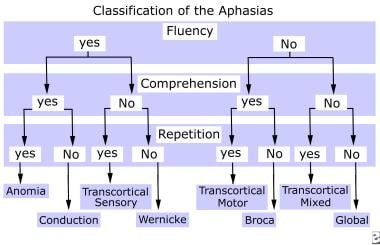
Athetosis
Paroxysmal kinesigenic choreoathetosis associated with frontotemporal arachnoid cyst--case report. (1/50)
A 17-year-old male presented with paroxysmal kinesigenic choreoathetosis (PKC) associated with frontotemporal arachnoid cyst. Xenon-133 single photon emission computed tomography detected a slight but equivocal decrease in regional cerebral blood flow in the vicinity of basal ganglia associated with the PKC episodes. PKC continued after surgical removal of the cyst but was well controlled by oral administration of carbamazepine. Whether the pathogenesis of symptomatic PKC was associated with the cortical lesion could not be determined in the present case. (+info)A new neurological syndrome with mental retardation, choreoathetosis, and abnormal behavior maps to chromosome Xp11. (2/50)
Choreoathetosis is a major clinical feature in only a small number of hereditary neurological disorders. We define a new X-linked syndrome with a unique clinical picture characterized by mild mental retardation, choreoathetosis, and abnormal behavior. We mapped the disease in a four-generation pedigree to chromosome Xp11 by linkage analysis and defined a candidate region containing a number of genes possibly involved in neuronal signaling, including a potassium channel gene and a neuronal G protein-coupled receptor. (+info)A second paroxysmal kinesigenic choreoathetosis locus (EKD2) mapping on 16q13-q22.1 indicates a family of genes which give rise to paroxysmal disorders on human chromosome 16. (3/50)
Paroxysmal kinesigenic choreoathetosis (PKC) is a rare paroxysmal movement disorder characterized by recurrent and brief attacks of choreiform or dystonic movements triggered or exacerbated by sudden voluntary movements. Some patients with PKC also have a history of infantile afebrile convulsions. PKC can be sporadic, or familial with autosomal dominant inheritance. PKC has been mapped to the pericentromeric region of human chromosome 16 in several Japanese families and in an African-American family, to regions which overlap by 9.8 cM (centiMorgan). Both regions overlap by 3.4 cM with a region containing a gene responsible for 'infantile convulsions and paroxysmal choreoathetosis' (ICCA). We have identified a second PKC locus (EKD2) on the long arm of chromosome 16 in a large Indian family with PKC. A maximum two-point LOD score of 3.66 (recombination fraction = 0.00, penetrance = 0.80) was obtained between PKC and D16S419. Haplotype and recombinant analysis localized EKD2 to a 15.8 cM region between D16S685 and D16S503. This region does not overlap with that identified in Japanese families, or with the ICCA locus. These results exclude one locus on chromosome 16 which causes both the ICCA and PKC syndromes; this suggests that there may be a cluster of genes on human chromosome 16 which lead to paroxysmal disorders. (+info)Delayed onset mixed involuntary movements after thalamic stroke: clinical, radiological and pathophysiological findings. (4/50)
Although occurrence of involuntary movements after thalamic stroke has occasionally been reported, studies using a sufficiently large number of patients and a control population are not available. Between 1995 and 1999, the author prospectively identified 35 patients with post-thalamic stroke delayed-onset involuntary movements, which included all or some degree of dystonia-athetosis-chorea-action tremor, occasionally associated with jerky, myoclonic components. A control group included 58 patients examined by the author during the same period who had lateral thalamic stroke but no involuntary movements. Demography, clinical features and imaging study results were compared. There were no differences in gender, age, risk factors, side of the lesion and follow-up periods. During the acute stage of stroke, the patients who had involuntary movements significantly more often had severe (< or = III/V) hemiparesis (50 versus 20%, P < 0.05) and severe sensory loss (in all modalities, P < 0.01) than the control group. At the time of assessment of involuntary movements, the patients with involuntary movements significantly more often had severe sensory deficit (in all modalities, P < 0.01) and severe limb ataxia (60 versus 5%, P < 0.01) than the control patients, but neither more severe motor dysfunction (7 versus 0%) nor more painful sensory symptoms (57 versus 57%). The patients with involuntary movements had a higher frequency of haemorrhagic (versus ischaemic) stroke (63 versus 31%, P < 0.05). Further analysis showed that dystonia-athetosis-chorea was closely associated with position sensory loss, whereas the tremor/myoclonic movements were related to cerebellar ataxia. Recovery of severe limb weakness seemed to augment the instability of the involuntary movements. Persistent failure of the proprioceptive sensory and cerebellar inputs in addition to successful, but unbalanced, recovery of the motor dysfunction seemed to result in a pathological motor integrative system and consequent involuntary movements in patients with relatively severe lateral-posterior thalamic strokes simultaneously damaging the lemniscal sensory pathway, the cerebellar-rubrothalamic tract and, relatively less severely, the pyramidal tract. (+info)Familial hyperargininaemia. (5/50)
A third case of hyperargininaemia occurring in one family was studied from birth. In cord blood serum arginine concentration was only slightly raised, but arginase activity in red blood cell haemolysates was very low. In the urine on day 2 a typical cystinuria pattern was present. Arginine concentration in serum increased to 158 mumol/100 ml on the 41st day of life. Later determinations of the arginase activity in peripheral blood showed values below the sensitivity of the method. Blood ammonia was consistently high, and cystinuria was present. The enzymatic defect was further displayed by intravenous loading tests with arginine. Serum urea values were predominantly normal or near the lower limit of normal, suggesting the presence of other metabolic pathways of urea synthesis. In urine there was no excretion of guanidinosuccinic acid, while the excretion of other monosubstituted guanidine derivatives was increased, pointing to a connexion with hyperargininaemia. Owing to parental attitude, a low protein diet (1-5 g/kg) was introduced only late. The infant developed severe mental retardation, athetosis, and spasticity. (+info)Persistent hemichorea associated with thyrotoxicosis. (6/50)
We describe a case with unilateral chorea associated with thyrotoxicosis. A 23-year-old female with no family history of neurological diseases acutely developed choreic movements of the left extremities during gross thyrotoxicosis. CT scan and MRI study demonstrated no abnormality. Single-photon emission CT with technetium Tc 99m-labeled hexamethylpropyleneamine oxime revealed normal cerebral perfusion. Although the choreic movements were partially improved by dopamine antagonist, they persisted for two months until successful treatment of the thyrotoxicosis finally abolished these movements. Increased sensitivity of dopamine receptors may be responsible for persistent choreic movements in thyrotoxicosis. (+info)DIAZEPAM: A PRELIMINARY STUDY OF ITS EFFECTS ON PATIENTS WITH ATHETOID CEREBRAL PALSY. (7/50)
Diazepam was administered to seven severely affected athetoid children for a period of two to three months to determine whether beneficial effects could be demonstrated from its use. All patients were started on a daily dose of 2.5 mg. and the dose was increased as tolerated. The patients were assessed by a neurologist, an occupational therapist, a physiotherapist and a speech therapist before and after the trial.The dose of diazepam cannot be determined in advance. The optimum dose must be established by trial in each individual patient. No beneficial effects were noted in four of these children. One of those who showed improvement became significantly worse when the drug was withdrawn and it was necessary to reinstitute the drug. The response in any individual patient is unpredictable. The most significant side effect was drowsiness. (+info)Follow-up study of patients with cerebral palsy. (8/50)
Of 319 patients with cerebral palsy recalled for reevaluation 15 years after the initial visit, 10 percent had died. Of the living, 55 percent had spasticity, 32 percent had athetosis, 4 percent had ataxia and 9 percent had mixed spasticity and athetosis; 38 percent had an intelligence quotient (IQ) less than 50, 24 percent between 50 and 79, and 38 percent had IQ above 80. There was a high correlation between overall functional outcome and intellectual level. Severity of physical disability, as measured by hand use, mobility and speech, also correlated with dependence, in part because increased severity of the disability was associated with decreased intellectual capacity generally.Twenty-five years after the initial visit, parental attitudes and personality intactness were evaluated (using the Minnesota Multiphasic Personality Inventory [MMPI]) and were correlated with satisfaction with status in life in 28 persons predicted to be independent on the 15-year study. Twenty (72 percent) of the 28 were satisfied with their status in life and of these, 16 were evaluated (with the MMPI) with 70 percent scoring in the normal range; 13 (65 percent) had parents with a positive attitude. Positive attitude was defined as parental feelings that the handicapped child was a worthy, valuable person, to be encouraged and assisted but not isolated from the world of nonhandicapped people. Careful serial assessment by professional teams combined with repeated long-term counseling of families can result in optimal outcome for the disability level involved, due to the primary role parents play in the development of a child's character and behavior. (+info)Athetosis is a medical term that describes a type of involuntary muscle movement. It is characterized by slow, writhing, and continuous movements that can affect the hands, feet, arms, or legs. These movements are not rhythmic and can be interrupted by other voluntary movements. Athetosis is often seen in individuals with certain neurological conditions, such as cerebral palsy or brain injury. It can also be a side effect of some medications. The exact cause of athetosis is not fully understood, but it is believed to result from damage to the basal ganglia, a part of the brain that helps regulate movement. Treatment for athetosis may include physical therapy, medication, or surgery, depending on the underlying cause and severity of the symptoms.
 Athetosis - Wikipedia
Athetosis - Wikipedia Athetosis
Athetosis Athetosis resulting from basal ganglia injury - Health Video: MedlinePlus Medical Encyclopedia
Athetosis resulting from basal ganglia injury - Health Video: MedlinePlus Medical Encyclopedia athetosis Archives - TheraTogs
athetosis Archives - TheraTogs Involuntary Movements: Causes, Diagnosis, and Treatment
Involuntary Movements: Causes, Diagnosis, and Treatment Athetosis - What does it mean?
Athetosis - What does it mean? Glossary of Terms | Parkinson's Disease
Glossary of Terms | Parkinson's Disease Chorea, Athetosis, and Hemiballismus - Neurologic Disorders - MSD Manual Professional Edition
Chorea, Athetosis, and Hemiballismus - Neurologic Disorders - MSD Manual Professional Edition Spasticity Management - Cerebral Palsy | UCLA Health
Spasticity Management - Cerebral Palsy | UCLA Health PoE - Sales figures STEAM - Page 8 - Pillars of Eternity: General Discussion (NO SPOILERS) - Obsidian Forum Community
PoE - Sales figures STEAM - Page 8 - Pillars of Eternity: General Discussion (NO SPOILERS) - Obsidian Forum Community About us | Cerebral Palsy Sport | CP Sport
About us | Cerebral Palsy Sport | CP Sport About Para Classification
About Para Classification Communication Disorders: Overview, The Normal Communication Process, Voice Disorders (Dysphonia)
Communication Disorders: Overview, The Normal Communication Process, Voice Disorders (Dysphonia) Fahr's Syndrome | National Institute of Neurological Disorders and Stroke
Fahr's Syndrome | National Institute of Neurological Disorders and Stroke Dyskinetic Cerebral Palsy | Rady Children's Hospital
Dyskinetic Cerebral Palsy | Rady Children's Hospital Cycling Classification & Categories
Cycling Classification & Categories Para-triathlon
Para-triathlon Movement Disorders: What They Are, Symptoms & Types
Movement Disorders: What They Are, Symptoms & Types Hereditary methemoglobinemia - About the Disease - Genetic and Rare Diseases Information Center
Hereditary methemoglobinemia - About the Disease - Genetic and Rare Diseases Information Center Frontsheet
Frontsheet Apply for funding from the PNZ Cyril Smith Legacy Fund - Paralympics New Zealand
Apply for funding from the PNZ Cyril Smith Legacy Fund - Paralympics New Zealand  DailyMed - PROMETHAZINE HYDROCHLORIDE tablet
DailyMed - PROMETHAZINE HYDROCHLORIDE tablet Athletics for disabled people - England Athletics
Athletics for disabled people - England Athletics