLA are used in a variety of applications, including diagnostic testing, tissue typing for transplantation, and research into the genetic basis of diseases that are associated with particular HLA types.
The Founder Effect is a concept in population genetics that refers to the loss of genetic variation that occurs when a new colony is established by a small number of individuals from a larger population. This decrease in genetic diversity can lead to an increase in homozygosity, which can in turn result in a higher frequency of certain genetic disorders or traits within the founding population and its descendants. The Founder Effect is named after the "founding" members of the new colony who carry and pass on their particular set of genes to the next generations. It is one of the mechanisms that can lead to the formation of distinct populations or even new species over time.
Medical Definition:
Magnetic Resonance Imaging (MRI) is a non-invasive diagnostic imaging technique that uses a strong magnetic field and radio waves to create detailed cross-sectional or three-dimensional images of the internal structures of the body. The patient lies within a large, cylindrical magnet, and the scanner detects changes in the direction of the magnetic field caused by protons in the body. These changes are then converted into detailed images that help medical professionals to diagnose and monitor various medical conditions, such as tumors, injuries, or diseases affecting the brain, spinal cord, heart, blood vessels, joints, and other internal organs. MRI does not use radiation like computed tomography (CT) scans.
Human chromosome pair 19 refers to a group of 19 identical chromosomes that are present in every cell of the human body, except for the sperm and egg cells which contain only 23 chromosomes. Chromosomes are thread-like structures that carry genetic information in the form of DNA (deoxyribonucleic acid) molecules.
Each chromosome is made up of two arms, a shorter p arm and a longer q arm, separated by a centromere. Human chromosome pair 19 is an acrocentric chromosome, which means that the centromere is located very close to the end of the short arm (p arm).
Chromosome pair 19 contains approximately 58 million base pairs of DNA and encodes for around 1,400 genes. It is one of the most gene-dense chromosomes in the human genome, with many genes involved in important biological processes such as metabolism, immunity, and neurological function.
Abnormalities in chromosome pair 19 have been associated with various genetic disorders, including Sotos syndrome, which is characterized by overgrowth, developmental delay, and distinctive facial features, and Smith-Magenis syndrome, which is marked by intellectual disability, behavioral problems, and distinct physical features.
A patent, in the context of medicine and healthcare, generally refers to a government-granted exclusive right for an inventor to manufacture, use, or sell their invention for a certain period of time, typically 20 years from the filing date. In the medical field, patents may cover a wide range of inventions, including new drugs, medical devices, diagnostic methods, and even genetic sequences.
The purpose of patents is to provide incentives for innovation by allowing inventors to profit from their inventions. However, patents can also have significant implications for access to medical technologies and healthcare costs. For example, a patent on a life-saving drug may give the patent holder the exclusive right to manufacture and sell the drug, potentially limiting access and driving up prices.
It's worth noting that the patent system is complex and varies from country to country. In some cases, there may be ways to challenge or circumvent patents in order to increase access to medical technologies, such as through compulsory licensing or generic substitution.
"Family Health" is not a term that has a single, widely accepted medical definition. However, in the context of healthcare and public health, "family health" often refers to the physical, mental, and social well-being of all members of a family unit. It includes the assessment, promotion, and prevention of health conditions that affect individual family members as well as the family as a whole.
Family health may also encompass interventions and programs that aim to strengthen family relationships, communication, and functioning, as these factors can have a significant impact on overall health outcomes. Additionally, family health may involve addressing social determinants of health, such as poverty, housing, and access to healthcare, which can affect the health of families and communities.
Overall, family health is a holistic approach to healthcare that recognizes the importance of considering the needs and experiences of all family members in promoting and maintaining good health.
Genetic linkage is the phenomenon where two or more genetic loci (locations on a chromosome) tend to be inherited together because they are close to each other on the same chromosome. This occurs during the process of sexual reproduction, where homologous chromosomes pair up and exchange genetic material through a process called crossing over.
The closer two loci are to each other on a chromosome, the lower the probability that they will be separated by a crossover event. As a result, they are more likely to be inherited together and are said to be linked. The degree of linkage between two loci can be measured by their recombination frequency, which is the percentage of meiotic events in which a crossover occurs between them.
Linkage analysis is an important tool in genetic research, as it allows researchers to identify and map genes that are associated with specific traits or diseases. By analyzing patterns of linkage between markers (identifiable DNA sequences) and phenotypes (observable traits), researchers can infer the location of genes that contribute to those traits or diseases on chromosomes.
Progressive Myoclonic Epilepsies (PME) is a group of rare, genetic disorders characterized by myoclonus (rapid, involuntary muscle jerks), tonic-clonic seizures (also known as grand mal seizures), and progressive neurological deterioration. The term "progressive" refers to the worsening of symptoms over time.
The myoclonic epilepsies are classified as progressive due to the underlying neurodegenerative process that affects the brain, leading to a decline in cognitive abilities, motor skills, and overall functioning. These disorders usually begin in childhood or adolescence and tend to worsen with age.
Examples of PMEs include:
1. Lafora disease: A genetic disorder caused by mutations in the EPM2A or NHLRC1 genes, leading to the accumulation of abnormal protein aggregates called Lafora bodies in neurons. Symptoms typically start between ages 6 and 16 and include myoclonus, seizures, and progressive neurological decline.
2. Unverricht-Lundborg disease: Also known as Baltic myoclonus, this is an autosomal recessive disorder caused by mutations in the CSTB gene. It is characterized by progressive myoclonic epilepsy, ataxia (loss of coordination), and cognitive decline. Symptoms usually begin between ages 6 and 18.
3. Neuronal Ceroid Lipofuscinoses (NCLs): A group of inherited neurodegenerative disorders characterized by the accumulation of lipopigments in neurons. Several types of NCLs can present with progressive myoclonic epilepsy, including CLN2 (late-infantile NCL), CLN3 (juvenile NCL), and CLN6 (early juvenile NCL).
4. Myoclonus Epilepsy Associated with Ragged Red Fibers (MERRF): A mitochondrial disorder caused by mutations in the MT-TK gene, leading to myoclonic epilepsy, ataxia, and ragged red fibers on muscle biopsy.
5. Dentatorubral-Pallidoluysian Atrophy (DRPLA): An autosomal dominant disorder caused by mutations in the ATN1 gene, characterized by myoclonic epilepsy, ataxia, chorea (involuntary movements), and dementia.
These are just a few examples of disorders that can present with progressive myoclonic epilepsy. It is essential to consult a neurologist or epileptologist for proper diagnosis and management.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Transgenic mice are genetically modified rodents that have incorporated foreign DNA (exogenous DNA) into their own genome. This is typically done through the use of recombinant DNA technology, where a specific gene or genetic sequence of interest is isolated and then introduced into the mouse embryo. The resulting transgenic mice can then express the protein encoded by the foreign gene, allowing researchers to study its function in a living organism.
The process of creating transgenic mice usually involves microinjecting the exogenous DNA into the pronucleus of a fertilized egg, which is then implanted into a surrogate mother. The offspring that result from this procedure are screened for the presence of the foreign DNA, and those that carry the desired genetic modification are used to establish a transgenic mouse line.
Transgenic mice have been widely used in biomedical research to model human diseases, study gene function, and test new therapies. They provide a valuable tool for understanding complex biological processes and developing new treatments for a variety of medical conditions.
Calcium channels, P-type, are a specific type of voltage-gated calcium channel found in excitable cells such as neurons and muscle cells. They are named "P-type" because they were initially identified in Purkinje cells of the cerebellum. These channels play a crucial role in various cellular processes, including neurotransmitter release, muscle contraction, and gene expression.
P-type calcium channels are characterized by their unique biophysical properties, such as slow voltage-dependent activation and inactivation, as well as sensitivity to the drug felodipine. They are composed of several subunits, including the pore-forming α1 subunit, which contains the voltage sensor and the selectivity filter for calcium ions. The α1 subunit is associated with accessory subunits, such as β, γ, and δ, that modulate the channel's properties and trafficking to the cell membrane.
P-type calcium channels are important targets for therapeutic interventions in various diseases, including neurological disorders, cardiovascular diseases, and cancer. For example, drugs that block P-type calcium channels have been used to treat hypertension and angina, while activators of these channels have shown promise in treating neurodegenerative disorders such as Parkinson's disease.
The brain is the central organ of the nervous system, responsible for receiving and processing sensory information, regulating vital functions, and controlling behavior, movement, and cognition. It is divided into several distinct regions, each with specific functions:
1. Cerebrum: The largest part of the brain, responsible for higher cognitive functions such as thinking, learning, memory, language, and perception. It is divided into two hemispheres, each controlling the opposite side of the body.
2. Cerebellum: Located at the back of the brain, it is responsible for coordinating muscle movements, maintaining balance, and fine-tuning motor skills.
3. Brainstem: Connects the cerebrum and cerebellum to the spinal cord, controlling vital functions such as breathing, heart rate, and blood pressure. It also serves as a relay center for sensory information and motor commands between the brain and the rest of the body.
4. Diencephalon: A region that includes the thalamus (a major sensory relay station) and hypothalamus (regulates hormones, temperature, hunger, thirst, and sleep).
5. Limbic system: A group of structures involved in emotional processing, memory formation, and motivation, including the hippocampus, amygdala, and cingulate gyrus.
The brain is composed of billions of interconnected neurons that communicate through electrical and chemical signals. It is protected by the skull and surrounded by three layers of membranes called meninges, as well as cerebrospinal fluid that provides cushioning and nutrients.
Cerebellar diseases refer to a group of medical conditions that affect the cerebellum, which is the part of the brain located at the back of the head, below the occipital lobe and above the brainstem. The cerebellum plays a crucial role in motor control, coordination, balance, and some cognitive functions.
Cerebellar diseases can be caused by various factors, including genetics, infections, tumors, stroke, trauma, or degenerative processes. These conditions can result in a wide range of symptoms, such as:
1. Ataxia: Loss of coordination and unsteady gait
2. Dysmetria: Inability to judge distance and force while performing movements
3. Intention tremors: Shaking or trembling that worsens during purposeful movements
4. Nystagmus: Rapid, involuntary eye movement
5. Dysarthria: Speech difficulty due to muscle weakness or incoordination
6. Hypotonia: Decreased muscle tone
7. Titubation: Rhythmic, involuntary oscillations of the head and neck
8. Cognitive impairment: Problems with memory, attention, and executive functions
Some examples of cerebellar diseases include:
1. Ataxia-telangiectasia
2. Friedrich's ataxia
3. Multiple system atrophy (MSA)
4. Spinocerebellar ataxias (SCAs)
5. Cerebellar tumors, such as medulloblastomas or astrocytomas
6. Infarctions or hemorrhages in the cerebellum due to stroke or trauma
7. Infections, such as viral encephalitis or bacterial meningitis
8. Autoimmune disorders, like multiple sclerosis (MS) or paraneoplastic syndromes
9. Metabolic disorders, such as Wilson's disease or phenylketonuria (PKU)
10. Chronic alcoholism and withdrawal
Treatment for cerebellar diseases depends on the underlying cause and may involve medications, physical therapy, surgery, or supportive care to manage symptoms and improve quality of life.
The Rotarod performance test is not a medical diagnosis or condition, but rather a laboratory test used in both preclinical research and clinical settings to evaluate various aspects of motor function and balance in animals, including mice and rats. The test is often used to assess the neurological status, sensorimotor function, and coordination abilities of animals following drug treatments, surgical interventions, or in models of neurodegenerative diseases.
In this test, a rodent is placed on a rotating rod with a diameter that allows the animal to comfortably grip it. The rotation speed gradually increases over time, and the researcher records how long the animal can maintain its balance and stay on the rod without falling off. This duration is referred to as the "latency to fall" or "rotarod performance."
The Rotarod performance test offers several advantages, such as its sensitivity to various neurological impairments, ease of use, and ability to provide quantitative data for statistical analysis. It can help researchers evaluate potential therapeutic interventions, monitor disease progression, and investigate the underlying mechanisms of motor function and balance in health and disease.
Neurologic mutant mice are genetically engineered or spontaneously mutated rodents that are used as models to study various neurological disorders and conditions. These mice have specific genetic modifications or mutations that affect their nervous system, leading to phenotypes that resemble human neurological diseases.
Some examples of neurologic mutant mice include:
1. Alzheimer's disease models: Mice that overexpress genes associated with Alzheimer's disease, such as the amyloid precursor protein (APP) or presenilin 1 (PS1), to study the pathogenesis and potential treatments of this disorder.
2. Parkinson's disease models: Mice that have genetic mutations in genes associated with Parkinson's disease, such as alpha-synuclein or parkin, to investigate the mechanisms underlying this condition and develop new therapies.
3. Huntington's disease models: Mice that carry an expanded CAG repeat in the huntingtin gene to replicate the genetic defect seen in humans with Huntington's disease and study disease progression and treatment strategies.
4. Epilepsy models: Mice with genetic mutations that cause spontaneous seizures or increased susceptibility to seizures, used to investigate the underlying mechanisms of epilepsy and develop new treatments.
5. Stroke models: Mice that have surgical induction of stroke or genetic modifications that increase the risk of stroke, used to study the pathophysiology of stroke and identify potential therapeutic targets.
Neurologic mutant mice are essential tools in biomedical research, allowing scientists to investigate the complex interactions between genes and the environment that contribute to neurological disorders. These models help researchers better understand disease mechanisms, develop new therapies, and test their safety and efficacy before moving on to clinical trials in humans.
An allele is a variant form of a gene that is located at a specific position on a specific chromosome. Alleles are alternative forms of the same gene that arise by mutation and are found at the same locus or position on homologous chromosomes.
Each person typically inherits two copies of each gene, one from each parent. If the two alleles are identical, a person is said to be homozygous for that trait. If the alleles are different, the person is heterozygous.
For example, the ABO blood group system has three alleles, A, B, and O, which determine a person's blood type. If a person inherits two A alleles, they will have type A blood; if they inherit one A and one B allele, they will have type AB blood; if they inherit two B alleles, they will have type B blood; and if they inherit two O alleles, they will have type O blood.
Alleles can also influence traits such as eye color, hair color, height, and other physical characteristics. Some alleles are dominant, meaning that only one copy of the allele is needed to express the trait, while others are recessive, meaning that two copies of the allele are needed to express the trait.
A missense mutation is a type of point mutation in which a single nucleotide change results in the substitution of a different amino acid in the protein that is encoded by the affected gene. This occurs when the altered codon (a sequence of three nucleotides that corresponds to a specific amino acid) specifies a different amino acid than the original one. The function and/or stability of the resulting protein may be affected, depending on the type and location of the missense mutation. Missense mutations can have various effects, ranging from benign to severe, depending on the importance of the changed amino acid for the protein's structure or function.
Animal disease models are specialized animals, typically rodents such as mice or rats, that have been genetically engineered or exposed to certain conditions to develop symptoms and physiological changes similar to those seen in human diseases. These models are used in medical research to study the pathophysiology of diseases, identify potential therapeutic targets, test drug efficacy and safety, and understand disease mechanisms.
The genetic modifications can include knockout or knock-in mutations, transgenic expression of specific genes, or RNA interference techniques. The animals may also be exposed to environmental factors such as chemicals, radiation, or infectious agents to induce the disease state.
Examples of animal disease models include:
1. Mouse models of cancer: Genetically engineered mice that develop various types of tumors, allowing researchers to study cancer initiation, progression, and metastasis.
2. Alzheimer's disease models: Transgenic mice expressing mutant human genes associated with Alzheimer's disease, which exhibit amyloid plaque formation and cognitive decline.
3. Diabetes models: Obese and diabetic mouse strains like the NOD (non-obese diabetic) or db/db mice, used to study the development of type 1 and type 2 diabetes, respectively.
4. Cardiovascular disease models: Atherosclerosis-prone mice, such as ApoE-deficient or LDLR-deficient mice, that develop plaque buildup in their arteries when fed a high-fat diet.
5. Inflammatory bowel disease models: Mice with genetic mutations affecting intestinal barrier function and immune response, such as IL-10 knockout or SAMP1/YitFc mice, which develop colitis.
Animal disease models are essential tools in preclinical research, but it is important to recognize their limitations. Differences between species can affect the translatability of results from animal studies to human patients. Therefore, researchers must carefully consider the choice of model and interpret findings cautiously when applying them to human diseases.
Ocular motility disorders refer to a group of conditions that affect the movement of the eyes. These disorders can result from nerve damage, muscle dysfunction, or brain injuries. They can cause abnormal eye alignment, limited range of motion, and difficulty coordinating eye movements. Common symptoms include double vision, blurry vision, strabismus (crossed eyes), nystagmus (involuntary eye movement), and difficulty tracking moving objects. Ocular motility disorders can be congenital or acquired and may require medical intervention to correct or manage the condition.
Repetitive sequences in nucleic acid refer to repeated stretches of DNA or RNA nucleotide bases that are present in a genome. These sequences can vary in length and can be arranged in different patterns such as direct repeats, inverted repeats, or tandem repeats. In some cases, these repetitive sequences do not code for proteins and are often found in non-coding regions of the genome. They can play a role in genetic instability, regulation of gene expression, and evolutionary processes. However, certain types of repeat expansions have been associated with various neurodegenerative disorders and other human diseases.
Neurons, also known as nerve cells or neurocytes, are specialized cells that constitute the basic unit of the nervous system. They are responsible for receiving, processing, and transmitting information and signals within the body. Neurons have three main parts: the dendrites, the cell body (soma), and the axon. The dendrites receive signals from other neurons or sensory receptors, while the axon transmits these signals to other neurons, muscles, or glands. The junction between two neurons is called a synapse, where neurotransmitters are released to transmit the signal across the gap (synaptic cleft) to the next neuron. Neurons vary in size, shape, and structure depending on their function and location within the nervous system.
Huntington Disease (HD) is a genetic neurodegenerative disorder that affects both cognitive and motor functions. It is characterized by the progressive loss of neurons in various areas of the brain, particularly in the striatum and cortex. The disease is caused by an autosomal dominant mutation in the HTT gene, which codes for the huntingtin protein. The most common mutation is a CAG repeat expansion in this gene, leading to the production of an abnormal form of the huntingtin protein that is toxic to nerve cells.
The symptoms of HD typically appear between the ages of 30 and 50, but they can start earlier or later in life. The early signs of HD may include subtle changes in mood, cognition, and coordination. As the disease progresses, individuals with HD experience uncontrolled movements (chorea), emotional disturbances, cognitive decline, and difficulties with communication and swallowing. Eventually, they become dependent on others for their daily needs and lose their ability to walk, talk, and care for themselves.
There is currently no cure for HD, but medications and therapies can help manage the symptoms of the disease and improve quality of life. Genetic testing is available to confirm the diagnosis and provide information about the risk of passing the disease on to future generations.
The brainstem is the lower part of the brain that connects to the spinal cord. It consists of the midbrain, pons, and medulla oblongata. The brainstem controls many vital functions such as heart rate, breathing, and blood pressure. It also serves as a relay center for sensory and motor information between the cerebral cortex and the rest of the body. Additionally, several cranial nerves originate from the brainstem, including those that control eye movements, facial movements, and hearing.
Licensure is the process by which a government regulatory agency grants a license to a physician (or other healthcare professional) to practice medicine (or provide healthcare services) in a given jurisdiction. The licensing process typically requires the completion of specific educational and training requirements, passing written and/or practical exams, and meeting other state-specific criteria.
The purpose of licensure is to ensure that healthcare professionals meet minimum standards of competence and safety in order to protect the public. Licensure laws vary by state, so a physician who is licensed to practice medicine in one state may not be able to practice in another state without obtaining additional licensure.
A haplotype is a group of genes or DNA sequences that are inherited together from a single parent. It refers to a combination of alleles (variant forms of a gene) that are located on the same chromosome and are usually transmitted as a unit. Haplotypes can be useful in tracing genetic ancestry, understanding the genetic basis of diseases, and developing personalized medical treatments.
In population genetics, haplotypes are often used to study patterns of genetic variation within and between populations. By comparing haplotype frequencies across populations, researchers can infer historical events such as migrations, population expansions, and bottlenecks. Additionally, haplotypes can provide information about the evolutionary history of genes and genomic regions.
In clinical genetics, haplotypes can be used to identify genetic risk factors for diseases or to predict an individual's response to certain medications. For example, specific haplotypes in the HLA gene region have been associated with increased susceptibility to certain autoimmune diseases, while other haplotypes in the CYP450 gene family can affect how individuals metabolize drugs.
Overall, haplotypes provide a powerful tool for understanding the genetic basis of complex traits and diseases, as well as for developing personalized medical treatments based on an individual's genetic makeup.
Molecular diagnostic techniques are a group of laboratory methods used to analyze biological markers in DNA, RNA, and proteins to identify specific health conditions or diseases at the molecular level. These techniques include various methods such as polymerase chain reaction (PCR), DNA sequencing, gene expression analysis, fluorescence in situ hybridization (FISH), and mass spectrometry.
Molecular diagnostic techniques are used to detect genetic mutations, chromosomal abnormalities, viral and bacterial infections, and other molecular changes associated with various diseases, including cancer, genetic disorders, infectious diseases, and neurological disorders. These techniques provide valuable information for disease diagnosis, prognosis, treatment planning, and monitoring of treatment response.
Compared to traditional diagnostic methods, molecular diagnostic techniques offer several advantages, such as higher sensitivity, specificity, and speed. They can detect small amounts of genetic material or proteins, even in early stages of the disease, and provide accurate results with a lower risk of false positives or negatives. Additionally, molecular diagnostic techniques can be automated, standardized, and performed in high-throughput formats, making them suitable for large-scale screening and research applications.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are a type of calcium ion channel found in the endoplasmic reticulum (ER) membrane of many cell types. They play a crucial role in intracellular calcium signaling and are activated by the second messenger molecule, inositol 1,4,5-trisphosphate (IP3).
IP3 is produced by enzymatic cleavage of the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) in response to extracellular signals such as hormones and neurotransmitters. When IP3 binds to the IP3R, it triggers a conformational change that opens the channel, allowing calcium ions to flow from the ER into the cytosol. This increase in cytosolic calcium can then activate various cellular processes such as gene expression, protein synthesis, and cell survival or death pathways.
There are three isoforms of IP3Rs (IP3R1, IP3R2, and IP3R3) that differ in their tissue distribution, regulation, and sensitivity to IP3. Dysregulation of IP3R-mediated calcium signaling has been implicated in various pathological conditions, including neurological disorders, cardiovascular diseases, and cancer.
Calcium channels are specialized proteins that span the membrane of cells and allow calcium ions (Ca²+) to flow in and out of the cell. They are crucial for many physiological processes, including muscle contraction, neurotransmitter release, hormone secretion, and gene expression.
There are several types of calcium channels, classified based on their biophysical and pharmacological properties. The most well-known are:
1. Voltage-gated calcium channels (VGCCs): These channels are activated by changes in the membrane potential. They are further divided into several subtypes, including L-type, P/Q-type, N-type, R-type, and T-type. VGCCs play a critical role in excitation-contraction coupling in muscle cells and neurotransmitter release in neurons.
2. Receptor-operated calcium channels (ROCCs): These channels are activated by the binding of an extracellular ligand, such as a hormone or neurotransmitter, to a specific receptor on the cell surface. ROCCs are involved in various physiological processes, including smooth muscle contraction and platelet activation.
3. Store-operated calcium channels (SOCCs): These channels are activated by the depletion of intracellular calcium stores, such as those found in the endoplasmic reticulum. SOCCs play a critical role in maintaining calcium homeostasis and signaling within cells.
Dysregulation of calcium channel function has been implicated in various diseases, including hypertension, arrhythmias, migraine, epilepsy, and neurodegenerative disorders. Therefore, calcium channels are an important target for drug development and therapy.
Genetically modified animals (GMAs) are those whose genetic makeup has been altered using biotechnological techniques. This is typically done by introducing one or more genes from another species into the animal's genome, resulting in a new trait or characteristic that does not naturally occur in that species. The introduced gene is often referred to as a transgene.
The process of creating GMAs involves several steps:
1. Isolation: The desired gene is isolated from the DNA of another organism.
2. Transfer: The isolated gene is transferred into the target animal's cells, usually using a vector such as a virus or bacterium.
3. Integration: The transgene integrates into the animal's chromosome, becoming a permanent part of its genetic makeup.
4. Selection: The modified cells are allowed to multiply, and those that contain the transgene are selected for further growth and development.
5. Breeding: The genetically modified individuals are bred to produce offspring that carry the desired trait.
GMAs have various applications in research, agriculture, and medicine. In research, they can serve as models for studying human diseases or testing new therapies. In agriculture, GMAs can be developed to exhibit enhanced growth rates, improved disease resistance, or increased nutritional value. In medicine, GMAs may be used to produce pharmaceuticals or other therapeutic agents within their bodies.
Examples of genetically modified animals include mice with added genes for specific proteins that make them useful models for studying human diseases, goats that produce a human protein in their milk to treat hemophilia, and pigs with enhanced resistance to certain viruses that could potentially be used as organ donors for humans.
It is important to note that the use of genetically modified animals raises ethical concerns related to animal welfare, environmental impact, and potential risks to human health. These issues must be carefully considered and addressed when developing and implementing GMA technologies.
The spinal cord is a major part of the nervous system, extending from the brainstem and continuing down to the lower back. It is a slender, tubular bundle of nerve fibers (axons) and support cells (glial cells) that carries signals between the brain and the rest of the body. The spinal cord primarily serves as a conduit for motor information, which travels from the brain to the muscles, and sensory information, which travels from the body to the brain. It also contains neurons that can independently process and respond to information within the spinal cord without direct input from the brain.
The spinal cord is protected by the bony vertebral column (spine) and is divided into 31 segments: 8 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 1 coccygeal. Each segment corresponds to a specific region of the body and gives rise to pairs of spinal nerves that exit through the intervertebral foramina at each level.
The spinal cord is responsible for several vital functions, including:
1. Reflexes: Simple reflex actions, such as the withdrawal reflex when touching a hot surface, are mediated by the spinal cord without involving the brain.
2. Muscle control: The spinal cord carries motor signals from the brain to the muscles, enabling voluntary movement and muscle tone regulation.
3. Sensory perception: The spinal cord transmits sensory information, such as touch, temperature, pain, and vibration, from the body to the brain for processing and awareness.
4. Autonomic functions: The sympathetic and parasympathetic divisions of the autonomic nervous system originate in the thoracolumbar and sacral regions of the spinal cord, respectively, controlling involuntary physiological responses like heart rate, blood pressure, digestion, and respiration.
Damage to the spinal cord can result in various degrees of paralysis or loss of sensation below the level of injury, depending on the severity and location of the damage.
Genetic testing is a type of medical test that identifies changes in chromosomes, genes, or proteins. The results of a genetic test can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Genetic tests are performed on a sample of blood, hair, skin, amniotic fluid (the fluid that surrounds a fetus during pregnancy), or other tissue. For example, a physician may recommend genetic testing to help diagnose a genetic condition, confirm the presence of a gene mutation known to increase the risk of developing certain cancers, or determine the chance for a couple to have a child with a genetic disorder.
There are several types of genetic tests, including:
* Diagnostic testing: This type of test is used to identify or confirm a suspected genetic condition in an individual. It may be performed before birth (prenatal testing) or at any time during a person's life.
* Predictive testing: This type of test is used to determine the likelihood that a person will develop a genetic disorder. It is typically offered to individuals who have a family history of a genetic condition but do not show any symptoms themselves.
* Carrier testing: This type of test is used to determine whether a person carries a gene mutation for a genetic disorder. It is often offered to couples who are planning to have children and have a family history of a genetic condition or belong to a population that has an increased risk of certain genetic disorders.
* Preimplantation genetic testing: This type of test is used in conjunction with in vitro fertilization (IVF) to identify genetic changes in embryos before they are implanted in the uterus. It can help couples who have a family history of a genetic disorder or who are at risk of having a child with a genetic condition to conceive a child who is free of the genetic change in question.
* Pharmacogenetic testing: This type of test is used to determine how an individual's genes may affect their response to certain medications. It can help healthcare providers choose the most effective medication and dosage for a patient, reducing the risk of adverse drug reactions.
It is important to note that genetic testing should be performed under the guidance of a qualified healthcare professional who can interpret the results and provide appropriate counseling and support.
Chromosome mapping, also known as physical mapping, is the process of determining the location and order of specific genes or genetic markers on a chromosome. This is typically done by using various laboratory techniques to identify landmarks along the chromosome, such as restriction enzyme cutting sites or patterns of DNA sequence repeats. The resulting map provides important information about the organization and structure of the genome, and can be used for a variety of purposes, including identifying the location of genes associated with genetic diseases, studying evolutionary relationships between organisms, and developing genetic markers for use in breeding or forensic applications.
Single-stranded DNA breaks (SSBs) refer to a type of DNA damage in which one strand of the double-helix structure is cleaved or broken. This kind of damage can occur spontaneously due to cellular metabolism or can be induced by various genotoxic agents, such as ionizing radiation and certain chemicals.
SSBs are typically repaired rapidly and efficiently by enzymes known as DNA repair proteins. However, if left unrepaired or misrepaired, they can lead to mutations, genomic instability, and increased risk of diseases, including cancer. In some cases, single-stranded breaks may also precede the formation of more severe double-stranded DNA breaks (DSBs).
It is important to note that while SSBs are less catastrophic than DSBs, they still play a significant role in genome maintenance and cellular health.
The TATA-box binding protein (TBP) is a general transcription factor that plays a crucial role in the initiation of transcription of protein-coding genes in archaea and eukaryotes. It is named after its ability to bind to the TATA box, a conserved DNA sequence found in the promoter regions of many genes.
TBP is a key component of the transcription preinitiation complex (PIC), which also includes other general transcription factors and RNA polymerase II in eukaryotes. The TBP protein has a unique structure, characterized by a saddle-shaped DNA-binding domain that allows it to recognize and bind to the TATA box in a sequence-specific manner.
By binding to the TATA box, TBP helps to position the RNA polymerase II complex at the start site of transcription, allowing for the initiation of RNA synthesis. TBP also plays a role in regulating gene expression by interacting with various coactivators and corepressors that modulate its activity.
Mutations in the TBP gene have been associated with several human diseases, including some forms of cancer and neurodevelopmental disorders.
 Spinocerebellar ataxia
Spinocerebellar ataxia
Spinocerebellar ataxia type-13
Spinocerebellar ataxia type 1
Spinocerebellar ataxia type 6
X-linked sideroblastic anemia and spinocerebellar ataxia
TTBK2
Autosomal dominant cerebellar ataxia
Cerebellar ataxia
Inferior olivary nucleus
Rubicon (protein)
Dysmetria
Repeated sequence (DNA)
John David Spillane
Machado-Joseph disease
Cav2.1
Cerebellar degeneration
Ataxin 1
QRICH1
KCNC3
Ataxin-2
Twinkle (protein)
Vocal cord paresis
Cerebellum
Trinucleotide repeat disorder
Ataxia
Tocopherol
Health of Abraham Lincoln
SPTBN2
SCYL1
Oculomotor apraxia
 The spinocerebellar ataxias
The spinocerebellar ataxias
Spinocerebellar ataxia - Wikipedia
 Spinocerebellar ataxia type 6: MedlinePlus Genetics
Spinocerebellar ataxia type 6: MedlinePlus Genetics
 Spinocerebellar Ataxia - National Organization for Rare Disorders
Spinocerebellar Ataxia - National Organization for Rare Disorders
 Identification of biomarkers of Spinocerebellar Ataxia Type 3 - National Ataxia Foundation
Identification of biomarkers of Spinocerebellar Ataxia Type 3 - National Ataxia Foundation
 Neurocognitive and cerebellar function in ADHD, autism and spinocerebellar ataxia | Lund University Publications
Neurocognitive and cerebellar function in ADHD, autism and spinocerebellar ataxia | Lund University Publications
 SciELO - Brazil - Patients with autosomal dominant spinocerebellar ataxia have more risk of falls, important balance...
SciELO - Brazil - Patients with autosomal dominant spinocerebellar ataxia have more risk of falls, important balance...
 "Spinocerebellar Ataxia With a Novel Chromosomal Defect: Del 5q34 - A c" by AYŞE FİLİZ KOÇ, YAKUP SARICA et al.
"Spinocerebellar Ataxia With a Novel Chromosomal Defect: Del 5q34 - A c" by AYŞE FİLİZ KOÇ, YAKUP SARICA et al.
Anemia sideroblastic and spinocerebellar ataxia - CheckOrphan
 USF study: Smoking cessation drug improves walking function in patients with spinocerebellar ataxia - USF Health NewsUSF Health...
USF study: Smoking cessation drug improves walking function in patients with spinocerebellar ataxia - USF Health NewsUSF Health...
 Childhood-onset autosomal recessive slowly progressive spinocerebellar ataxia - Living with the Disease - Genetic and Rare...
Childhood-onset autosomal recessive slowly progressive spinocerebellar ataxia - Living with the Disease - Genetic and Rare...
 Spino Cerebellar Ataxia Type 3 Archives - The Stratford Observer
Spino Cerebellar Ataxia Type 3 Archives - The Stratford Observer
 Biohaven Completes Randomization In Phase 2/3 Trial In Spinocerebellar Ataxia: Expected Topline Data Advanced To Fourth Quarter...
Biohaven Completes Randomization In Phase 2/3 Trial In Spinocerebellar Ataxia: Expected Topline Data Advanced To Fourth Quarter...
 Spinocerebellar ataxia (NORD): Video & Anatomy | Osmosis
Spinocerebellar ataxia (NORD): Video & Anatomy | Osmosis
 Spinocerebellar Ataxias - MeSH - NCBI
Spinocerebellar Ataxias - MeSH - NCBI
 Altmetric - Phenotype variability and early onset ataxia symptoms in spinocerebellar ataxia type 7: comparison and correlation...
Altmetric - Phenotype variability and early onset ataxia symptoms in spinocerebellar ataxia type 7: comparison and correlation...
 Spinocerebellar Ataxia Type 2 via the ATXN2 CAG Repeat Expansion Test - PreventionGenetics
Spinocerebellar Ataxia Type 2 via the ATXN2 CAG Repeat Expansion Test - PreventionGenetics
 Spinocerebellar Ataxia Archives - Global Genes
Spinocerebellar Ataxia Archives - Global Genes
 Spinocerebellar ataxia - Medical Cannabis Doctors
Spinocerebellar ataxia - Medical Cannabis Doctors
Evolution of Outcome Measures in Spinocerebellar Ataxias: centogene.com
 Spinocerebellar ataxia 23 | Rare Diseases | RareGuru
Spinocerebellar ataxia 23 | Rare Diseases | RareGuru
Hong Kong Spinocerebellar Ataxia Association (HKSCAA)
 Spinocerebellar ataxia (SCA) type 12 - Bioarray
Spinocerebellar ataxia (SCA) type 12 - Bioarray
 Spinocerebellar Ataxia Archives | Canine Genetic Testing
Spinocerebellar Ataxia Archives | Canine Genetic Testing
Spinocerebellar ataxia (SCA) type 2 - Bioarray
 Spinocerebellar Ataxia Precision Panel - Middle East
Spinocerebellar Ataxia Precision Panel - Middle East
Hong Kong Spinocerebellar Ataxia Association (HKSCAA)
 spinocerebellar ataxia - Inspire - Women of Dartmouth Stories
spinocerebellar ataxia - Inspire - Women of Dartmouth Stories
 Spinocerebellar Ataxia (SCA): An Overview | Syndromes: Rapid Recognition and Perioperative Implications, 2e |...
Spinocerebellar Ataxia (SCA): An Overview | Syndromes: Rapid Recognition and Perioperative Implications, 2e |...
Spinocerebellar ataxia 46 (Concept Id: C4540404)
- MedGen - NCBI
National Ataxia Foundation2
- We would like to thank and acknowledge all of those who are working with us, including the patients who enrolled in this trial, their families and caregivers, the National Ataxia Foundation, and the many academic clinicians, investigators and trial sites who have helped us to advance trigriluzole. (businessinsider.com)
- Damaris receives Young Investigator Award for Spinocerebellar Ataxia Research from the National Ataxia Foundation. (lorenzolab.org)
SCAs11
- The spinocerebellar ataxias (SCAs) are diseases characterized by the progressive degeneration and subsequent loss of neurons accompanied by reactive gliosis, degeneration of fibers from the deteriorating neurons, and clinical symptoms reflecting the locations of the lost neurons. (nih.gov)
- There are numerous types of autosomal-dominant cerebellar ataxias There are five typical autosomal recessive disorders in which ataxia is a prominent feature A few SCAs remain unspecified and can not be precisely diagnosed, but in the last decade[as of? (wikipedia.org)
- To study the long-term evolution of patient-reported outcome measures (PROMs) in the most common spinocerebellar ataxias (SCAs), we analyzed 8 years follow-up data of the EUROSCA Natural History Study, a cohort study of 526 patients with SCA1, SCA2, SCA3 and SCA6. (centogene.com)
- The wide spectrum of spinocerebellar ataxias (SCAs). (igenomix.net)
- The autosomal dominant cerebellar degenerative disorders are generally referred to as 'spinocerebellar ataxias,' (SCAs) even though 'spinocerebellar' is a hybrid term, referring to both clinical signs and neuroanatomical regions (Margolis, 2003). (beds.ac.uk)
- Neuropathologists have defined SCAs as cerebellar ataxias with variable involvement of the brainstem and spinal cord, and the clinical features of the disorders are caused by degeneration of the cerebellum and its afferent and efferent connections, which involve the brainstem and spinal cord (Schols et al. (beds.ac.uk)
- Introduction: The aim of this study is to reappraise the progression of the five most common spinocerebellar ataxias (SCAs) in the Chinese population and to establish a much-needed critical comparison with that in other ethnic groups. (tmu.edu.tw)
- Therefore, this study demonstrated multiple neurochemical alterations in SCAs and MSA-C relative to controls and the potential for these neurochemical levels to differentiate ataxia types. (umn.edu)
- Neurodegenerative disorders such as for example spinocerebellar ataxias (SCAs) and Alzheimer's disease (AD) represent an enormous medical and medical question however the molecular mechanisms of the diseases remain not yet determined. (ampkpathway.com)
- ataxias Spinocerebellar ataxias (SCAs) represent several intensifying hereditary neurodegenerative illnesses that change from one another in clinical demonstration and hereditary basis. (ampkpathway.com)
- Spinocerebellar ataxias (SCAs) are the main autosomal dominant ataxias. (msdmanuals.com)
SCA36
- Subjects diagnosed with Spinocerebellar Ataxia Type 3 (SCA3) and healthy controls will be asked to undergo three lumbar punctures to donate cerebral spinal fluid and three blood draws to obtain samples for the purposes of our study. (ataxia.org)
- Several neuropsychiatric disorders such as attention deficit-hyperactivity disorder (ADHD), autism spectrum disorder (ASD), as well as neurological diseases such as spinocerebellar ataxia type 3 (SCA3) are associated with differences in cerebellar function. (lu.se)
- The randomized controlled clinical trial investigated the effectiveness of varenicline (Chantix®) in treating spinocerebellar ataxia type 3, or SCA3. (usf.edu)
- Tsimpanouli et al ask h ow neural circuits function differenetly during sleep in a model of SCA3-type spinocerebellar ataxia? (google.com)
- Methods: Patients with SCA1, SCA2, SCA3, SCA6 or SCA17 were consecutively assessed using the scale for the assessment and rating of ataxia (SARA) for five years. (tmu.edu.tw)
- Despite undeniable progresses in the knowledge concerning the molecular pathology of Machado- Joseph disease (MJD)/Spinocerebellar ataxia type 3 (SCA3), therapeutic compounds remain to be discovered and validated. (ataxia.org)
SCA12
- The autosomal dominant ataxias are, however, most often referred to as the spinocerebellar ataxias, identified as SCA1 through SCA45. (mhmedical.com)
- Spinocerebellar ataxia type 1 (SCA1) is characterized by progressive cerebellar ataxia, dysarthria, and eventual deterioration of bulbar functions. (beds.ac.uk)
Characterized by cerebellar ataxia3
- Clinically characterized by cerebellar ataxia of gait and limbs, invariably associated with supranuclear ophthalmoplegia, pyramidal or extrapyramidal signs, mild dementia, and peripheral neuropathy. (mhmedical.com)
- ADCA I was characterized by cerebellar ataxia in combination with various associated neurologic features, such as ophthalmoplegia, pyramidal and extrapyramidal signs, peripheral neuropathy, and dementia, among others. (beds.ac.uk)
- The spinocerebellar ataxia type 17 (SCA17) is characterized by cerebellar ataxia, dementia, and involuntary movements, including chorea and dystonia. (uni-luebeck.de)
Function in patients with spinocerebellar ataxia1
- To assess balance and ability to function in patients with spinocerebellar ataxia. (scielo.br)
Type17
- The symptoms of an ataxia vary with the specific type and with the individual patient. (wikipedia.org)
- Spinocerebellar ataxia type 6 (SCA6) is a condition characterized by progressive problems with movement. (medlineplus.gov)
- Casey HL, Gomez CM. Spinocerebellar Ataxia Type 6. (medlineplus.gov)
- Spinocerebellar ataxia type 2 (SCA2) is an autosomal dominant form of SCA. (preventiongenetics.com)
- Table S-2 reports the autosomal dominant hereditary ataxias and each type is indicated as SCA#. (mhmedical.com)
- A type of ataxia characterized by the inability to carry out movements with the correct range and motion across the plane of more than one joint related to incorrect estimation of the distances required for targeted movements. (nih.gov)
- A type of ataxia characterized by the impairment of the ability to coordinate the movements required for normal walking. (nih.gov)
- Genotype-phenotype correlations, dystonia and disease progression in spinocerebellar ataxia type 14. (ox.ac.uk)
- Spinocerebellar ataxia type 35 (SCA35) is a rare, autosomal dominant neurodegenerative disorder associated with mutations in TGM6 gene that encode the protein transglutaminase 6 (TG6). (ataxia.org)
- Profiles of 10-13 neurochemical concentrations obtained in vivo by high field proton magnetic resonance spectroscopy ( 1 H MRS) can potentially provide ataxia-type specific biomarkers. (umn.edu)
- Background: Spinocerebellar ataxia type 3 (SCA) is a cerebellum-dominant degenerative disorder that is characterized primarily by infratentorial damage, although less severe supratentorial involvement may contribute to the clinical manifestation. (figshare.com)
- Dominant Mutations in GRM1 Cause Spinocerebellar Ataxia Type 44. (ox.ac.uk)
- This test is for the assessment of one type of the specified spinocerebellar ataxias (SCA), including types 1, 2, 3, 6, or 7. (mayocliniclabs.com)
- 1.1 Spinocerebellar ataxia type 2 pathogenesis In this section we will discuss SCA pathogenesis by the example of SCA2. (ampkpathway.com)
- Zühlke, C & Bürk, K 2007, ' Spinocerebellar ataxia type 17 is caused by mutations in the TATA-box binding protein ', Cerebellum , vol. 6, no. 4, pp. 300-307. (uni-luebeck.de)
- spinocerebellar ataxia type 34 [SCA34]) (OMIM # 133190). (medscape.com)
- Psychological aspects of presymptomatic diagnosis of spinocerebellar Ataxia type 2 in Cuba. (bvsalud.org)
Hereditary ataxias4
- citation needed] The hereditary ataxias are categorized by mode of inheritance and causative gene or chromosomal locus. (wikipedia.org)
- The hereditary ataxias can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. (wikipedia.org)
- There are fewer autosomal recessive ataxias than autosomal dominant hereditary ataxias. (mhmedical.com)
- Hereditary ataxias may be autosomal recessive or autosomal dominant. (msdmanuals.com)
SCA25
- Testing for ATXN2 repeat expansions is recommended for individuals suspected to have SCA2 (presentation of progressive ataxia and dysarthria, nystagmus, and slow saccadic eye movement) in addition to a family history consistent with autosomal dominant inheritance. (preventiongenetics.com)
- Expansion of the poly-glutamine track in ATXN2 has been associated with several conditions including spinocerebellar ataxia 2 (SCA2), amyotrophic lateral sclerosis (ALS), and progressive encephalopathy with autonomic dysfunction (Pulst. (preventiongenetics.com)
- All individuals with SCA2 demonstrate cerebellar dysfunction characterized by progressive ataxia and dysarthria (Pulst et al. (preventiongenetics.com)
- SCA2 is the second most common subtype of autosomal dominant cerebellar ataxias worldwide. (preventiongenetics.com)
- Spinocerebellar ataxia aype-2 (SCA2), the most common SCA of India, is caused by expansion of uninterrupted CAG triplet repeats in first exon of ATXN2 gene. (mdsabstracts.org)
Dysarthria3
- Interruption of afferent and efferent connections within the spinocerebellar system results in ataxic gait, scanning dysarthria, explosive speech, intention tremor, dysdiadochokinesia, dysmetria, and abnormalities of eye movements. (mhmedical.com)
- This Mirabegron disorder is accompanied by a wide spectrum of severe clinical symptoms such as ataxia of gait and stance ataxia of limb movements dysarthria ophthalmoplegia pyramidal and extrapyramidal disorders muscular rigidity and other severe neurological symptoms [2-4]. (ampkpathway.com)
- it is followed by upper-extremity ataxia, dysarthria, and paresis, particularly of the lower extremities. (msdmanuals.com)
Gait ataxia1
- Gait ataxia is characteirzed by a wide-based staggering gait with a tendency to fall. (nih.gov)
Symptoms5
- As with other forms of ataxia, SCA frequently results in atrophy of the cerebellum, loss of fine coordination of muscle movements leading to unsteady and clumsy motion, and other symptoms. (wikipedia.org)
- This is the first clinical trial in patients with ataxia showing that nicotinic acetycholine agonists improve symptoms associated with the ability to stand straight and walk," said Dr. Zesiewicz, professor of neurology and director of the USF Ataxia Research Center. (usf.edu)
- The SARA is an eight-item clinician-administered semi-qualitative performance-based assessment of cerebellar ataxia symptoms that measures impairment on a scale of zero to 40, with a higher score indicating more severe ataxia. (businessinsider.com)
- Severity of ataxia was assessed using the Scale for the Assessment and Rating of Ataxia (SARA) and neurological symptoms other than ataxia with the Inventory of Non-Ataxia Signs (INAS). (centogene.com)
- Symptoms vary with the cause but typically include ataxia (impaired muscle coordination). (msdmanuals.com)
Atrophy1
- Synonyms for autosomal-dominant cerebellar ataxias (ADCA) used prior to the current understanding of the molecular genetics were Marie's ataxia, inherited olivopontocerebellar atrophy, cerebello-olivary atrophy, or the more generic term "spinocerebellar degeneration. (wikipedia.org)
Cerebellum4
- Spinocerebellar ataxias (SCA) comprise a group of autosomal dominant neurodegenerative disorders with involvement of the cerebellum and its afferent and efferent pathways 1 1. (scielo.br)
- Spinocerebellar ataxia (SCA) is a group of neurological disorders that affect the cerebellum, a part of the brain that controls movement, coordination, and balance. (cannabisdoctors.com)
- Hereditary and sporadic neurodegenerative ataxias are movement disorders that affect the cerebellum. (umn.edu)
- Cerebellum & ataxias , 2020;7:11. (afpm.org.my)
Phenotype3
- Spinocerebellar ataxia: relationship between phenotype and genotype - a review. (igenomix.net)
- Their clinical features most closely resemble the phenotype of a single consanguineous Japanese family with four individuals affected by spinocerebellar ataxia, peripheral neuropathy, raised AFP and hypercholesterolaemia. (ox.ac.uk)
- Fifteen patients with SCA 3 had detailed clinical phenotype evaluation using Inventory for Non -Ataxia Signs (INAS) and Ataxia Severity evaluation using the Scale for Assessment and Rating of Ataxia (SARA). (afpm.org.my)
Pathogenesis1
- The main goal of our project is the development of a powerful genetic model to investigate pathogenesis of spinocerebellar ataxia with axonal neuropathy-1 (SCAN-1) disease. (ataxia.org)
Espinocerebelosas1
- La expansión de repeticiones del trinucleótido CAG en los genes que codifican Ataxinas se asocia con las ATAXIAS ESPINOCEREBELOSAS (AEC). (bvsalud.org)
Clinical8
- A nicotinic drug approved for smoking cessation significantly improved the walking ability of patients suffering from an inherited form of ataxia, reports a new clinical study led by University of South Florida researchers. (usf.edu)
- Further preclinical research is needed to understand how nicotinic acetylcholine agonists improve ataxia, and larger clinical studies with more patients are needed to identify other neurodegenerative diseases that may benefit from nicotinic medications, the authors conclude. (usf.edu)
- A group of predominately late-onset, cerebellar ataxias which have been divided into multiple subtypes based on clinical features and genetic mapping. (beds.ac.uk)
- Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. (igenomix.net)
- Historically, Harding (1982) proposed a clinical classification for autosomal dominant cerebellar ataxias (ADCAs). (beds.ac.uk)
- Although they all had raised serum AFP levels, their clinical, immunological, biochemical, cytogenetic and molecular genetic studies failed to support a diagnosis of Ataxia Telangiectasia. (ox.ac.uk)
- In addition, such biomarkers may help discriminate between ataxia subtypes because these diseases display substantial overlap in clinical presentation and conventional MRI. (umn.edu)
- Studies with higher numbers of patients and other ataxias are warranted to further investigate the clinical utility of neurochemical levels as measured by high-field MRS as ataxia biomarkers. (umn.edu)
Friedreich3
- citation needed] There are five typical autosomal-recessive disorders in which ataxia is a prominent feature: Friedreich ataxia, ataxia-telangiectasia, ataxia with vitamin E deficiency, ataxia with oculomotor apraxia (AOA), spastic ataxia. (wikipedia.org)
- Previously, all autosomal dominant ataxias were called Marie ataxia and all autosomal recessive ataxias were called Friedreich ataxia. (mhmedical.com)
- Autosomal recessive ataxias include Friedreich ataxia (the most prevalent), ataxia-telangiectasia, abetalipoproteinemia, ataxia with isolated vitamin E deficiency, and cerebrotendinous xanthomatosis. (msdmanuals.com)
Genes1
- The expansion of CAG trinucleotide repeats in genes that encode Ataxins is associated with SPINOCEREBELLAR ATAXIAS (SCA). (bvsalud.org)
Neurological5
- Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. (wikipedia.org)
- Spinocerebellar degeneration is a rare inherited neurological disorder of the central nervous system characterized by the slow degeneration of certain areas of the brain. (wikipedia.org)
- The uncoordinated movements, or ataxia, is a neurological symptom with no treatment or cure and can lead to serious fall-related injuries. (usf.edu)
- Spinocerebellar ataxia represents a group of slow and progressive neurodegenerative diseases of varying inherited degrees of rarity, which is in contrast to a related group of neurological disorders that are acquired following traumatic injuries or other external agents. (mhmedical.com)
- Acknowledgements This work was supported by the National Institute of Neurological Disorders and Stroke grant R21 NS056172 (GÖ) and Jay D. Schlueter Ataxia Research Fund. (umn.edu)
Genetic6
- Spinocerebellar ataxia (SCA) is one of a group of genetic disorders characterized by slowly progressive incoordination of gait and is often associated with poor coordination of hands, speech, and eye movements. (wikipedia.org)
- citation needed] Many types of autosomal dominant cerebellar ataxias for which specific genetic information is available are now known. (wikipedia.org)
- In 2008, a genetic ataxia blood test developed to test for 12 types of SCA, Friedreich's ataxia, and several others. (wikipedia.org)
- Spinocerebellar A taxia (SCA) refers to a heterogeneous gro up of progressive neurodegenerative diseases of genetic origin. (igenomix.net)
- Extensive investigation including imaging, biochemical and genetic studies excluded other known ataxias. (ox.ac.uk)
- Genetic Variant in GRM1 Underlies Congenital Cerebellar Ataxia with No Obvious Intellectual Disability. (nih.gov)
Movements4
- Balance disturbance in ataxias results in increased postural sway, excessive or reduced response to disturbances, poor balance control during body movements, and unusual body oscillation. (scielo.br)
- A rare inherited condition characterized by anemia at birth as well as spinocerebellar ataxia (impaired ability to control voluntary movements). (checkorphan.org)
- Spinocerebellar ataxia impairs the brain and spinal cord causing progressive difficulty with coordination of movements, including walking. (usf.edu)
- A kind of ataxia that affects movements of the extremities. (nih.gov)
ADCA2
- ADCA II was characterized by the cerebellar ataxia, associated neurologic features, and the additional findings of macular and retinal degeneration. (beds.ac.uk)
- ADCA III was a pure form of late-onset cerebellar ataxia without additional features. (beds.ac.uk)
Severe ataxia1
- Furthermore, the more severe ataxia is, the more compromised are postural balance, risk of falls, and ability to function. (scielo.br)
Disorders1
- Heterogeneous group of disorders in which progressive cerebellar ataxia is the primary feature. (mhmedical.com)
Patients5
- A total of 44 patients with different spinocerebellar ataxia types 1, 2, 3, and 6 were evaluated using the Tinetti balance and gait assessment and the functional independence measure. (scielo.br)
- Patients with spinocerebellar ataxia have important balance impairment and risk of falls that influence the ability to function such as self-care, transfers, and locomotion. (scielo.br)
- NEW HAVEN, Conn. , Aug. 7, 2017 /PRNewswire/ -- Biohaven Pharmaceutical Holding Company Ltd. (NYSE: BHVN) announced today that it has commenced dosing of all 141 randomized patients with spinocerebellar ataxia (SCA) in its Phase 2/3 trial of trigriluzole (previously known as BHV-4157). (businessinsider.com)
- Connect with other caregivers and patients with Spinocerebellar ataxia 23 and get the support you need. (rareguru.com)
- We describe three patients from two families with progressive spinocerebellar ataxia, peripheral neuropathy, raised alpha-fetoprotein (AFP) and hypercholesterolaemia. (ox.ac.uk)
Diseases1
- Spinocerebellar ataxias (SCA) are highly heterogenous group of neurodegenerative diseases causing progressive cerebellar dysfunction. (afpm.org.my)
Neurologic1
- The development of ataxia is a neurologic sign that may provide a clue to the nature of the underlying disorder. (mhmedical.com)
Degeneration1
- There are three forms of spinocerebellar degeneration: Types 1, 2, 3. (wikipedia.org)
Biomarkers1
- Robust and objective biomarkers are critical for treatment trials of ataxias. (umn.edu)
Diagnosis2
- An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. (wikipedia.org)
- The Igenomix Spinocerebellar Ataxia Precision Panel can be used as a tool for an accurate diagnosis and differential diagnosis of loss of balance and coordination ultimately leading to a better management and prognosis of the disease. (igenomix.net)
Disease2
Late-onset1
- A pure form of late-onset cerebellar ataxia. (mhmedical.com)
Funcional4
- Avaliar o equilíbrio e a capacidade funcional em pacientes com ataxia espinocerebelar. (scielo.br)
- Quarenta e quatro pacientes com diferentes tipos de ataxia espinocerebelar foram avaliados usando os testes de equilíbrio e de marcha de Tinetti, e da Medida de Independência Funcional. (scielo.br)
- Os pacientes com ataxia espinocerebelar possuem comprometimento importante do equilíbrio e risco de quedas, que influenciam a capacidade funcional, como por exemplo: auto-cuidado, transferências e locomoção. (scielo.br)
- Além disso, quanto mais grave a ataxia, maior o comprometimento do equilíbrio postural, do risco de quedas, e da capacidade funcional. (scielo.br)
Enfermedad1
- En pacientes de AEC, el número de repeticiones de CAG se correlaciona con la gravedad de la enfermedad e inversamente correlacionada con la edad de aparición de la enfermedad. (bvsalud.org)