Chondrodysplasia Punctata
Chondrodysplasia Punctata, Rhizomelic
Plasmalogens
Phytanic Acid
Peroxisomal Disorders
Refsum Disease
Arylsulfatases
Zellweger Syndrome
Acetyl-CoA C-Acetyltransferase
Microbodies
Acyltransferases
Peroxisomes
Exostoses, Multiple Hereditary
Dwarfism
Bone Diseases, Developmental
Cockroaches
The Conradi-Hunermann-Happle syndrome (CDPX2) and emopamil binding protein: novel mutations, and somatic and gonadal mosaicism. (1/46)
The Conradi-Hunermann-Happle (CHH) syndrome (X-chromosomal dominant chondrodysplasia punctata type II; MIM 302960) is an X-linked dominant disorder that is characterized by ichthyosis, chondrodysplasia punctata, cataracts and short stature. The disease occurs almost exclusively in females and shows increased disease expression in successive generations (anticipation). Recently, causative mutations in the emopamil binding protein (EBP) have been identified. To better appreciate the genetics of this syndrome we analyzed the EBP gene in seven independent families using PCR, conformation-sensitive gel electrophoresis, direct sequencing and restriction enzyme analysis. We found five novel mutations: three nonsense mutations in exon 2 and exon 3 and two frameshift mutations, one deletion in exon 4 and an insertion in exon 5. In two families, known mutations affecting exon 2 were identified. Surprisingly, we failed to detect the mutation in a grandmother exhibiting minor disease symptoms such as sectorial cataract and attribute this to gonadal and somatic mosaicism. Gonadal mosaicism appeared also to be involved in the case of healthy parents having two affected girls, one of whom died due to the disease. We conclude that gonadal mosaicism has to be considered when dealing with seemingly sporadic cases. (+info)X-Linked dominant disorders of cholesterol biosynthesis in man and mouse. (2/46)
The X-linked dominant male-lethal mouse mutations tattered and bare patches are homologous to human X-linked dominant chondrodysplasia punctata and CHILD syndrome, rare human skeletal dysplasias. These disorders also affect the skin and can cause cataracts and microphthalmia in surviving, affected heterozygous females. They have recently been shown to result from mutations in genes encoding enzymes involved in sequential steps in the conversion of lanosterol to cholesterol. This review will summarize clinical features of the disorders and describe recent biochemical and molecular investigations that have resulted in the elucidation of the involved genes and their metabolic pathway. Finally, speculations about possible mechanisms of pathogenesis will be provided. (+info)A case of chondrodysplasia punctata with features of osteogenesis imperfecta type II. (3/46)
The osteogenesis imperfecta syndromes constitute a group of heterogeneous, heritable skeletal dysplasias. Of the 4 types, type II is the most severe, with an incidence of 1 per 55,000. It is characterized by malformed bones secondary to abnormal collagen type I synthesis. Affected fetuses are divided into 3 groups: A, B, and C. All groups have long bones described as "wrinkled" or "crumpled" secondary to repeated fractures. Many bones also show evidence of demineralization, which is especially evident in the bones of the face and calvaria. In groups A and C, the chest is generally small, with thickened and shortened ribs, and each rib has characteristic "beading" patterns secondary to repeated fracturing. Sonography has traditionally been successful in the diagnosis of osteogenesis imperfecta at an early gestational age. Chondrodysplasia punctata describes a heterogeneous group of skeletal disorders characterized by abnormal mineralization of bones during gestation. There are many different causes of it, but some of the specific subtypes include rhizomelic, X-linked dominant (also known as Conradi-Hunermann syndrome), X-linked recessive, and tibia-metacarpal. We report a case of severe X-linked dominant chondrodysplasia punctata, which sonographically had common features with osteogenesis imperfecta type II. (+info)Gas chromatography-mass spectrometry and molecular genetic studies in families with the Conradi-Hunermann-Happle syndrome. (4/46)
The Conradi-Hunermann-Happle syndrome is an X-linked dominant disease that is due to mutations in the gene for emopamil binding protein. Emopamil binding protein is a Delta8-Delta7 sterol isomerase and plays a pivotal role in the final steps of cholesterol biosynthesis. We wanted to know to what extent this X-linked dominant enzyme defect has functional consequences at the biochemical level and whether it is possible to predict the clinical phenotype from serum sterol measurements. Therefore we performed sterol biochemical studies in 11 Conradi-Hunermann-Happle syndrome families and compared the results obtained to the clinical and molecular genetic findings. To assess disease severity a score considering bone and skin involvement and further features was used. For evaluation of the functional consequences we studied serum samples using gas chromatography-mass spectrometry analysis. For mutation screening we analyzed the emopamil binding protein gene using polymerase chain reaction, heteroduplex analysis of all exons, direct sequencing, and restriction enzyme analysis. Mutations in the emopamil binding protein gene were found in all 11 families including seven novel mutations affecting exons 2, 4, and 5. Gas chromatography-mass spectrometry analysis revealed markedly elevated levels of 8-dehydrocholesterol and of cholest-8(9)-en-3beta-ol and helped to identify somatic mosaicism in a clinically unaffected man. The extent of the metabolic alterations in the serum, however, do not allow prediction of the clinical phenotype, nor the genotype. This lack of correlation may be due to differences in X-inactivation between different tissues of the same patient and/or loss of the mutant clone by outgrowth of proficient clones after some time. (+info)Fetal musculoskeletal malformations with a poor outcome: ultrasonographic, pathologic, and radiographic findings. (5/46)
The early and accurate antenatal diagnosis of fetal musculoskeletal malfomations with a poor outcome has important implications for the management of a pregnancy. Careful ultrasonographic examination of a fetus helps detect such anomalies, and a number of characteristic features may suggest possible differential diagnoses. During the last five years, we have encountered 39 cases of such anomalies, and the typical prenatal ultrasonographic and pathologic findings of a number of those are described in this article. (+info)Vitreoretinopathy with phalangeal epiphyseal dysplasia, a type II collagenopathy resulting from a novel mutation in the C-propeptide region of the molecule. (6/46)
A large family with dominantly inherited rhegmatogenous retinal detachment, premature arthropathy, and development of phalangeal epiphyseal dysplasia, resulting in brachydactyly was linked to COL2A1, the gene encoding proalpha1(II) collagen. Mutational analysis of the gene by exon sequencing identified a novel mutation in the C-propeptide region of the molecule. The glycine to aspartic acid change occurred in a region that is highly conserved in all fibrillar collagen molecules. The resulting phenotype does not fit easily into pre-existing subgroups of the type II collagenopathies, which includes spondyloepiphyseal dysplasia, and the Kniest, Strudwick, and Stickler dysplasias. (+info)Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. (7/46)
Since 1998, five disorders involving enzyme defects in post-squalene cholesterol biosynthesis have been identified-desmosterolosis, X-linked dominant chondrodysplasia punctata, CHILD syndrome, lathosterolosis, and hydrops-ectopic calcification-moth-eaten skeletal dysplasia. They join the most common cholesterol biosynthetic disorder, Smith-Lemli-Opitz syndrome, whose underlying defect was identified in 1993. All are associated with major developmental malformations that are unusual for metabolic disorders. The existence of mouse models for five of these disorders is beginning to enable more detailed developmental and in vitro studies examining the mechanisms involved in disease pathogenesis. In this review, an overview of the cholesterol biosynthetic pathway will be presented. Clinical features of the human disorders and mouse models of post-squalene cholesterol biosynthesis will then be discussed. (+info)Subcellular localisation and processing of non-specific lipid transfer protein are not aberrant in Rhizomelic Chondrodysplasia Punctata fibroblasts. (8/46)
The import into peroxisomes and maturation of peroxisomal 3-oxoacyl-CoA thiolase are impaired in patients with the Rhizomelic form of Chondrodysplasia Punctata (RCDP). Here we show by means of immunoblotting and subcellular fractionation that non-specific lipid transfer protein (nsLTP), another peroxisomal protein synthesised as a larger precursor, is localised in peroxisomes and is present as the mature protein in RCDP fibroblasts. Thus the component of the import machinery defective in RCDP is not required for the import of nsLTP into peroxisomes. (+info)Chondrodysplasia punctata is a group of genetic disorders that affect the development of bones and cartilage. The condition is characterized by stippled calcifications, or spots of calcium deposits, in the cartilage that can be seen on X-rays. These spots are typically found at the ends of long bones, in the sternum, and in the pelvis.
The symptoms of chondrodysplasia punctata can vary widely depending on the specific type of the disorder. Some people with the condition may have short stature, bowed legs, and other skeletal abnormalities, while others may have only mild symptoms or no symptoms at all. The condition can also be associated with developmental delays, intellectual disability, and other health problems.
There are several different types of chondrodysplasia punctata, each caused by a different genetic mutation. Some forms of the disorder are inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) in order to develop the condition. Other forms of chondrodysplasia punctata are inherited in an X-linked dominant manner, meaning that a single copy of the mutated gene (on the X chromosome) is enough to cause the disorder in females. Males, who have only one X chromosome, will typically be more severely affected by X-linked dominant disorders.
There is no cure for chondrodysplasia punctata, and treatment is focused on managing the symptoms of the condition. This may include physical therapy, bracing or surgery to correct skeletal abnormalities, and medications to manage pain or other health problems.
Chondrodysplasia punctata, rhizomelic is a rare genetic disorder that affects the development of bones and cartilage. The condition is characterized by shortened limbs (rhizomelia), particularly the upper arms and thighs, and multiple small punctate calcifications in the cartilage of the body, including the ears, nose, and other areas.
The disorder is caused by mutations in the gene PEX7, which is involved in the transport of enzymes to peroxisomes, cellular organelles that break down fatty acids and other substances. Without functional PEX7, these enzymes cannot reach the peroxisomes, leading to abnormal accumulation of lipids and other substances in various tissues, including bone and cartilage.
Chondrodysplasia punctata, rhizomelic is typically diagnosed in infancy or early childhood based on clinical features and imaging studies. The condition can be associated with a range of complications, including developmental delays, respiratory problems, hearing loss, and visual impairment. There is no cure for the disorder, and treatment is focused on managing symptoms and addressing specific complications as they arise.
Plasmalogens are a type of complex lipid called glycerophospholipids, which are essential components of cell membranes. They are characterized by having a unique chemical structure that includes a vinyl ether bond at the sn-1 position of the glycerol backbone and an ester bond at the sn-2 position, with the majority of them containing polyunsaturated fatty acids. The headgroup attached to the sn-3 position is typically choline or ethanolamine.
Plasmalogens are abundant in certain tissues, such as the brain, heart, and skeletal muscle. They have been suggested to play important roles in cellular functions, including membrane fluidity, signal transduction, and protection against oxidative stress. Reduced levels of plasmalogens have been associated with various diseases, including neurological disorders, cardiovascular diseases, and aging-related conditions.
Phytanic acid is a branched-chain fatty acid that is primarily found in animal products, such as dairy foods and meat, but can also be present in some plants. It is a secondary plant metabolite that originates from the breakdown of phytol, a component of chlorophyll.
Phytanic acid is unique because it contains a methyl group branching off from the middle of the carbon chain, making it difficult for the body to break down and metabolize. Instead, it must be degraded through a process called α-oxidation, which takes place in peroxisomes.
In some cases, impaired phytanic acid metabolism can lead to a rare genetic disorder known as Refsum disease, which is characterized by the accumulation of phytanic acid in various tissues and organs, leading to neurological symptoms, retinal degeneration, and cardiac dysfunction.
Osteochondrodysplasias are a group of genetic disorders that affect the development of bones and cartilage. These conditions can result in dwarfism or short stature, as well as other skeletal abnormalities. Osteochondrodysplasias can be caused by mutations in genes that regulate bone and cartilage growth, and they are often characterized by abnormalities in the shape, size, and/or structure of the bones and cartilage.
There are many different types of osteochondrodysplasias, each with its own specific symptoms and patterns of inheritance. Some common examples include achondroplasia, thanatophoric dysplasia, and spondyloepiphyseal dysplasia. These conditions can vary in severity, and some may be associated with other health problems, such as respiratory difficulties or neurological issues.
Treatment for osteochondrodysplasias typically focuses on managing the symptoms and addressing any related health concerns. This may involve physical therapy, bracing or surgery to correct skeletal abnormalities, and treatment for any associated medical conditions. In some cases, genetic counseling may also be recommended for individuals with osteochondrodysplasias and their families.
Peroxisomal disorders are a group of inherited metabolic diseases caused by defects in the function or structure of peroxisomes, which are specialized subcellular organelles found in the cells of animals, plants, and humans. These disorders can affect various aspects of metabolism, including fatty acid oxidation, bile acid synthesis, and plasma cholesterol levels.
Peroxisomal disorders can be classified into two main categories: single peroxisomal enzyme deficiencies and peroxisome biogenesis disorders (PBDs). Single peroxisomal enzyme deficiencies are characterized by a defect in a specific enzyme found within the peroxisome, while PBDs are caused by problems with the formation or assembly of the peroxisome itself.
Examples of single peroxisomal enzyme deficiencies include X-linked adrenoleukodystrophy (X-ALD), Refsum disease, and acyl-CoA oxidase deficiency. PBDs include Zellweger spectrum disorders, such as Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease.
Symptoms of peroxisomal disorders can vary widely depending on the specific disorder and the severity of the enzyme or biogenesis defect. They may include neurological problems, vision and hearing loss, developmental delays, liver dysfunction, and skeletal abnormalities. Treatment typically focuses on managing symptoms and addressing any underlying metabolic imbalances.
Refsum Disease is a rare inherited neurological disorder characterized by the accumulation of phytanic acid in various tissues of the body due to impaired breakdown of this fatty acid. This is caused by a deficiency in the enzyme phytanoyl-CoA hydroxylase or the transporter protein peroxisomal biogenesis factor 7 (PEX7).
The symptoms of Refsum Disease can vary but often include progressive neurological dysfunction, retinitis pigmentosa leading to decreased vision and night blindness, hearing loss, ichthyosis (dry, scaly skin), and cardiac abnormalities. The onset of symptoms is usually in childhood or adolescence, but milder cases may not become apparent until later in life.
The treatment for Refsum Disease involves a strict diet that limits the intake of phytanic acid, which is found in dairy products, beef, and certain fish. Plasmapheresis, a procedure to remove harmful substances from the blood, may also be used to reduce the levels of phytanic acid in the body. Early diagnosis and treatment can help slow down or prevent the progression of the disease.
Arylsulfatases are a group of enzymes that play a role in the breakdown and recycling of complex molecules in the body. Specifically, they catalyze the hydrolysis of sulfate ester bonds in certain types of large sugar molecules called glycosaminoglycans (GAGs).
There are several different types of arylsulfatases, each of which targets a specific type of sulfate ester bond. For example, arylsulfatase A is responsible for breaking down sulfate esters in a GAG called cerebroside sulfate, while arylsulfatase B targets a different GAG called dermatan sulfate.
Deficiencies in certain arylsulfatases can lead to genetic disorders. For example, a deficiency in arylsulfatase A can cause metachromatic leukodystrophy, a progressive neurological disorder that affects the nervous system and causes a range of symptoms including muscle weakness, developmental delays, and cognitive decline. Similarly, a deficiency in arylsulfatase B can lead to Maroteaux-Lamy syndrome, a rare genetic disorder that affects the skeleton, eyes, ears, heart, and other organs.
Zellweger Syndrome is a rare genetic disorder that affects the development and function of multiple organ systems in the body. It is part of a group of conditions known as peroxisome biogenesis disorders (PBDs), which are characterized by abnormalities in the structure and function of peroxisomes, which are cellular structures that break down fatty acids and other substances in the body.
Zellweger Syndrome is caused by mutations in one or more genes involved in the formation and maintenance of peroxisomes. As a result, people with this condition have reduced levels of certain enzymes that are necessary for normal brain development, as well as for the breakdown of fats and other substances in the body.
Symptoms of Zellweger Syndrome typically appear within the first few months of life and may include:
* Severe developmental delays and intellectual disability
* Hypotonia (low muscle tone) and poor motor skills
* Vision and hearing problems
* Facial abnormalities, such as a high forehead, wide-set eyes, and a prominent nasal bridge
* Liver dysfunction and jaundice
* Seizures
* Feeding difficulties and failure to thrive
There is no cure for Zellweger Syndrome, and treatment is focused on managing the symptoms of the condition. The prognosis for people with this disorder is generally poor, with most individuals not surviving beyond the first year of life. However, some individuals with milder forms of the condition may live into early childhood or adolescence.
Acetyl-CoA C-acetyltransferase (also known as acetoacetyl-CoA thiolase or just thiolase) is an enzyme involved in the metabolism of fatty acids and ketone bodies. Specifically, it catalyzes the reaction that converts two molecules of acetyl-CoA into acetoacetyl-CoA, which is a key step in the breakdown of fatty acids through beta-oxidation.
The enzyme works by bringing together two acetyl-CoA molecules and removing a coenzyme A (CoA) group from one of them, forming a carbon-carbon bond between the two molecules to create acetoacetyl-CoA. This reaction is reversible, meaning that the enzyme can also catalyze the breakdown of acetoacetyl-CoA into two molecules of acetyl-CoA.
There are several different isoforms of Acetyl-CoA C-acetyltransferase found in various tissues throughout the body, with differing roles and regulation. For example, one isoform is highly expressed in the liver and plays a key role in ketone body metabolism, while another isoform is found in mitochondria and is involved in fatty acid synthesis.
Microbodies are small, membrane-bound organelles found in the cells of eukaryotic organisms. They typically measure between 0.2 to 0.5 micrometers in diameter and play a crucial role in various metabolic processes, particularly in the detoxification of harmful substances and the synthesis of lipids.
There are several types of microbodies, including:
1. Peroxisomes: These are the most common type of microbody. They contain enzymes that help break down fatty acids and amino acids, producing hydrogen peroxide as a byproduct. Another set of enzymes within peroxisomes then converts the harmful hydrogen peroxide into water and oxygen, thus detoxifying the cell.
2. Glyoxysomes: These microbodies are primarily found in plants and some fungi. They contain enzymes involved in the glyoxylate cycle, a metabolic pathway that helps convert stored fats into carbohydrates during germination.
3. Microbody-like particles (MLPs): These are smaller organelles found in certain protists and algae. Their functions are not well understood but are believed to be involved in lipid metabolism.
It is important to note that microbodies do not have a uniform structure or function across all eukaryotic cells, and their specific roles can vary depending on the organism and cell type.
Acyltransferases are a group of enzymes that catalyze the transfer of an acyl group (a functional group consisting of a carbon atom double-bonded to an oxygen atom and single-bonded to a hydrogen atom) from one molecule to another. This transfer involves the formation of an ester bond between the acyl group donor and the acyl group acceptor.
Acyltransferases play important roles in various biological processes, including the biosynthesis of lipids, fatty acids, and other metabolites. They are also involved in the detoxification of xenobiotics (foreign substances) by catalyzing the addition of an acyl group to these compounds, making them more water-soluble and easier to excrete from the body.
Examples of acyltransferases include serine palmitoyltransferase, which is involved in the biosynthesis of sphingolipids, and cholesteryl ester transfer protein (CETP), which facilitates the transfer of cholesteryl esters between lipoproteins.
Acyltransferases are classified based on the type of acyl group they transfer and the nature of the acyl group donor and acceptor molecules. They can be further categorized into subclasses based on their sequence similarities, three-dimensional structures, and evolutionary relationships.
Peroxisomes are membrane-bound subcellular organelles found in the cytoplasm of eukaryotic cells. They play a crucial role in various cellular processes, including the breakdown of fatty acids and the detoxification of harmful substances such as hydrogen peroxide (H2O2). Peroxisomes contain numerous enzymes, including catalase, which converts H2O2 into water and oxygen, thus preventing oxidative damage to cellular components. They also participate in the biosynthesis of ether phospholipids, a type of lipid essential for the structure and function of cell membranes. Additionally, peroxisomes are involved in the metabolism of reactive oxygen species (ROS) and contribute to the regulation of intracellular redox homeostasis. Dysfunction or impairment of peroxisome function has been linked to several diseases, including neurological disorders, developmental abnormalities, and metabolic conditions.
Multiple hereditary exostoses (MHE) is a genetic condition characterized by the growth of multiple benign tumors known as osteochondromas. These tumors typically develop at the ends of long bones near the growth plates and can cause various skeletal deformities, limitations in mobility, and other health issues.
MHE is usually inherited in an autosomal dominant pattern, meaning that a child has a 50% chance of inheriting the condition if one parent has it. However, some cases may result from spontaneous mutations. The condition typically becomes apparent during childhood or adolescence and can affect both sexes equally.
The primary diagnostic feature of MHE is the presence of multiple osteochondromas, which are made up of bone and cartilage. These growths can cause a range of symptoms, including pain, swelling, decreased mobility, and an increased risk of fractures. In some cases, they may also lead to complications such as nerve compression or vascular damage.
Treatment for MHE typically involves surgical removal of the osteochondromas, particularly if they are causing significant symptoms or complications. Regular monitoring is also important to detect any new growths and assess their potential impact on health. In addition, physical therapy and other supportive measures may be recommended to help manage symptoms and maintain mobility.
Dwarfism is a medical condition that is characterized by short stature, typically with an adult height of 4 feet 10 inches (147 centimeters) or less. It is caused by a variety of genetic and medical conditions that affect bone growth, including skeletal dysplasias, hormonal deficiencies, and chromosomal abnormalities.
Skeletal dysplasias are the most common cause of dwarfism and are characterized by abnormalities in the development and growth of bones and cartilage. Achondroplasia is the most common form of skeletal dysplasia, accounting for about 70% of all cases of dwarfism. It is caused by a mutation in the fibroblast growth factor receptor 3 (FGFR3) gene and results in short limbs, a large head, and a prominent forehead.
Hormonal deficiencies, such as growth hormone deficiency or hypothyroidism, can also cause dwarfism if they are not diagnosed and treated early. Chromosomal abnormalities, such as Turner syndrome (monosomy X) or Down syndrome (trisomy 21), can also result in short stature and other features of dwarfism.
It is important to note that people with dwarfism are not "dwarves" - the term "dwarf" is a medical and sociological term used to describe individuals with this condition, while "dwarves" is a term often used in fantasy literature and media to refer to mythical beings. The use of the term "dwarf" can be considered disrespectful or offensive to some people with dwarfism, so it is important to use respectful language when referring to individuals with this condition.
Developmental bone diseases are a group of medical conditions that affect the growth and development of bones. These diseases are present at birth or develop during childhood and adolescence, when bones are growing rapidly. They can result from genetic mutations, hormonal imbalances, or environmental factors such as poor nutrition.
Some examples of developmental bone diseases include:
1. Osteogenesis imperfecta (OI): Also known as brittle bone disease, OI is a genetic disorder that affects the body's production of collagen, a protein necessary for healthy bones. People with OI have fragile bones that break easily and may also experience other symptoms such as blue sclerae (whites of the eyes), hearing loss, and joint laxity.
2. Achondroplasia: This is the most common form of dwarfism, caused by a genetic mutation that affects bone growth. People with achondroplasia have short limbs and a large head relative to their body size.
3. Rickets: A condition caused by vitamin D deficiency or an inability to absorb or use vitamin D properly. This leads to weak, soft bones that can bow or bend easily, particularly in children.
4. Fibrous dysplasia: A rare bone disorder where normal bone is replaced with fibrous tissue, leading to weakened bones and deformities.
5. Scoliosis: An abnormal curvature of the spine that can develop during childhood or adolescence. While not strictly a developmental bone disease, scoliosis can be caused by various underlying conditions such as cerebral palsy, muscular dystrophy, or spina bifida.
Treatment for developmental bone diseases varies depending on the specific condition and its severity. Treatment may include medication, physical therapy, bracing, or surgery to correct deformities and improve function. Regular follow-up with a healthcare provider is essential to monitor growth, manage symptoms, and prevent complications.
Cockroaches are not a medical condition or disease. They are a type of insect that can be found in many parts of the world. Some species of cockroaches are known to carry diseases and allergens, which can cause health problems for some people. Cockroach allergens can trigger asthma symptoms, especially in children. Additionally, cockroaches can contaminate food and surfaces with bacteria and other germs, which can lead to illnesses such as salmonellosis and gastroenteritis.
If you have a problem with cockroaches in your home or workplace, it is important to take steps to eliminate them to reduce the risk of health problems. This may include cleaning up food and water sources, sealing entry points, and using pesticides or hiring a professional pest control service.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
 Chondrodysplasia punctata
Chondrodysplasia punctata
Rhizomelic chondrodysplasia punctata
X-linked recessive chondrodysplasia punctata
Emopamil binding protein
Plasmalogen
List of OMIM disorder codes
Hypercoagulability in pregnancy
Rhizomelia
Stippled epiphyses
Conradi-Hünermann syndrome
Glyceronephosphate O-acyltransferase
Congenital vertebral anomaly
Arylsulfatase E
Peroxisomal disorder
Keutel syndrome
Peroxin-7
Refsum disease
Steroid sulfatase
List of diseases (C)
List of skin conditions
List of MeSH codes (C05)
Alkylglycerone phosphate synthase
ADCY2
List of MeSH codes (C18)
List of MeSH codes (C16)
Ocular albinism late onset sensorineural deafness
Zellweger syndrome
List of diseases (P)
Chondrodysplasia punctata - Wikipedia
 X-linked chondrodysplasia punctata 1: MedlinePlus Genetics
X-linked chondrodysplasia punctata 1: MedlinePlus Genetics
 Chondrodysplasia punctata durch X-chromosomale Deletion - Deutsch Wörterbuch
Chondrodysplasia punctata durch X-chromosomale Deletion - Deutsch Wörterbuch
 Chondrodysplasia punctata - ULTRASOUNDPAEDIA
Chondrodysplasia punctata - ULTRASOUNDPAEDIA
GNPAT gene: MedlinePlus Genetics
 Chondrodysplasia Punctata 1, X-Linked - GeneReviews® - NCBI Bookshelf
Chondrodysplasia Punctata 1, X-Linked - GeneReviews® - NCBI Bookshelf
 CHONDRODYSPLASIA PUNCTATA ASSOCIATED WITH TETRALOGY OF FALLOT IN A NEWBORN INFANT | AVESİS
CHONDRODYSPLASIA PUNCTATA ASSOCIATED WITH TETRALOGY OF FALLOT IN A NEWBORN INFANT | AVESİS
 Chondrodysplasia Punctata and Maternal Warfarin Use During Pregnancy | JAMA Pediatrics | JAMA Network
Chondrodysplasia Punctata and Maternal Warfarin Use During Pregnancy | JAMA Pediatrics | JAMA Network
BOA Meetings & Surveys: The Skeletal Dysplasia Group (UK) Spring Meeting: Chondrodysplasia punctata - 4th April 2011
BOA Meetings & Surveys: The Skeletal Dysplasia Group (UK) Spring Meeting: Chondrodysplasia punctata - 4th April 2011
 General Data Protection Regulation Compliance - 23andMe Europe
General Data Protection Regulation Compliance - 23andMe Europe
 Atlantoaxial Instability: Practice Essentials, Pathophysiology, Etiology
Atlantoaxial Instability: Practice Essentials, Pathophysiology, Etiology
 Cervical Spine Clinic | Crawford Spine Center
Cervical Spine Clinic | Crawford Spine Center
 Peroxisomal leukoencephalopathy
Peroxisomal leukoencephalopathy
CHILD Syndrome Differential Diagnoses
 Infections
Infections
Cord
Browse In Spine | jns Journals
Index by author - March 01, 1991, 12 (2) | American Journal of Neuroradiology
 urofacial syndrome - Ontology Browser - Rat Genome Database
urofacial syndrome - Ontology Browser - Rat Genome Database
 Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US
 SMART: PlsC domain annotation
SMART: PlsC domain annotation
 Disease Browser - Sample List
Disease Browser - Sample List
 Peroxisomal Disorders - Children's Health Issues - MSD Manual Consumer Version
Peroxisomal Disorders - Children's Health Issues - MSD Manual Consumer Version
Leiner disease
 Human Metabolome Database: Showing metabocard for Acetoacetyl-CoA (HMDB0001484)
Human Metabolome Database: Showing metabocard for Acetoacetyl-CoA (HMDB0001484)
SMART: WD40 domain annotation
SMART: WD40 domain annotation
 Ghent University Academic Bibliography
Ghent University Academic BibliographyRCDP5
- Rhizomelic chondrodysplasia punctata (RCDP) is a type of peroxisomal disorder which impairs the normal development of many parts of the body. (nih.gov)
- Cassidy McCrone suffered a rare form of dwarfism, rhizomelic chondrodysplasia punctata (RCDP). (dailyrecord.co.uk)
- The PBD group is comprised of four disorders: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). (ucsc.edu)
- Although the physiological role of plasmalogens is unclear, defects in their biosynthesis, which occur as a result of peroxisomal disorders, are associated with severe developmental conditions, including rhizomelic chondrodysplasia punctata (RCDP) and Zellweger syndrome . (britannica.com)
- In rhizomelic chondrodysplasia punctata (RCDP), characterised by proximal limb shortening and mental retardation, peroxisomes are present but of abnormal structure and there are defects of import of some proteins into the organelles. (sas-centre.org)
Associated with rhizomelic chondrod2
- The genes associated with rhizomelic chondrodysplasia punctata are involved in the formation and function of structures called peroxisomes . (medlineplus.gov)
- Mutations in this gene are associated with rhizomelic chondrodysplasia punctata. (oftalmic.ru)
Rhizomelic form of chondrodysplasia punctata2
- The metabolic defects associated with the impaired peroxisomes are present only in the rhizomelic form of chondrodysplasia punctata. (jefferson.edu)
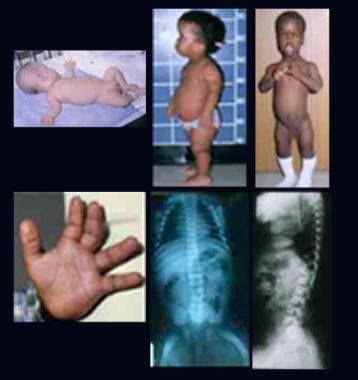
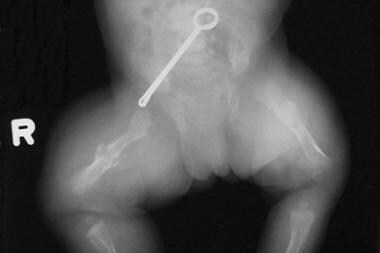
- Infant with rhizomelic form of chondrodysplasia punctata (left). (medscape.com)
Symptoms of Rhizomelic2
- When Do Symptoms of Rhizomelic chondrodysplasia punctata Begin? (nih.gov)
- Researchers are working to determine how problems with plasmalogen synthesis lead to the specific signs and symptoms of rhizomelic chondrodysplasia punctata. (medlineplus.gov)
Recessive chondrodysplasia punctata2
- 500 : 549 Rhizomelic chondrodysplasia punctata 215100, 222765, 600121 X-linked recessive chondrodysplasia punctata 302950 Conradi-Hünermann syndrome (chondrodysplasia punctata 2, x-linked dominant) 302960 Autosomal dominant chondrodysplasia punctata 118650 List of cutaneous conditions List of radiographic findings associated with cutaneous conditions Freedberg, et al. (wikipedia.org)
- X-linked recessive chondrodysplasia punctata (CDPX) is a congenital defect of bone and cartilage development characterized by aberrant bone mineralization, severe underdevelopment of nasal cartilage, and distal phalangeal hypoplasia. (units.it)
Epiphyses2
- Chondrodysplasia punctata is a clinically and genetically diverse group of rare diseases, first described by Erich Conradi (1882-1968), that share the features of stippled epiphyses and skeletal changes. (wikipedia.org)
- Non-rhizomelic types are characterized by asymmetric, dysplastic skeletal changes, punctate calcifications of the epiphyses, variably asymmetric limb lengths or normal limb lengths, nasal hypoplasia, variable upper airway compromise, skin changes, cataracts, and a generally favorable prognosis. (thefetus.net)
Zellweger2
- Mutations in this gene have been associated with Refsum disease (RD) and deficient protein activity has been associated with Zellweger syndrome and rhizomelic chondrodysplasia punctata. (abnova.com)
- It shares many features with other PBDs including those formerly called Zellweger syndrome ( 214100 ), rhizomelic chondrodysplasia punctata ( 215100 ), and neonatal adrenoleukodystrophy ( 601539 ). (arizona.edu)
CDPX23
- The findings in X-linked chondrodysplasia punctata 2 (CDPX2) range from fetal demise with multiple malformations and severe growth retardation to much milder manifestations, including females with no recognizable physical abnormalities. (nih.gov)
- Purpose: Human X-linked dominant chondrodysplasia punctata (CDPX2) or Happle syndrome is associated with mutations in the human emopamil binding protein (EBP), a Δ 8 -Δ 7 -sterol isomerase involved in cholesterol biosynthesis. (johnshopkins.edu)
- Other human malformation syndromes caused by inborn errors of cholesterol synthesis include Lathosterolosis, Desmosterolosis, X-linked dominant chondrodysplasia punctata type 2 (CDPX2), and Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects (CHILD syndrome). (nih.gov)
Dominant5
- Methods: Genomic sequencing of the coding exons of the human Δ 8 - Δ 7 -sterol isomerase gene was performed on DNA from 26 females with suspected X-linked dominant chondrodysplasia punctata. (johnshopkins.edu)
- Chondrodysplasia punctata has variable degrees of severity and outcomes, and variable genetic inheritance including X-linked recessive, X-linked dominant and autosomal recessive. (thefetus.net)
- For example in the dominant form, fine punctata are usually confined to the spine. (thefetus.net)
- The group includes a severe autosomal recessive form (CHONDRODYSPLASIA PUNCTATA, RHIZOMELIC), an autosomal dominant form (Conradi-Hunermann syndrome), and a milder X-linked form. (bvsalud.org)
- X-linked dominant chondrodysplasia Chassaing-Lacombe type is a rare genetic bone disorder characterized by chondrodysplasia, intrauterine growth retardation (IUGR), hydrocephaly and facial dysmorphism in the affected males. (nih.gov)
Coronal clefts1
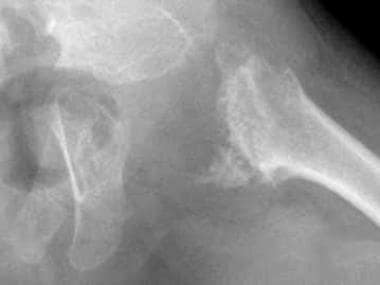
- Classic (severe) RCDP1 is characterized by proximal shortening of the humerus (rhizomelia) and to a lesser degree the femur, punctate calcifications in cartilage with epiphyseal and metaphyseal abnormalities (chondrodysplasia punctata, or CDP), coronal clefts of the vertebral bodies, and cataracts that are usually present at birth or appear in the first few months of life. (nih.gov)
Congenital1
- Congenital heart defects common in rhizomelic chondrodysplasia punctata. (jefferson.edu)
RCDP32
- Researchers have described three types of rhizomelic chondrodysplasia punctata: type 1 (RCDP1), type 2 (RCDP2), and type 3 (RCDP3). (medlineplus.gov)
- Rhizomelic chondrodysplasia punctata type 3 (RCDP3) is a rare genetic disorder that affects bone growth and development. (dnalabsindia.com)
RCDP11
- Rhizomelic chondrodysplasia punctata type 1 (RCDP1), a peroxisome biogenesis disorder (PBD) has a classic (severe) form and a nonclassic (mild) form. (nih.gov)
Syndrome1
- Warfarin inhibits ARSE activity, so there is a pathophysiological link between fetal warfarin syndrome and chondrodysplasia punctata. (thefetus.net)
Proximal2
- Rhizomelic form is autosomal recessive and characterized by rhizomelic shortening of the long bones (humeri and femora) and punctate calcifications of the cartilaginous portions of skeleton, particularly the proximal humeri and femora. (thefetus.net)
- The proximal femoral metaphyses sometimes show chondrodysplasia. (arizona.edu)
Type6
- Rhizomelic Chondrodysplasia Punctata Type 1. (medlineplus.gov)
- Clinical History of Patient who is going for AGPS Gene Rhizomelic chondrodysplasia punctata type 3 NGS Genetic DNA Test. (dnalabsindia.com)
- The cost of AGPS gene rhizomelic chondrodysplasia punctata type 3 NGS genetic DNA testing in India is approximately INR 20,000. (dnalabsindia.com)
- Rhizomelic chondrodysplasia punctata type 3 is a rare genetic disorder that can have significant physical and cognitive effects. (dnalabsindia.com)
- At DNA Labs India, we offer comprehensive genetic testing services, including AGPS gene rhizomelic chondrodysplasia punctata type 3 NGS genetic DNA testing, at an affordable cost. (dnalabsindia.com)
- Diseases associated with PEX5L include Rhizomelic Chondrodysplasia Punctata, Type 5 and Rhizomelic Chondrodysplasia Punctata, Type 2. (antibodiesinc.com)
Abnormality2
- Affected individuals also have a specific bone abnormality called chondrodysplasia punctata, which affects the growth of the long bones and can be seen on x-rays. (medlineplus.gov)
- Chondrodysplasia punctata is a skeletal abnormality characterized by premature foci of calcification within the cartilage, referred to as stippling. (thefetus.net)
Descriptor1
- Chondrodysplasia Punctata, Rhizomelic" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (jefferson.edu)
Mutations1
- Rhizomelic chondrodysplasia punctata results from mutations in one of three genes. (medlineplus.gov)
Distinctive1
- Distinctive facial features are also seen with rhizomelic chondrodysplasia punctata. (medlineplus.gov)
Severe2
- Rhizomelic chondrodysplasia punctata is associated with significantly delayed development and severe intellectual disability. (medlineplus.gov)
- Because of their severe health problems, most people with rhizomelic chondrodysplasia punctata survive only into childhood. (medlineplus.gov)
Findings1
- Ultrasound findings can help identify the subtype of chondrodysplasia punctata based on the location of the stippling and the associated malformations. (thefetus.net)
100,0001
- Rhizomelic chondrodysplasia punctata affects fewer than 1 in 100,000 people worldwide. (medlineplus.gov)
Form1
- An autosomal recessive form of CHONDRODYSPLASIA PUNCTATA characterized by defective plasmalogen biosynthesis and impaired peroxisomes. (jefferson.edu)
Growth1
- Growth charts for individuals with rhizomelic chondrodysplasia punctata. (jefferson.edu)
Website1
- This graph shows the total number of publications written about "Chondrodysplasia Punctata, Rhizomelic" by people in this website by year, and whether "Chondrodysplasia Punctata, Rhizomelic" was a major or minor topic of these publications. (jefferson.edu)
Types1
- Rhizomelic chondrodysplasia punctata can be subclassified into types 1, 2, and 3 according to the affected gene (PEX7, DHAPAT and ADAPS) and is generally lethal. (thefetus.net)
People2
- People with rhizomelic chondrodysplasia punctata often develop joint deformities (contractures) that make the joints stiff and painful. (medlineplus.gov)
- Below are the most recent publications written about "Chondrodysplasia Punctata, Rhizomelic" by people in Profiles. (jefferson.edu)