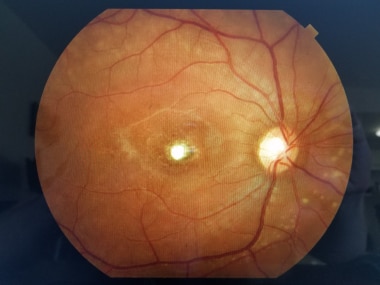
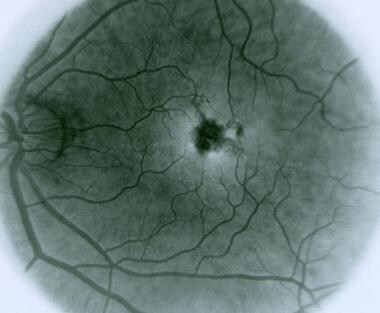
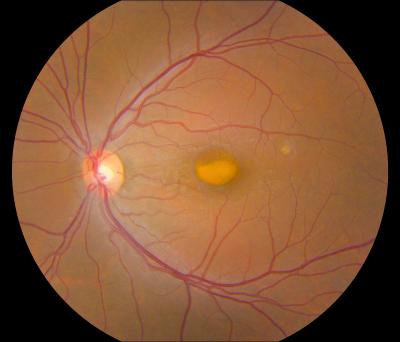
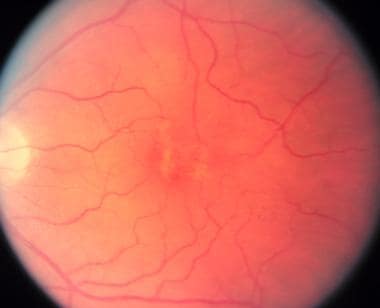
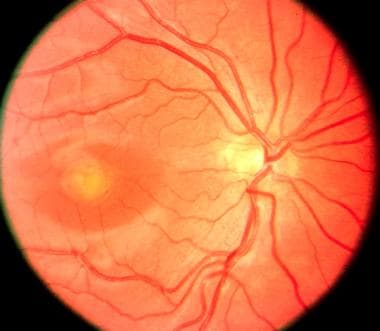
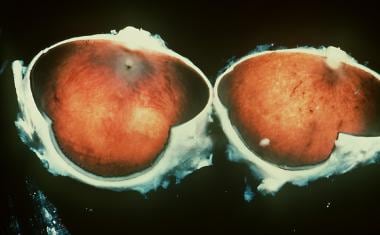
Vitelliform Macular Dystrophy
Electrooculography
Eye Diseases, Hereditary
Macular Degeneration
Eye Proteins
Chloride Channels
Fluorescein Angiography
Pedigree
Macula Lutea
Chromosomes, Human, Pair 11
Tomography, Optical Coherence
Muscular Dystrophies
Paraneoplastic Syndromes, Ocular
Corneal Dystrophies, Hereditary
Myotonic Dystrophy
Fundus Oculi
Mutation
Muscular Dystrophy, Duchenne
Retinal Diseases
Visual Acuity
Retinal Degeneration
Peripherins
Muscular Dystrophy, Animal
Genes, Dominant
Best's macular dystrophy in Australia: phenotypic profile and identification of novel BEST1 mutations. (1/16)
(+info)Clinicopathologic findings in Best vitelliform macular dystrophy. (2/16)
(+info)Phenotypic variability in a French family with a novel mutation in the BEST1 gene causing multifocal best vitelliform macular dystrophy. (3/16)
AIMS: To describe genetic and clinical findings in a French family affected by best vitelliform macular dystrophy (BVMD). METHODS: We screened eight at-risk members of a family, including a BVMD-affected proband, by direct sequencing of 11 bestrophin-1 (BEST1) exons. Individuals underwent ophthalmic examination and autofluorescent fundus imaging, indocyanine green angiography, electro-oculogram (EOG), electroretinogram (ERG), multifocal ERG, optical coherence tomography (OCT), and where possible, spectral domain OCT. RESULTS: The sequence analysis of the BEST1 gene revealed one previously unknown mutation, c.15C>A (p.Y5X), in two family members and one recently described mutation, c.430A>G (p.S144G), in five family members. Fundus examination and electrophysiological responses provided no evidence of the disease in the patient carrying only the p.Y5X mutation. Three patients with the p.S144G mutation did not show any preclinical sign of BVMD except altered EOGs. Two individuals of the family exhibited a particularly severe phenotype of multifocal BVMD-one individual carrying the p.S144G mutation heterozygously and one individual harboring both BEST1 mutations (p.S144G inherited from his mother and p.Y5X from his father). Both of these family members had multifocal vitelliform autofluorescent lesions combined with abnormal EOG, and the spectral domain OCT displayed a serous retinal detachment. In addition, ERGs demonstrated widespread retinal degeneration and multifocal ERGs showed a reduction in the central retina function, which could be correlated with the decreased visual acuity and visual field scotomas. CONCLUSIONS: A thorough clinical evaluation found no pathological phenotype in the patient carrying the isolated p.Y5X mutation. The patients carrying the p.S144G variation in the protein exhibited considerable intrafamilial phenotypic variability. Two young affected patients in this family exhibited an early onset, severe, multifocal BVMD with a diffuse distribution of autofluorescent deposits throughout the retina and rapid evolution toward the loss of central vision. The other genetically affected relatives had only abnormal EOGs and displayed no or extremely slow electrophysiological evolution. (+info)The spectrum of subclinical Best vitelliform macular dystrophy in subjects with mutations in BEST1 gene. (4/16)
(+info)A homozygous frameshift mutation in BEST1 causes the classical form of Best disease in an autosomal recessive mode. (5/16)
(+info)Clinical evaluation of two consanguineous families with homozygous mutations in BEST1. (6/16)
PURPOSE: To describe the clinical and genetic findings in two consanguineous families with Best vitelliform macular dystrophy (BVMD) and homozygous mutations in the bestrophin-1 (BEST1) gene. METHODS: Ophthalmologic examination was performed in eight members of two families originating from Spain and Denmark. Mutation screening was performed using the Vitelliform Macular Dystrophy mutation array from Asper Biotech, and by the directed genomic sequencing of BEST1. RESULTS: Two homozygous mutations were detected in these families. Mutation c.936C>A (p.Asp312Glu) has been reported previously in a Danish family; here, we describe four additional individuals in this family demonstrating findings compatible with a severe dominant BVMD, albeit with reduced penetrance in heterozygotes. In the Spanish family, a novel homozygous missense mutation in exon 4, c.388 C>A (p.Arg130Ser), was identified in the siblings. Homozygous siblings demonstrated evidence of multifocal vitelliform retinopathy, whereas heterozygous family members presented findings ranging from isolated reduction of the electrooculogram Arden ratio to normal values on all clinical parameters. CONCLUSIONS: As demonstrated in these consanguineous families, a great clinical variability is associated with homozygous mutations in BEST1, ranging from severe dominant BVMD with reduced penetrance in heterozygotes to autosomal recessive bestrophinopathy. (+info)Autosomal dominant Best disease with an unusual electrooculographic light rise and risk of angle-closure glaucoma: a clinical and molecular genetic study. (7/16)
PURPOSE: To describe the clinical and molecular characteristics of two families with autosomal dominant Best disease and atypical electrooculography (EOG). METHODS: Four affected individuals from two families were ascertained. Detailed ophthalmic examinations, refraction, and biometry (anterior chamber depth [ACD] and axial length [AL]), gonioscopy, optical coherence tomography of the anterior segment and retina, retinal imaging, and electrophysiological assessment were performed. Arden ratios from EOG testing were calculated by direct measurement of the light peak to dark trough amplitudes. Mutations in bestrophin 1 (BEST1) were identified by bidirectional Sanger sequencing. In family 1, segregation of BEST1 alleles was performed by assaying four microsatellite markers (D11S935, D11S4102, D11S987, and D11S4162) that flank BEST1. RESULTS: The proband from family 1 (three of four siblings affected with Best disease) was 42 years old with bilateral macular vitelliform lesions, advanced angle closure glaucoma (ACG), a normal electroretinogram, and no EOG light rise. Her 44-year-old brother had similar fundus appearances and an EOG light rise of 170%. Their 48-year-old sister had a normal left fundus, whereas the right fundus showed a vitelliform lesion and subretinal thickening. There was no EOG light rise detectable from either eye. Mutation analysis of BEST1 showed all affected siblings to be heterozygous for a missense mutation, c.914T>C, p.Phe305Ser. Their unaffected sister had an EOG light rise of 200%, a normal fundus appearance, and did not harbor the BEST1 mutation. Haplotype analysis of family 1 showed that the affected brother with the 170% EOG light rise had inherited the same nondiseased parental BEST1 allele as his unaffected sister. The other two affected sisters with undetectable EOG light rises shared a different nondiseased parental BEST1 allele. An unrelated 53-year-old female carrying the same c.914T>C, p.Phe305Ser mutation showed typical features of Best disease and an EOG light rise of 180%. All four siblings from family 1 had shorter axial biometry (ACD range 2.06-2.74 mm; AL range 20.46-22.60 mm) than the normal population, contributing to their risk of ACG development. Proband 2 had deeper ACDs (2.83 mm OD and 2.85 mm OS), but similar ALs (21.52 mm OD and 21.42 mm OS) compared to family 1. She had no gonioscopic evidence of angle closure. CONCLUSIONS: A near normal EOG light rise is uncommon in molecularly confirmed Best disease, and in the present report is associated with the same mutation in two families, suggesting a specific role for this amino acid in the retinal pigment epithelium dysfunction associated with this disorder. Haplotype analysis in family 1 was consistent with an effect of the nondisease allele in mediating the presence of an EOG light rise. Clinical assessment of ACG risk is recommended for BEST1 mutation carriers and their first degree relatives. (+info)Ocular phenotypes associated with biallelic mutations in BEST1 in Italian patients. (8/16)
PURPOSE: To report on the phenotype and the genotype of Italian patients carrying BEST1 mutations on both alleles. METHODS: Five Italian patients from four independent pedigrees with retinal dystrophy associated with biallelic BEST1 variants were recruited from different parts of Italy. Molecular genetic analysis of the BEST1 gene was performed with direct sequencing techniques. All the subjects included in the study were clinically evaluated with a standard ophthalmologic examination, fundus photography, optical coherence tomography scan, and electrophysiological investigations. RESULTS: Six BEST1 variants were identified. Three, c.1699del (p.Glu557AsnfsX52), c.625delAAC (p.Asn179del), and c.139C>T (p.Arg47Cys), were novel, and three had already been reported in the literature, c.301C>A(p.Pro101Thr), c.934G>A (p.Asp312Asn), and c.638A>G (p.Glu213Gly). Four were missense mutations, and two were deletions. Only one BEST1 mutation was located within one of the four mutational clusters described in typical autosomal dominant Best vitelliform macular dystrophy (BVMD). Four patients showed a BVMD phenotype while one patient presented a clinical picture consistent with autosomal recessive bestrophinopathy (ARB). CONCLUSIONS: Biallelic BEST1 sequence variants can be associated with at least two different phenotypes: BVMD and ARB. The phenotypic result of the molecular changes probably depends on the characteristics and the combination of the different BEST1 mutations, but unknown modifying factors such as other genes or the environment may also play a role. (+info)Vitelliform Macular Dystrophy is a genetic eye condition that affects the macula, which is the central part of the retina responsible for sharp, detailed vision. This disorder is characterized by the formation of yellowish deposits or lesions beneath the retina at the macula, giving it an appearance similar to an egg yolk (hence the name "vitelliform"). These deposits can disturb vision and may lead to progressive vision loss over time.
There are different types of Vitelliform Macular Dystrophy, with the most common being Best's Disease or Vitelliform Macular Dystrophy type 1 (VMD1). This form is caused by mutations in the BEST1 gene and typically manifests in childhood or early adulthood. The condition can progress through various stages, including the appearance of a yellowish lesion, followed by atrophy and scarring of the retina, which can result in significant vision loss.
Another form is Vitelliform Macular Dystrophy type 2 (VMD2), caused by mutations in the PRPH2 gene. This condition tends to progress more slowly than VMD1 and may not lead to severe vision loss.
Early diagnosis, monitoring, and low-vision rehabilitation can help manage the symptoms of Vitelliform Macular Dystrophy and maintain visual function as much as possible.
Electrooculography (EOG) is a technique for measuring the resting potential of the eye and the changes in this potential that occur with eye movements. It involves placing electrodes near the eyes to detect the small electric fields generated by the movement of the eyeball within the surrounding socket. This technique is used in research and clinical settings to study eye movements and their control, as well as in certain diagnostic applications such as assessing the function of the oculomotor system in patients with neurological disorders.
Hereditary eye diseases refer to conditions that affect the eyes and are passed down from parents to their offspring through genetics. These diseases are caused by mutations or changes in an individual's DNA that are inherited from their parents. The mutations can occur in any of the genes associated with eye development, function, or health.
There are many different types of hereditary eye diseases, some of which include:
1. Retinitis Pigmentosa - a group of rare, genetic disorders that involve a breakdown and loss of cells in the retina.
2. Macular Degeneration - a progressive disease that damages the central portion of the retina, impairing vision.
3. Glaucoma - a group of eye conditions that damage the optic nerve, often caused by an increase in pressure inside the eye.
4. Cataracts - clouding of the lens inside the eye, which can lead to blurry vision and blindness.
5. Keratoconus - a progressive eye disease that causes the cornea to thin and bulge outward into a cone shape.
6. Color Blindness - a condition where an individual has difficulty distinguishing between certain colors.
7. Optic Neuropathy - damage to the optic nerve, which can result in vision loss.
The symptoms and severity of hereditary eye diseases can vary widely depending on the specific condition and the individual's genetic makeup. Some conditions may be present at birth or develop in early childhood, while others may not appear until later in life. Treatment options for these conditions may include medication, surgery, or lifestyle changes, and are often most effective when started early.
Macular degeneration, also known as age-related macular degeneration (AMD), is a medical condition that affects the central part of the retina, called the macula. The macula is responsible for sharp, detailed vision, which is necessary for activities such as reading, driving, and recognizing faces.
In AMD, there is a breakdown or deterioration of the macula, leading to gradual loss of central vision. There are two main types of AMD: dry (atrophic) and wet (exudative). Dry AMD is more common and progresses more slowly, while wet AMD is less common but can cause rapid and severe vision loss if left untreated.
The exact causes of AMD are not fully understood, but risk factors include age, smoking, family history, high blood pressure, obesity, and exposure to sunlight. While there is no cure for AMD, treatments such as vitamin supplements, laser therapy, and medication injections can help slow its progression and reduce the risk of vision loss.
Eye proteins, also known as ocular proteins, are specific proteins that are found within the eye and play crucial roles in maintaining proper eye function and health. These proteins can be found in various parts of the eye, including the cornea, iris, lens, retina, and other structures. They perform a wide range of functions, such as:
1. Structural support: Proteins like collagen and elastin provide strength and flexibility to the eye's tissues, enabling them to maintain their shape and withstand mechanical stress.
2. Light absorption and transmission: Proteins like opsins and crystallins are involved in capturing and transmitting light signals within the eye, which is essential for vision.
3. Protection against damage: Some eye proteins, such as antioxidant enzymes and heat shock proteins, help protect the eye from oxidative stress, UV radiation, and other environmental factors that can cause damage.
4. Regulation of eye growth and development: Various growth factors and signaling molecules, which are protein-based, contribute to the proper growth, differentiation, and maintenance of eye tissues during embryonic development and throughout adulthood.
5. Immune defense: Proteins involved in the immune response, such as complement components and immunoglobulins, help protect the eye from infection and inflammation.
6. Maintenance of transparency: Crystallin proteins in the lens maintain its transparency, allowing light to pass through unobstructed for clear vision.
7. Neuroprotection: Certain eye proteins, like brain-derived neurotrophic factor (BDNF), support the survival and function of neurons within the retina, helping to preserve vision.
Dysfunction or damage to these eye proteins can contribute to various eye disorders and diseases, such as cataracts, age-related macular degeneration, glaucoma, diabetic retinopathy, and others.
Chloride channels are membrane proteins that form hydrophilic pores or gaps, allowing the selective passage of chloride ions (Cl-) across the lipid bilayer of cell membranes. They play crucial roles in various physiological processes, including regulation of neuronal excitability, maintenance of resting membrane potential, fluid and electrolyte transport, and pH and volume regulation of cells.
Chloride channels can be categorized into several groups based on their structure, function, and mechanism of activation. Some of the major classes include:
1. Voltage-gated chloride channels (ClC): These channels are activated by changes in membrane potential and have a variety of functions, such as regulating neuronal excitability and transepithelial transport.
2. Ligand-gated chloride channels: These channels are activated by the binding of specific ligands or messenger molecules, like GABA (gamma-aminobutyric acid) or glycine, and are involved in neurotransmission and neuromodulation.
3. Cystic fibrosis transmembrane conductance regulator (CFTR): This is a chloride channel primarily located in the apical membrane of epithelial cells, responsible for secreting chloride ions and water to maintain proper hydration and mucociliary clearance in various organs, including the lungs and pancreas.
4. Calcium-activated chloride channels (CaCCs): These channels are activated by increased intracellular calcium concentrations and participate in various physiological processes, such as smooth muscle contraction, neurotransmitter release, and cell volume regulation.
5. Swelling-activated chloride channels (ClSwells): Also known as volume-regulated anion channels (VRACs), these channels are activated by cell swelling or osmotic stress and help regulate cell volume and ionic homeostasis.
Dysfunction of chloride channels has been implicated in various human diseases, such as cystic fibrosis, myotonia congenita, epilepsy, and certain forms of cancer.
Fluorescein angiography is a medical diagnostic procedure used in ophthalmology to examine the blood flow in the retina and choroid, which are the inner layers of the eye. This test involves injecting a fluorescent dye, Fluorescein, into a patient's arm vein. As the dye reaches the blood vessels in the eye, a specialized camera takes rapid sequences of photographs to capture the dye's circulation through the retina and choroid.
The images produced by fluorescein angiography can help doctors identify any damage to the blood vessels, leakage, or abnormal growth of new blood vessels. This information is crucial in diagnosing and managing various eye conditions such as age-related macular degeneration, diabetic retinopathy, retinal vein occlusions, and inflammatory eye diseases.
It's important to note that while fluorescein angiography is a valuable diagnostic tool, it does carry some risks, including temporary side effects like nausea, vomiting, or allergic reactions to the dye. In rare cases, severe adverse reactions can occur, so patients should discuss these potential risks with their healthcare provider before undergoing the procedure.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
The macula lutea, often simply referred to as the macula or fovea centralis, is a part of the eye that is responsible for central vision and color perception. It's located in the center of the retina, the light-sensitive tissue at the back of the eye. The macula contains a high concentration of pigments called xanthophylls, which give it a yellowish color and protect the photoreceptor cells in this area from damage by blue light.
The central part of the macula is called the fovea, which is a small depression that contains only cones, the photoreceptor cells responsible for color vision and high visual acuity. The fovea is surrounded by the parafovea and the perifovea, which contain both cones and rods, the photoreceptor cells responsible for low-light vision and peripheral vision.
Damage to the macula can result in a loss of central vision and color perception, a condition known as age-related macular degeneration (AMD), which is a leading cause of blindness in older adults. Other conditions that can affect the macula include macular edema, macular holes, and macular pucker.
Human chromosome pair 11 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each member of the pair is a single chromosome, and together they contain the genetic material that is inherited from both parents. They are located on the eleventh position in the standard karyotype, which is a visual representation of the 23 pairs of human chromosomes.
Chromosome 11 is one of the largest human chromosomes and contains an estimated 135 million base pairs. It contains approximately 1,400 genes that provide instructions for making proteins, as well as many non-coding RNA molecules that play a role in regulating gene expression.
Chromosome 11 is known to contain several important genes and genetic regions associated with various human diseases and conditions. For example, it contains the Wilms' tumor 1 (WT1) gene, which is associated with kidney cancer in children, and the neurofibromatosis type 1 (NF1) gene, which is associated with a genetic disorder that causes benign tumors to grow on nerves throughout the body. Additionally, chromosome 11 contains the region where the ABO blood group genes are located, which determine a person's blood type.
It's worth noting that human chromosomes come in pairs because they contain two copies of each gene, one inherited from the mother and one from the father. This redundancy allows for genetic diversity and provides a backup copy of essential genes, ensuring their proper function and maintaining the stability of the genome.
Optical coherence tomography (OCT) is a non-invasive imaging technique that uses low-coherence light to capture high-resolution cross-sectional images of biological tissues, particularly the retina and other ocular structures. OCT works by measuring the echo time delay of light scattered back from different depths within the tissue, creating a detailed map of the tissue's structure. This technique is widely used in ophthalmology to diagnose and monitor various eye conditions such as macular degeneration, diabetic retinopathy, and glaucoma.
Muscular dystrophies are a group of genetic disorders that primarily affect skeletal muscles, causing progressive weakness and degeneration. They are characterized by the lack or deficiency of a protein called dystrophin, which is essential for maintaining the integrity of muscle fibers. The most common form is Duchenne muscular dystrophy (DMD), but there are many other types with varying symptoms and severity. Over time, muscle wasting and weakness can lead to disability and shortened lifespan, depending on the type and progression of the disease. Treatment typically focuses on managing symptoms, maintaining mobility, and supporting quality of life.
Paraneoplastic syndromes are a group of rare disorders that occur in some individuals with cancer. These syndromes are caused by substances produced by the tumor or the body's immune response to the tumor, which can affect distant organs and cause various symptoms.
Ocular paraneoplastic syndromes refer to a subset of these disorders that specifically affect the eyes. They are caused by an abnormal immune response directed against antigens shared by both the tumor and the nervous tissue of the eye. This results in damage to the nerve cells and can lead to various visual symptoms, such as:
1. Visual loss or blurring
2. Double vision (diplopia)
3. Light sensitivity (photophobia)
4. Abnormalities in pupil size or reactivity
5. Jerky eye movements (nystagmus)
6. Loss of peripheral vision (visual field defects)
7. Impaired color vision
8. Deterioration of the optic nerve (optic neuropathy)
Some examples of ocular paraneoplastic syndromes include:
1. Paraneoplastic retinopathy: A condition characterized by damage to the light-sensitive cells in the retina, leading to visual loss and other visual disturbances.
2. Paraneoplastic optic neuropathy: Damage to the optic nerve that can result in visual loss and visual field defects.
3. Cancer-associated retinopathy (CAR): A condition characterized by progressive vision loss, night blindness, and abnormalities in the electroretinogram (ERG), a test used to assess retinal function.
4. Melanoma-associated retinopathy (MAR): Similar to CAR but specifically associated with melanoma, this condition can cause visual loss, night blindness, and abnormal ERG results.
5. Opsoclonus-myoclonus syndrome: A rare disorder characterized by rapid, involuntary eye movements (opsoclonus) and muscle jerks (myoclonus), which can be associated with various types of cancer, including breast, lung, and ovarian cancer.
It is important to note that these conditions are relatively rare but can significantly impact a patient's quality of life. Early diagnosis and treatment of the underlying cancer can help improve outcomes for patients with ocular paraneoplastic syndromes.
Corneal dystrophies, hereditary are a group of genetic disorders that affect the cornea, which is the clear, outermost layer at the front of the eye. These conditions are characterized by the buildup of abnormal material in the cornea, leading to decreased vision, pain, or cloudiness in the eye.
There are many different types of corneal dystrophies, each affecting a specific layer of the cornea and having its own pattern of inheritance. Some common types include:
1. Fuchs' endothelial dystrophy: This affects the inner lining of the cornea (endothelium) and causes swelling and cloudiness in the cornea. It is typically inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the condition if one parent has it.
2. Granular dystrophy: This affects the stroma, which is the middle layer of the cornea. It causes the formation of opaque, grayish-white deposits in the cornea that can affect vision. It is typically inherited in an autosomal dominant or recessive manner.
3. Lattice dystrophy: This also affects the stroma and is characterized by the formation of a lattice-like pattern of fine, whitish lines in the cornea. It is typically inherited in an autosomal dominant manner.
4. Macular dystrophy: This affects the central part of the cornea (macula) and can cause cloudiness, leading to decreased vision. It is typically inherited in an autosomal recessive manner.
Treatment for corneal dystrophies may include eyedrops, medications, or surgery, depending on the severity of the condition and its impact on vision. In some cases, a corneal transplant may be necessary to restore vision.
Myotonic dystrophy is a genetic disorder characterized by progressive muscle weakness, myotonia (delayed relaxation of muscles after contraction), and other symptoms. It is caused by an expansion of repetitive DNA sequences in the DMPK gene on chromosome 19 (type 1) or the ZNF9 gene on chromosome 3 (type 2). These expansions result in abnormal protein production and accumulation, which disrupt muscle function and can also affect other organs such as the heart, eyes, and endocrine system. Myotonic dystrophy is a progressive disease, meaning that symptoms tend to worsen over time. It is typically divided into two types: myotonic dystrophy type 1 (DM1), which is more common and severe, and myotonic dystrophy type 2 (DM2), which tends to be milder with a later onset of symptoms.
"Fundus Oculi" is a medical term that refers to the back part of the interior of the eye, including the optic disc, macula, fovea, retinal vasculature, and peripheral retina. It is the area where light is focused and then transmitted to the brain via the optic nerve, forming visual images. Examinations of the fundus oculi are crucial for detecting various eye conditions such as diabetic retinopathy, macular degeneration, glaucoma, and other retinal diseases. The examination is typically performed using an ophthalmoscope or a specialized camera called a retinal camera.
Electroretinography (ERG) is a medical test used to evaluate the functioning of the retina, which is the light-sensitive tissue located at the back of the eye. The test measures the electrical responses of the retina to light stimulation.
During the procedure, a special contact lens or electrode is placed on the surface of the eye to record the electrical activity generated by the retina's light-sensitive cells (rods and cones) and other cells in the retina. The test typically involves presenting different levels of flashes of light to the eye while the electrical responses are recorded.
The resulting ERG waveform provides information about the overall health and function of the retina, including the condition of the photoreceptors, the integrity of the inner retinal layers, and the health of the retinal ganglion cells. This test is often used to diagnose and monitor various retinal disorders, such as retinitis pigmentosa, macular degeneration, and diabetic retinopathy.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by progressive muscle weakness and degeneration. It is caused by the absence of dystrophin, a protein that helps keep muscle cells intact. Without dystrophin, the muscle cells break down and are replaced with scar tissue, leading to loss of muscle function over time.
DMD primarily affects boys, as it is inherited in an X-linked recessive pattern, meaning that females who carry one affected X chromosome typically do not show symptoms but can pass the gene on to their offspring. Symptoms usually begin in early childhood and include difficulty with motor skills such as walking, running, and climbing stairs. Over time, the muscle weakness progresses and can lead to loss of ambulation, respiratory and cardiac complications, and ultimately, premature death.
Currently, there is no cure for DMD, but various treatments such as corticosteroids, physical therapy, and assisted ventilation can help manage symptoms and improve quality of life. Gene therapy approaches are also being investigated as potential treatments for this disorder.
Retinal diseases refer to a group of conditions that affect the retina, which is the light-sensitive tissue located at the back of the eye. The retina is responsible for converting light into electrical signals that are sent to the brain and interpreted as visual images. Retinal diseases can cause vision loss or even blindness, depending on their severity and location in the retina.
Some common retinal diseases include:
1. Age-related macular degeneration (AMD): A progressive disease that affects the central part of the retina called the macula, causing blurred or distorted vision.
2. Diabetic retinopathy: A complication of diabetes that can damage the blood vessels in the retina, leading to vision loss.
3. Retinal detachment: A serious condition where the retina becomes separated from its underlying tissue, requiring immediate medical attention.
4. Macular edema: Swelling or thickening of the macula due to fluid accumulation, which can cause blurred vision.
5. Retinitis pigmentosa: A group of inherited eye disorders that affect the retina's ability to respond to light, causing progressive vision loss.
6. Macular hole: A small break in the macula that can cause distorted or blurry vision.
7. Retinal vein occlusion: Blockage of the retinal veins that can lead to bleeding, swelling, and potential vision loss.
Treatment for retinal diseases varies depending on the specific condition and its severity. Some treatments include medication, laser therapy, surgery, or a combination of these options. Regular eye exams are essential for early detection and treatment of retinal diseases.
Hypotrichosis is a medical term that refers to a condition characterized by an abnormal lack or sparseness of hair growth. This can apply to the eyebrows, eyelashes, or scalp hair. It's important to note that this is not a complete loss of hair, but rather a significant reduction in hair density. The onset and severity can vary greatly, and it can be inherited or acquired later in life due to various factors such as diseases, burns, or certain medications.
Visual acuity is a measure of the sharpness or clarity of vision. It is usually tested by reading an eye chart from a specific distance, such as 20 feet (6 meters). The standard eye chart used for this purpose is called the Snellen chart, which contains rows of letters that decrease in size as you read down the chart.
Visual acuity is typically expressed as a fraction, with the numerator representing the testing distance and the denominator indicating the smallest line of type that can be read clearly. For example, if a person can read the line on the eye chart that corresponds to a visual acuity of 20/20, it means they have normal vision at 20 feet. If their visual acuity is 20/40, it means they must be as close as 20 feet to see what someone with normal vision can see at 40 feet.
It's important to note that visual acuity is just one aspect of overall vision and does not necessarily reflect other important factors such as peripheral vision, depth perception, color vision, or contrast sensitivity.
Retinal degeneration is a broad term that refers to the progressive loss of photoreceptor cells (rods and cones) in the retina, which are responsible for converting light into electrical signals that are sent to the brain. This process can lead to vision loss or blindness. There are many different types of retinal degeneration, including age-related macular degeneration, retinitis pigmentosa, and Stargardt's disease, among others. These conditions can have varying causes, such as genetic mutations, environmental factors, or a combination of both. Treatment options vary depending on the specific type and progression of the condition.
Peripherins are a family of neuron-specific type III intermediate filament proteins that are expressed in the peripheral nervous system. They play crucial roles in maintaining the structural integrity and stability of nerve cells, particularly during development and regeneration. Peripherins have also been implicated in various neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS) and Charcot-Marie-Tooth disease (CMT). There are several isoforms of peripherins, with peripherin 2 being the most widely studied. Mutations in the gene encoding peripherin 2 have been linked to certain forms of CMT.
I'm sorry for any confusion, but "Muscular Dystrophy, Animal" is not a standard medical term. Muscular Dystrophy is a group of genetic disorders that cause progressive weakness and loss of muscle mass. They are primarily human diseases and there are no known animal models of muscular dystrophy that directly correspond to any type of muscular dystrophy in humans.
However, scientists often use animals (like mice, dogs, and cats) as models for human diseases, including various types of muscular dystrophies. These animal models are used to study the disease process and to test potential treatments. For example, the mdx mouse is a well-known model of Duchenne Muscular Dystrophy (DMD), which is caused by a mutation in the dystrophin gene. This mouse lacks the muscle protein dystrophin, similar to humans with DMD, and shows many of the same symptoms, making it a valuable tool for research.
Dominant genes refer to the alleles (versions of a gene) that are fully expressed in an individual's phenotype, even if only one copy of the gene is present. In dominant inheritance patterns, an individual needs only to receive one dominant allele from either parent to express the associated trait. This is in contrast to recessive genes, where both copies of the gene must be the recessive allele for the trait to be expressed. Dominant genes are represented by uppercase letters (e.g., 'A') and recessive genes by lowercase letters (e.g., 'a'). If an individual inherits one dominant allele (A) from either parent, they will express the dominant trait (A).
The pigment epithelium of the eye, also known as the retinal pigment epithelium (RPE), is a layer of cells located between the photoreceptor cells of the retina and the choroid, which is the vascular layer of the eye. The RPE plays a crucial role in maintaining the health and function of the photoreceptors by providing them with nutrients, removing waste products, and helping to regulate the light that enters the eye.
The RPE cells contain pigment granules that absorb excess light, preventing it from scattering within the eye and improving visual acuity. They also help to create a barrier between the retina and the choroid, which is important for maintaining the proper functioning of the photoreceptors. Additionally, the RPE plays a role in the regeneration of visual pigments in the photoreceptor cells, allowing us to see in different light conditions.
Damage to the RPE can lead to various eye diseases and conditions, including age-related macular degeneration (AMD), which is a leading cause of vision loss in older adults.
 Vitelliform macular dystrophy - Wikipedia
Vitelliform macular dystrophy - Wikipedia Ophthal Bytes - Best Vitelliform Macular Dystrophy - EyeToday
Ophthal Bytes - Best Vitelliform Macular Dystrophy - EyeToday Association of optic nerve head drusen with best vitelliform macular dystrophy: A case series<...
Association of optic nerve head drusen with best vitelliform macular dystrophy: A case series<... Outer Retinal Structure in Best Vitelliform Macular Dystrophy
-
UCL Discovery
Outer Retinal Structure in Best Vitelliform Macular Dystrophy
-
UCL Discovery Retinal Disorders | Retina | Macular Degeneration | MedlinePlus
Retinal Disorders | Retina | Macular Degeneration | MedlinePlus Ghent University Academic Bibliography
Ghent University Academic Bibliography Best Disease: Background, Pathophysiology, Epidemiology
Best Disease: Background, Pathophysiology, Epidemiology High-tech imaging reveals details about rare eye disorder | National Eye Institute
High-tech imaging reveals details about rare eye disorder | National Eye Institute PubMed (OMIM) for id: 411553 - Search Results - PubMed
PubMed (OMIM) for id: 411553 - Search Results - PubMed Publications
Publications Related Articles | Annals Singapore
Related Articles | Annals Singapore Alfredo Dubra, PhD's Profile | Stanford Profiles
Alfredo Dubra, PhD's Profile | Stanford Profiles Molecular mechanisms of gating in the calcium-activated chloride channel bestrophin | eLife
Molecular mechanisms of gating in the calcium-activated chloride channel bestrophin | eLife Gwyneth Farrar'Department of Genetics - Trinity College Dublin
Gwyneth Farrar'Department of Genetics - Trinity College Dublin Molecular Vision: Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes...
Molecular Vision: Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes... vitelliform macula | Hereditary Ocular Diseases
vitelliform macula | Hereditary Ocular Diseases Johns Hopkins scientists elected to Institute of Medicine : Gazette Archives
Johns Hopkins scientists elected to Institute of Medicine : Gazette Archives Blog - Low Vision Aids
Blog - Low Vision Aids Sten Andréasson - Forskningsoutput
- Lunds universitet
Sten Andréasson - Forskningsoutput
- Lunds universitet Department of Ophthalmology and Visual Sciences - Research output
- Research Profiles at Washington University School of...
Department of Ophthalmology and Visual Sciences - Research output
- Research Profiles at Washington University School of... Acronyms about medicine
Acronyms about medicine Paraneoplastic and Related Retinopathies
Paraneoplastic and Related Retinopathies