DiGeorge Syndrome
Chromosomes, Human, Pair 22
Cardiovascular Abnormalities
T-Box Domain Proteins
Abnormalities, Multiple
Truncus Arteriosus, Persistent
Branchial Region
Hypocalcemia
Hypoparathyroidism
In Situ Hybridization, Fluorescence
Heart Defects, Congenital
Immunologic Deficiency Syndromes
Monosomy
Thymus Gland
Parathyroid Glands
Gene Deletion
Neural Crest
Phenotype
Chromosome Mapping
Translocation, Genetic
Microdeletion 22q11 and oesophageal atresia. (1/344)
Oesophageal atresia (OA) is a congenital defect associated with additional malformations in 30-70% of the cases. In particular, OA is a component of the VACTERL association. Since some major features of the VACTERL association, including conotruncal heart defect, radial aplasia, and anal atresia, have been found in patients with microdeletion 22q11.2 (del(22q11.2)), we have screened for del(22q11.2) by fluorescent in situ hybridisation (FISH) in 15 syndromic patients with OA. Del(22q11.2) was detected in one of them, presenting with OA, tetralogy of Fallot, anal atresia, neonatal hypocalcaemia, and subtle facial anomalies resembling those of velocardiofacial syndrome. The occurrence of del(22q11.2) in our series of patients with OA is low (1/15), but this chromosomal anomaly should be included among causative factors of malformation complexes with OA. In addition, clinical variability of del(22q11.2) syndrome is further corroborated with inclusion of OA in the list of the findings associated with the deletion. (+info)Der(22) syndrome and velo-cardio-facial syndrome/DiGeorge syndrome share a 1.5-Mb region of overlap on chromosome 22q11. (2/344)
Derivative 22 (der[22]) syndrome is a rare disorder associated with multiple congenital anomalies, including profound mental retardation, preauricular skin tags or pits, and conotruncal heart defects. It can occur in offspring of carriers of the constitutional t(11;22)(q23;q11) translocation, owing to a 3:1 meiotic malsegregation event resulting in partial trisomy of chromosomes 11 and 22. The trisomic region on chromosome 22 overlaps the region hemizygously deleted in another congenital anomaly disorder, velo-cardio-facial syndrome/DiGeorge syndrome (VCFS/DGS). Most patients with VCFS/DGS have a similar 3-Mb deletion, whereas some have a nested distal deletion endpoint resulting in a 1.5-Mb deletion, and a few rare patients have unique deletions. To define the interval on 22q11 containing the t(11;22) breakpoint, haplotype analysis and FISH mapping were performed for five patients with der(22) syndrome. Analysis of all the patients was consistent with 3:1 meiotic malsegregation in the t(11;22) carrier parent. FISH-mapping studies showed that the t(11;22) breakpoint occurred in the same interval as the 1.5-Mb distal deletion breakpoint for VCFS. The deletion breakpoint of one VCFS patient with an unbalanced t(18;22) translocation also occurred in the same region. Hamster-human somatic hybrid cell lines from a patient with der(22) syndrome and a patient with VCFS showed that the breakpoints occurred in an interval containing low-copy repeats, distal to RANBP1 and proximal to ZNF74. The presence of low-copy repetitive sequences may confer susceptibility to chromosome rearrangements. A 1.5-Mb region of overlap on 22q11 in both syndromes suggests the presence of dosage-dependent genes in this interval. (+info)Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. (3/344)
Velo-cardio-facial syndrome (VCFS) is the most common microdeletion syndrome in humans. It occurs with an estimated frequency of 1 in 4, 000 live births. Most cases occur sporadically, indicating that the deletion is recurrent in the population. More than 90% of patients with VCFS and a 22q11 deletion have a similar 3-Mb hemizygous deletion, suggesting that sequences at the breakpoints confer susceptibility to rearrangements. To define the region containing the chromosome breakpoints, we constructed an 8-kb-resolution physical map. We identified a low-copy repeat in the vicinity of both breakpoints. A set of genetic markers were integrated into the physical map to determine whether the deletions occur within the repeat. Haplotype analysis with genetic markers that flank the repeats showed that most patients with VCFS had deletion breakpoints in the repeat. Within the repeat is a 200-kb duplication of sequences, including a tandem repeat of genes/pseudogenes, surrounding the breakpoints. The genes in the repeat are GGT, BCRL, V7-rel, POM121-like, and GGT-rel. Physical mapping and genomic fingerprint analysis showed that the repeats are virtually identical in the 200-kb region, suggesting that the deletion is mediated by homologous recombination. Examination of two three-generation families showed that meiotic intrachromosomal recombination mediated the deletion. (+info)Molecular genetic analysis of the DiGeorge syndrome among Korean patients with congenital heart disease. (4/344)
The DiGeorge syndrome (DGS) is a developmental defect of the third and fourth pharyngeal pouches, which is associated with congenital heart defects, hypoparathyroidism, cell-mediated immunodeficiency, velo-pharyngeal insufficiency and craniofacial dysmorphism. The aetiological factor in a great majority of DGS cases is monosomy for the chromosomal region 22q11. To analyze DGS at the molecular level, a new molecular probe (DGCR680) encompassing the ADU balanced translocation breakpoint was prepared. When 13 Korean patients with DGS-type congenital heart disease were analyzed with this probe, 9 turned out to have a deletion at this locus, and all of them except one exhibited a typical facial dysmorphism associated DGS. Though only 9 independent patients were detected to have a deletion at the locus using the commercial probe N25 (D22S75), which maps at about 160 kb from the ADU breakpoint to the telomeric end, results from fluorescence in situ hybridization revealed a deletion in all cases tested at this locus. Two patients who had a deletion at the locus D22S75 but not at DGCR680 did not exhibit any DGS-type facial abnormalities. This result implies that the 680 bp probe covering the ADU translocation breakpoint might be a candidate for a molecular marker that can distinguish a specific phenotype, such as facial features associated with the DiGeorge syndrome. This study also suggested that systematic approaches with several small DNA probes along the DGCR could help to dissect the complex phenotypes associated with the DiGeorge syndrome, such as cardiac defects, abnormal faces, thymic hypoplasia, cleft palate, and hypocalcemia, etc. (+info)A common molecular basis for rearrangement disorders on chromosome 22q11. (5/344)
The chromosome 22q11 region is susceptible to rearrangements that are associated with congenital anomaly disorders and malignant tumors. Three congenital anomaly disorders, cat-eye syndrome, der() syndrome and velo-cardio-facial syndrome/DiGeorge syndrome (VCFS/DGS) are associated with tetrasomy, trisomy or monosomy, respectively, for part of chromosome 22q11. VCFS/DGS is the most common syndrome associated with 22q11 rearrangements. In order to determine whether there are particular regions on 22q11 that are prone to rearrangements, the deletion end-points in a large number of VCFS/DGS patients were defined by haplotype analysis. Most VCFS/DGS patients have a similar 3 Mb deletion, some have a nested distal deletion breakpoint resulting in a 1.5 Mb deletion and a few rare patients have unique deletions or translocations. The high prevalence of the disorder in the population and the fact that most cases occur sporadically suggest that sequences at or near the breakpoints confer susceptibility to chromosome rearrangements. To investigate this hypothesis, we developed hamster-human somatic hybrid cell lines from VCFS/DGS patients with all three classes of deletions and we now show that the breakpoints occur within similar low copy repeats, termed LCR22s. To support this idea further, we identified a family that carries an interstitial duplication of the same 3 Mb region that is deleted in VCFS/DGS patients. We present models to explain how the LCR22s can mediate different homologous recombination events, thereby generating a number of rearrangements that are associated with congenital anomaly disorders. We identified five additional copies of the LCR22 on 22q11 that may mediate other rearrangements leading to disease. (+info)Comparative mapping of the DiGeorge region in the dog and exclusion of linkage to inherited canine conotruncal heart defects. (6/344)
Conotruncal defects (CTDs) of the heart are a frequent component of DiGeorge, velocardiofacial, or other syndromes caused by deletions of the human chromosome 22q11 region (HSA22q11). In addition, some human patients with isolated nonsyndromic CTDs have been reported to have deletions of this region. Taken together, these findings lead to the conclusion that deletions of an HSA22q11 locus or loci produce abnormalities in cardiac development leading to CTDs. A spontaneous model of isolated inherited conotruncal malformations occurs in the keeshond dog. We have previously shown in experimental matings that nonsyndromic CTDs in the keeshond are inherited in a manner consistent with a major underlying locus. In the studies described in this article we tested two hypotheses: (1) the region of HSA22q11 commonly deleted in DiGeorge and related syndromes is evolutionarily conserved in the dog, and (2) a locus in this region is linked to hereditary CTD in the keeshond. Two loci within the minimal DiGeorge critical region (MDGCR) and two loci that lie telomeric to the MDGCR, one of which is commonly deleted in DiGeorge patients, were mapped in the dog using a combination of linkage analysis and fluorescence in situ hybridization (FISH). The results confirm conserved synteny of the loci DGS-I, CTP, D22S788 (N41), and IGLC on the telomeric end of canine chromosome 26 (CFA26). The group of four syntenic gene loci, which spans a genetic distance of 2.5 cM is the first to be mapped to this small acrocentric canine chromosome and adds gene-associated polymorphic markers to the developing dog linkage map. Linkage of loci in this region to hereditary CTD in the keeshond was excluded. (+info)B cell non-Hodgkin's lymphoma in a girl with the DiGeorge anomaly. (7/344)
The DiGeorge anomaly (DGA) is occasionally associated with cellular immunodeficiency. We report a female infant diagnosed with complete DGA, who developed fatal, high grade, non-Hodgkin's lymphoma that expressed Epstein-Barr virus (EBV). Non-Hodgkin's lymphoma should be considered in children with DGA. (+info)Transplantation of thymus tissue in complete DiGeorge syndrome. (8/344)
BACKGROUND: The DiGeorge syndrome is a congenital disorder that affects the heart, parathyroid glands, and thymus. In complete DiGeorge syndrome, patients have severely reduced T-cell function. METHODS: We treated five infants (age, one to four months) with complete DiGeorge syndrome by transplantation of cultured postnatal thymus tissue. Follow-up evaluations included immune phenotyping and proliferative studies of peripheral-blood mononuclear cells plus biopsy of the thymus allograft. Thymic production of new T cells was assessed in peripheral blood by tests for T-cell-receptor recombination excision circles, which are formed from excised DNA during the rearrangement of T-cell-receptor genes. RESULTS: After the transplantation of thymus tissue, T-cell proliferative responses to mitogens developed in four of the five patients. Two of the patients survived with restoration of immune function; three patients died from infection or abnormalities unrelated to transplantation. Biopsies of grafted thymus in the surviving patients showed normal morphologic features and active T-cell production. In three patients, donor T cells could be detected about four weeks after transplantation, although there was no evidence of graft-versus-host disease on biopsy or at autopsy. In one patient, the T-cell development within the graft was demonstrated to accompany the appearance of recently developed T cells in the periphery and coincided with the onset of normal T-cell function. In one patient, there was evidence of thymus function and CD45RA+CD62L+ T cells more than five years after transplantation. CONCLUSIONS: In some infants with profound immunodeficiency and complete DiGeorge syndrome, the transplantation of thymus tissue can restore normal immune function. Early thymus transplantation - before the development of infectious complications - may promote successful immune reconstitution. (+info)DiGeorge syndrome is a genetic disorder caused by the deletion of a small piece of chromosome 22. It is also known as 22q11.2 deletion syndrome. The symptoms and severity can vary widely among affected individuals, but often include birth defects such as congenital heart disease, poor immune system function, and palatal abnormalities. Characteristic facial features, learning disabilities, and behavioral problems are also common. Some people with DiGeorge syndrome may have mild symptoms while others may be more severely affected. The condition is typically diagnosed through genetic testing. Treatment is focused on managing the specific symptoms and may include surgery, medications, and therapy.
Human chromosome pair 22 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex structure called a chromatin.
Chromosome pair 22 is one of the 22 autosomal pairs of human chromosomes, meaning they are not sex chromosomes (X or Y). Chromosome 22 is the second smallest human chromosome, with each arm of the chromosome designated as p and q. The short arm is labeled "p," and the long arm is labeled "q."
Chromosome 22 contains several genes that are associated with various genetic disorders, including DiGeorge syndrome, velocardiofacial syndrome, and cat-eye syndrome, which result from deletions or duplications of specific regions on the chromosome. Additionally, chromosome 22 is the location of the NRXN1 gene, which has been associated with an increased risk for autism spectrum disorder (ASD) and schizophrenia when deleted or disrupted.
Understanding the genetic makeup of human chromosome pair 22 can provide valuable insights into human genetics, evolution, and disease susceptibility, as well as inform medical diagnoses, treatments, and research.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
Cardiovascular abnormalities refer to structural or functional anomalies in the heart or blood vessels. These abnormalities can be present at birth (congenital) or acquired later in life. They can affect the heart's chambers, valves, walls, or blood vessels, leading to various complications such as heart failure, stroke, or even death if left untreated.
Examples of congenital cardiovascular abnormalities include:
1. Septal defects - holes in the walls separating the heart's chambers (atrial septal defect, ventricular septal defect)
2. Valvular stenosis or insufficiency - narrowing or leakage of the heart valves
3. Patent ductus arteriosus - a persistent opening between the aorta and pulmonary artery
4. Coarctation of the aorta - narrowing of the aorta
5. Tetralogy of Fallot - a combination of four heart defects, including ventricular septal defect, overriding aorta, pulmonary stenosis, and right ventricular hypertrophy
Examples of acquired cardiovascular abnormalities include:
1. Atherosclerosis - the buildup of plaque in the arteries, leading to narrowing or blockage
2. Cardiomyopathy - disease of the heart muscle, causing it to become enlarged, thickened, or stiffened
3. Hypertension - high blood pressure, which can damage the heart and blood vessels over time
4. Myocardial infarction (heart attack) - damage to the heart muscle due to blocked blood supply
5. Infective endocarditis - infection of the inner lining of the heart chambers and valves
These abnormalities can be diagnosed through various tests, such as echocardiography, electrocardiogram (ECG), stress testing, cardiac catheterization, or magnetic resonance imaging (MRI). Treatment options depend on the type and severity of the abnormality and may include medications, medical procedures, or surgery.
T-box domain proteins are a family of transcription factors that share a highly conserved DNA-binding domain, known as the T-box. The T-box domain is a DNA-binding motif that specifically recognizes and binds to T-box binding elements (TBEs) in the regulatory regions of target genes. These proteins play crucial roles during embryonic development, particularly in the formation of specific tissues and organs, such as the heart, limbs, and brain. Mutations in T-box domain proteins can lead to various congenital defects and developmental disorders. Some examples of T-box domain proteins include TBX1, TBX5, and TBX20.
'Abnormalities, Multiple' is a broad term that refers to the presence of two or more structural or functional anomalies in an individual. These abnormalities can be present at birth (congenital) or can develop later in life (acquired). They can affect various organs and systems of the body and can vary greatly in severity and impact on a person's health and well-being.
Multiple abnormalities can occur due to genetic factors, environmental influences, or a combination of both. Chromosomal abnormalities, gene mutations, exposure to teratogens (substances that cause birth defects), and maternal infections during pregnancy are some of the common causes of multiple congenital abnormalities.
Examples of multiple congenital abnormalities include Down syndrome, Turner syndrome, and VATER/VACTERL association. Acquired multiple abnormalities can result from conditions such as trauma, infection, degenerative diseases, or cancer.
The medical evaluation and management of individuals with multiple abnormalities depend on the specific abnormalities present and their impact on the individual's health and functioning. A multidisciplinary team of healthcare professionals is often involved in the care of these individuals to address their complex needs.
A chromosome deletion is a type of genetic abnormality that occurs when a portion of a chromosome is missing or deleted. Chromosomes are thread-like structures located in the nucleus of cells that contain our genetic material, which is organized into genes.
Chromosome deletions can occur spontaneously during the formation of reproductive cells (eggs or sperm) or can be inherited from a parent. They can affect any chromosome and can vary in size, from a small segment to a large portion of the chromosome.
The severity of the symptoms associated with a chromosome deletion depends on the size and location of the deleted segment. In some cases, the deletion may be so small that it does not cause any noticeable symptoms. However, larger deletions can lead to developmental delays, intellectual disabilities, physical abnormalities, and various medical conditions.
Chromosome deletions are typically detected through a genetic test called karyotyping, which involves analyzing the number and structure of an individual's chromosomes. Other more precise tests, such as fluorescence in situ hybridization (FISH) or chromosomal microarray analysis (CMA), may also be used to confirm the diagnosis and identify the specific location and size of the deletion.
Persistent Truncus Arteriosus is a rare congenital heart defect that is characterized by the failure of the truncus arteriosus to divide into the separate pulmonary artery and aorta during fetal development. This results in a single large vessel, the truncus arteriosus, which gives rise to both the systemic and pulmonary circulations.
The truncus arteriosus contains a single semilunar valve, instead of the two separate semilunar valves (pulmonary and aortic) found in a normal heart. Additionally, there is often a ventricular septal defect (VSD), a hole in the wall between the two lower chambers of the heart, present.
This condition leads to mixing of oxygenated and deoxygenated blood within the truncus arteriosus, resulting in cyanosis (bluish discoloration of the skin and mucous membranes) and decreased oxygen delivery to the body. Symptoms typically appear soon after birth and may include difficulty breathing, poor feeding, rapid heart rate, and failure to thrive.
Persistent truncus arteriosus is usually treated with surgical repair in infancy or early childhood to separate the pulmonary and systemic circulations, close the VSD, and reconstruct the great vessels as needed.
The branchial region, also known as the pharyngeal region or viscerocranium, is a term used in human anatomy to refer to the area of the developing embryo that gives rise to structures derived from the branchial (or pharyngeal) arches. The branchial arches are a series of paired, rod-like structures that appear early in embryonic development and give rise to various head and neck structures, including the bones and muscles of the face, jaws, and neck, as well as the associated nerves, blood vessels, and connective tissues.
The branchial region is divided into several subregions, each corresponding to a specific branchial arch. The first branchial arch gives rise to structures such as the mandible (lower jaw), maxilla (upper jaw), and muscles of mastication (chewing). The second branchial arch forms the stapes and styloid process in the ear, as well as some neck muscles. The third and fourth branchial arches contribute to the formation of the larynx, thyroid cartilage, and other structures in the neck.
Abnormalities in the development of the branchial region can lead to a variety of congenital defects, such as cleft palate, micrognathia (small jaw), and branchial cysts or sinuses. These conditions may require surgical intervention to correct.
Histone chaperones are a group of proteins that play a crucial role in the process of nucleosome assembly and disassembly. They facilitate the transfer of histones, the protein components of nucleosomes, to and from DNA during various cellular processes such as DNA replication, repair, transcription, and chromatin remodeling.
Histone chaperones bind to histones and prevent their nonspecific aggregation or association with DNA. They help in the ordered deposition of histone proteins onto DNA, forming nucleosomes, which are the fundamental units of chromatin structure. Additionally, they assist in the removal of histones from DNA during transcription, DNA repair, and replication. Histone chaperones contribute to the dynamic regulation of chromatin structure and function, thereby playing an essential role in epigenetic regulation and gene expression.
Hypocalcemia is a medical condition characterized by an abnormally low level of calcium in the blood. Calcium is a vital mineral that plays a crucial role in various bodily functions, including muscle contraction, nerve impulse transmission, and bone formation. Normal calcium levels in the blood usually range from 8.5 to 10.2 milligrams per deciliter (mg/dL). Hypocalcemia is typically defined as a serum calcium level below 8.5 mg/dL or, when adjusted for albumin (a protein that binds to calcium), below 8.4 mg/dL (ionized calcium).
Hypocalcemia can result from several factors, such as vitamin D deficiency, hypoparathyroidism (underactive parathyroid glands), kidney dysfunction, certain medications, and severe magnesium deficiency. Symptoms of hypocalcemia may include numbness or tingling in the fingers, toes, or lips; muscle cramps or spasms; seizures; and, in severe cases, cognitive impairment or cardiac arrhythmias. Treatment typically involves correcting the underlying cause and administering calcium and vitamin D supplements to restore normal calcium levels in the blood.
Hypoparathyroidism is a medical condition characterized by decreased levels or insufficient function of parathyroid hormone (PTH), which is produced and released by the parathyroid glands. These glands are located in the neck, near the thyroid gland, and play a crucial role in regulating calcium and phosphorus levels in the body.
In hypoparathyroidism, low PTH levels result in decreased absorption of calcium from the gut, increased excretion of calcium through the kidneys, and impaired regulation of bone metabolism. This leads to low serum calcium levels (hypocalcemia) and high serum phosphorus levels (hyperphosphatemia).
Symptoms of hypoparathyroidism can include muscle cramps, spasms, or tetany (involuntary muscle contractions), numbness or tingling sensations in the fingers, toes, and around the mouth, fatigue, weakness, anxiety, cognitive impairment, and in severe cases, seizures. Hypoparathyroidism can be caused by various factors, including surgical removal or damage to the parathyroid glands, autoimmune disorders, radiation therapy, genetic defects, or low magnesium levels. Treatment typically involves calcium and vitamin D supplementation to maintain normal serum calcium levels and alleviate symptoms. In some cases, recombinant PTH (Natpara) may be prescribed as well.
In situ hybridization, fluorescence (FISH) is a type of molecular cytogenetic technique used to detect and localize the presence or absence of specific DNA sequences on chromosomes through the use of fluorescent probes. This technique allows for the direct visualization of genetic material at a cellular level, making it possible to identify chromosomal abnormalities such as deletions, duplications, translocations, and other rearrangements.
The process involves denaturing the DNA in the sample to separate the double-stranded molecules into single strands, then adding fluorescently labeled probes that are complementary to the target DNA sequence. The probe hybridizes to the complementary sequence in the sample, and the location of the probe is detected by fluorescence microscopy.
FISH has a wide range of applications in both clinical and research settings, including prenatal diagnosis, cancer diagnosis and monitoring, and the study of gene expression and regulation. It is a powerful tool for identifying genetic abnormalities and understanding their role in human disease.
Congenital heart defects (CHDs) are structural abnormalities in the heart that are present at birth. They can affect any part of the heart's structure, including the walls of the heart, the valves inside the heart, and the major blood vessels that lead to and from the heart.
Congenital heart defects can range from mild to severe and can cause various symptoms depending on the type and severity of the defect. Some common symptoms of CHDs include cyanosis (a bluish tint to the skin, lips, and fingernails), shortness of breath, fatigue, poor feeding, and slow growth in infants and children.
There are many different types of congenital heart defects, including:
1. Septal defects: These are holes in the walls that separate the four chambers of the heart. The two most common septal defects are atrial septal defect (ASD) and ventricular septal defect (VSD).
2. Valve abnormalities: These include narrowed or leaky valves, which can affect blood flow through the heart.
3. Obstruction defects: These occur when blood flow is blocked or restricted due to narrowing or absence of a part of the heart's structure. Examples include pulmonary stenosis and coarctation of the aorta.
4. Cyanotic heart defects: These cause a lack of oxygen in the blood, leading to cyanosis. Examples include tetralogy of Fallot and transposition of the great arteries.
The causes of congenital heart defects are not fully understood, but genetic factors and environmental influences during pregnancy may play a role. Some CHDs can be detected before birth through prenatal testing, while others may not be diagnosed until after birth or later in childhood. Treatment for CHDs may include medication, surgery, or other interventions to improve blood flow and oxygenation of the body's tissues.
Immunologic deficiency syndromes refer to a group of disorders characterized by defective functioning of the immune system, leading to increased susceptibility to infections and malignancies. These deficiencies can be primary (genetic or congenital) or secondary (acquired due to environmental factors, medications, or diseases).
Primary immunodeficiency syndromes (PIDS) are caused by inherited genetic mutations that affect the development and function of immune cells, such as T cells, B cells, and phagocytes. Examples include severe combined immunodeficiency (SCID), common variable immunodeficiency (CVID), Wiskott-Aldrich syndrome, and X-linked agammaglobulinemia.
Secondary immunodeficiency syndromes can result from various factors, including:
1. HIV/AIDS: Human Immunodeficiency Virus infection leads to the depletion of CD4+ T cells, causing profound immune dysfunction and increased vulnerability to opportunistic infections and malignancies.
2. Medications: Certain medications, such as chemotherapy, immunosuppressive drugs, and long-term corticosteroid use, can impair immune function and increase infection risk.
3. Malnutrition: Deficiencies in essential nutrients like protein, vitamins, and minerals can weaken the immune system and make individuals more susceptible to infections.
4. Aging: The immune system naturally declines with age, leading to an increased incidence of infections and poorer vaccine responses in older adults.
5. Other medical conditions: Chronic diseases such as diabetes, cancer, and chronic kidney or liver disease can also compromise the immune system and contribute to immunodeficiency syndromes.
Immunologic deficiency syndromes require appropriate diagnosis and management strategies, which may include antimicrobial therapy, immunoglobulin replacement, hematopoietic stem cell transplantation, or targeted treatments for the underlying cause.
Monosomy is a type of chromosomal abnormality in which there is only one copy of a particular chromosome instead of the usual pair in a diploid cell. In monosomy, an individual has one less chromosome than the normal diploid number (46 chromosomes) due to the absence of one member of a chromosome pair. This condition arises from the loss of one chromosome in an egg or sperm during gamete formation or at conception.
Examples of monosomy include Turner syndrome, which is characterized by the presence of only one X chromosome (45,X), and Cri du Chat syndrome, which results from a deletion of a portion of the short arm of chromosome 5 (46,del(5)(p15.2)). Monosomy can lead to developmental abnormalities, physical defects, intellectual disabilities, and various health issues depending on the chromosome involved.
The thymus gland is an essential organ of the immune system, located in the upper chest, behind the sternum and surrounding the heart. It's primarily active until puberty and begins to shrink in size and activity thereafter. The main function of the thymus gland is the production and maturation of T-lymphocytes (T-cells), which are crucial for cell-mediated immunity, helping to protect the body from infection and cancer.
The thymus gland provides a protected environment where immune cells called pre-T cells develop into mature T cells. During this process, they learn to recognize and respond appropriately to foreign substances while remaining tolerant to self-tissues, which is crucial for preventing autoimmune diseases.
Additionally, the thymus gland produces hormones like thymosin that regulate immune cell activities and contribute to the overall immune response.
The parathyroid glands are four small endocrine glands located in the neck, usually near or behind the thyroid gland. They secrete parathyroid hormone (PTH), which plays a critical role in regulating calcium and phosphate levels in the blood and bones. PTH helps maintain the balance of these minerals by increasing the absorption of calcium from food in the intestines, promoting reabsorption of calcium in the kidneys, and stimulating the release of calcium from bones when needed. Additionally, PTH decreases the excretion of calcium through urine and reduces phosphate reabsorption in the kidneys, leading to increased phosphate excretion. Disorders of the parathyroid glands can result in conditions such as hyperparathyroidism (overactive glands) or hypoparathyroidism (underactive glands), which can have significant impacts on calcium and phosphate homeostasis and overall health.
Gene deletion is a type of mutation where a segment of DNA, containing one or more genes, is permanently lost or removed from a chromosome. This can occur due to various genetic mechanisms such as homologous recombination, non-homologous end joining, or other types of genomic rearrangements.
The deletion of a gene can have varying effects on the organism, depending on the function of the deleted gene and its importance for normal physiological processes. If the deleted gene is essential for survival, the deletion may result in embryonic lethality or developmental abnormalities. However, if the gene is non-essential or has redundant functions, the deletion may not have any noticeable effects on the organism's phenotype.
Gene deletions can also be used as a tool in genetic research to study the function of specific genes and their role in various biological processes. For example, researchers may use gene deletion techniques to create genetically modified animal models to investigate the impact of gene deletion on disease progression or development.
The neural crest is a transient, multipotent embryonic cell population that originates from the ectoderm (outermost layer) of the developing neural tube (precursor to the central nervous system). These cells undergo an epithelial-to-mesenchymal transition and migrate throughout the embryo, giving rise to a diverse array of cell types and structures.
Neural crest cells differentiate into various tissues, including:
1. Peripheral nervous system (PNS) components: sensory neurons, sympathetic and parasympathetic ganglia, and glial cells (e.g., Schwann cells).
2. Facial bones and cartilage, as well as connective tissue of the skull.
3. Melanocytes, which are pigment-producing cells in the skin.
4. Smooth muscle cells in major blood vessels, heart, gastrointestinal tract, and other organs.
5. Secretory cells in endocrine glands (e.g., chromaffin cells of the adrenal medulla).
6. Parts of the eye, such as the cornea and iris stroma.
7. Dental tissues, including dentin, cementum, and dental pulp.
Due to their wide-ranging contributions to various tissues and organs, neural crest cells play a crucial role in embryonic development and organogenesis. Abnormalities in neural crest cell migration or differentiation can lead to several congenital disorders, such as neurocristopathies.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Chromosome mapping, also known as physical mapping, is the process of determining the location and order of specific genes or genetic markers on a chromosome. This is typically done by using various laboratory techniques to identify landmarks along the chromosome, such as restriction enzyme cutting sites or patterns of DNA sequence repeats. The resulting map provides important information about the organization and structure of the genome, and can be used for a variety of purposes, including identifying the location of genes associated with genetic diseases, studying evolutionary relationships between organisms, and developing genetic markers for use in breeding or forensic applications.
Translocation, genetic, refers to a type of chromosomal abnormality in which a segment of a chromosome is transferred from one chromosome to another, resulting in an altered genome. This can occur between two non-homologous chromosomes (non-reciprocal translocation) or between two homologous chromosomes (reciprocal translocation). Genetic translocations can lead to various clinical consequences, depending on the genes involved and the location of the translocation. Some translocations may result in no apparent effects, while others can cause developmental abnormalities, cancer, or other genetic disorders. In some cases, translocations can also increase the risk of having offspring with genetic conditions.
The thoracic aorta is the segment of the largest artery in the human body (the aorta) that runs through the chest region (thorax). The thoracic aorta begins at the aortic arch, where it branches off from the ascending aorta, and extends down to the diaphragm, where it becomes the abdominal aorta.
The thoracic aorta is divided into three parts: the ascending aorta, the aortic arch, and the descending aorta. The ascending aorta rises from the left ventricle of the heart and is about 2 inches (5 centimeters) long. The aortic arch curves backward and to the left, giving rise to the brachiocephalic trunk, the left common carotid artery, and the left subclavian artery. The descending thoracic aorta runs downward through the chest, passing through the diaphragm to become the abdominal aorta.
The thoracic aorta supplies oxygenated blood to the upper body, including the head, neck, arms, and chest. It plays a critical role in maintaining blood flow and pressure throughout the body.
 DiGeorge syndrome
DiGeorge syndrome
LZTR1
Angelo DiGeorge
TBX1
Ultimopharyngeal body
Interrupted aortic arch
Quinn Bradlee
Thymus transplantation
DGCR14
Microprocessor complex subunit DGCR8
DGCR6
Genocopy
DGCR2
1968 in science
Transplantable organs and tissues
RVT-802
Fertilysin
Chromosome 22
Metirosine
HIRA
ZNF74
Microprocessor complex
Deborah F. Kelly
DGCR5
22q11.2 distal deletion syndrome
Tetralogy of Fallot
Retinoic acid
TANGO2
Vici syndrome
22q11.2 duplication syndrome
DiGeorge syndrome - Wikipedia
 DiGeorge Syndrome Demystifed | ScienceBlogs
DiGeorge Syndrome Demystifed | ScienceBlogs
 DiGeorge Syndrome: Practice Essentials, Background, Pathophysiology
DiGeorge Syndrome: Practice Essentials, Background, Pathophysiology
 WATCH: Sweet 16 parade for girl with DiGeorge Syndrome Video | All Good
WATCH: Sweet 16 parade for girl with DiGeorge Syndrome Video | All Good
A region of homozygosity within 22q11.2 associated with congenital heart disease: recessive DiGeorge/velocardiofacial syndrome?...
 Quiz: DiGeorge Syndrome - MSD Manual Consumer Version
Quiz: DiGeorge Syndrome - MSD Manual Consumer Version
 DiGeorge Syndrome Archives - Global Genes
DiGeorge Syndrome Archives - Global Genes
 Genetic testing - Deletion 22q11.2 (See DiGeorge syndrome ...). - IVAMI
Genetic testing - Deletion 22q11.2 (See DiGeorge syndrome ...). - IVAMI
 DIGEORGE SYNDROME (22q11.2)
DIGEORGE SYNDROME (22q11.2)
 DiGeorge Syndrome - WeHeal.org
DiGeorge Syndrome - WeHeal.org
 DiGeorge syndrome Archieven - Amsterdam UMC Genome Diagnostics
DiGeorge syndrome Archieven - Amsterdam UMC Genome Diagnostics
DiGeorge Syndrome - Immune Disorders - MSD Manual Consumer Version
 Microdeletion 22q11.2
Microdeletion 22q11.2
 DiGeorge (22q11.2 deletion) syndrome: Clinical features and diagnosis
DiGeorge (22q11.2 deletion) syndrome: Clinical features and diagnosis
Genetic dosage compensation in a family with velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome<...
 Disrupted Coordination of Hypoglossal Motor Control in a Mouse Model of Pediatric Dysphagia in DiGeorge/22q11.2 Deletion...
Disrupted Coordination of Hypoglossal Motor Control in a Mouse Model of Pediatric Dysphagia in DiGeorge/22q11.2 Deletion...
Structure of the DNA-bound T-box domain of human TBX1, a transcription factor associated with the DiGeorge syndrome - Wellcome...
 22Q11.2 Deletion Syndrome (Digeorge Syndrome, Velocardiofacial Syndrome) | Select 5-Minute Pediatrics Topics
22Q11.2 Deletion Syndrome (Digeorge Syndrome, Velocardiofacial Syndrome) | Select 5-Minute Pediatrics Topics
 Časopis Paediatrics - Článok DiGeorge syndrome in the paediatric clinical practice - one center experience
Časopis Paediatrics - Článok DiGeorge syndrome in the paediatric clinical practice - one center experience
 22q11.2 deletion syndrome: MedlinePlus Genetics
22q11.2 deletion syndrome: MedlinePlus Genetics
 D
D
 ACIP Altered Immunocompetence Guidelines for Immunizations | CDC
ACIP Altered Immunocompetence Guidelines for Immunizations | CDC
Low-copy-number repeat sequences flank the DiGeorge/velo-cardio-facial syndrome loci at 22q11. - Oxford Neuroscience
Intrauterine Growth Restriction (IUGR) Imaging: Practice Essentials, Ultrasonography
 Nutrients | Free Full-Text | Vegan Nutrition for Mothers and Children: Practical Tools for Healthcare Providers
Nutrients | Free Full-Text | Vegan Nutrition for Mothers and Children: Practical Tools for Healthcare Providers
Table A1 - Human Parechovirus Infections in Canada - Volume 12, Number 6-June 2006 - Emerging Infectious Diseases journal - CDC
 Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on...
Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on...
 Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. | Clin Immunol;147(1): 11...
Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. | Clin Immunol;147(1): 11...
 Cleft & Craniofacial Program: Conditions We Treat | Johns Hopkins Medicine
Cleft & Craniofacial Program: Conditions We Treat | Johns Hopkins Medicine
 "FISHED" OUT THE CORRECT DIAGNOSIS: A CASE OF DIGEORGE SYNDROME WITH MENTAL RETARDATION, SHORT STATURE AND DYSMORPHIC FACIAL...
"FISHED" OUT THE CORRECT DIAGNOSIS: A CASE OF DIGEORGE SYNDROME WITH MENTAL RETARDATION, SHORT STATURE AND DYSMORPHIC FACIAL...Velocardiofacial7
- DiGeorge syndrome (DGS) is one of a group of phenotypically similar disorders-including velocardiofacial syndrome (VCFS, or Shprintzen syndrome) and conotruncal anomaly face (CTAF) syndrome-that share a microdeletion of chromosome 22q11.2, a region known as the DGS critical region (see the image below). (medscape.com)
- A region of homozygosity within 22q11.2 associated with congenital heart disease: recessive DiGeorge/velocardiofacial syndrome? (bmj.com)
- E ditor -DiGeorge syndrome (DGS, MIM 18840) and velocardiofacial syndrome (VCFS, MIM 192430) are associated with interstitial deletions of chromosome 22q11.2 and are considered to be phenotypic variations of the same underlying genetic defect. (bmj.com)
- Chromosome 22q11.2 deletion syndrome (22qDS) includes DGS and other similar syndromes, such as velocardiofacial syndrome. (medilib.ir)
- 22q11.2 deletion syndrome, formerly known as DiGeorge or velocardiofacial syndrome, is a multisystem disorder with variable severity and number of associated features, classically including developmental delay, learning difficulties, congenital cardiac anomalies, palatal abnormalities, especially velopharyngeal insufficiency, hypocalcemia, and subtle facial dysmorphism. (unboundmedicine.com)
- Doctors named these conditions DiGeorge syndrome, velocardiofacial syndrome (also called Shprintzen syndrome), and conotruncal anomaly face syndrome. (medlineplus.gov)
- It encompasses several syndromes with overlapping abnormalities including the DIGEORGE SYNDROME, VELOCARDIOFACIAL SYNDROME, and CONOTRUNCAL AMOMALY FACE SYNDROME. (harvard.edu)
Velo-cardio-f3
- The original classifications included velo-cardio-facial syndrome, Shprintzen syndrome, 22q11 deletion syndrome, Sedlackova syndrome and conotruncal anomaly face syndrome. (scienceblogs.com)
- Low-copy-number repeat sequences flank the DiGeorge/velo-cardio-facial syndrome loci at 22q11. (ox.ac.uk)
- DiGeorge syndrome and velo-cardio-facial syndrome are associated with deletions within 22q11. (ox.ac.uk)
Thymus3
- Accordingly, I urge all DiGeorge researchers, "DiGeorge families", and the entire medical community to respect and honor the remarkable man who first described the syndrome and who discovered the role of the thymus gland in human function by continuing to call it what it is, DiGeorge Syndrome. (scienceblogs.com)
- Children with DiGeorge syndrome are born with several abnormalities, including heart defects, underdeveloped or absent parathyroid glands, an underdeveloped or absent thymus gland, and characteristic facial features. (msdmanuals.com)
- The syndrome is marked by absence or underdevelopment of the thymus and parathyroid glands. (healthofchildren.com)
22q11.2 deletion39
- DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22. (wikipedia.org)
- All these syndromes, because of their overlapping features, are now designated as a 22q11.2 deletion syndrome (22q11.2DS) and in the rest of the article will be referred to as 22q11.2DS. (medscape.com)
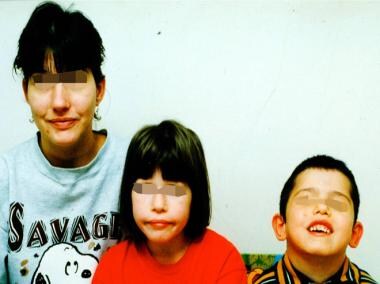
- Mother and children with 22q11.2 deletion syndrome. (medscape.com)
- 22q11.2 deletion syndrome has a prevalence of 1 in 2,000 -that is thought to be an underestimate due to the lack of syndrome familiarity and recognition by doctors, and the wide variability of symptoms. (medicover-genetics.com)
- It is the second most common genetic disorder after Down syndrome, the most common cause of cleft lip and palate, and the second most common cause of developmental delays and heart defects - 1 in 68 children born with a cardiovascular abnormality are diagnosed with a 22q11.2 deletion. (medicover-genetics.com)
- See "DiGeorge (22q11.2 deletion) syndrome: Epidemiology and pathogenesis" and "DiGeorge (22q11.2 deletion) syndrome: Management and prognosis" . (medilib.ir)
- Cytogenetic studies of a male child carrying the 22q11.2 deletion common in patients with velo-cardio-facial/DiGeorge syndrome showed an unexpected rearrangement of the 22q11.2 region in his normal appearing mother. (elsevierpure.com)
- 22q11.2 deletion syndrome has many possible signs and symptoms that can affect almost any part of the body. (medlineplus.gov)
- People with 22q11.2 deletion syndrome commonly have heart abnormalities that are often present from birth, recurrent infections caused by problems with the immune system, and distinctive facial features. (medlineplus.gov)
- Many children with 22q11.2 deletion syndrome have developmental delays, including delayed growth and speech development, and some have mild intellectual disability or learning disabilities. (medlineplus.gov)
- Additionally, affected children are more likely than children without 22q11.2 deletion syndrome to have attention-deficit/hyperactivity disorder (ADHD) and developmental conditions such as autism spectrum disorder that affect communication and social interaction. (medlineplus.gov)
- Because the signs and symptoms of 22q11.2 deletion syndrome are so varied, different groupings of features were once described as separate conditions. (medlineplus.gov)
- In addition, some children with the 22q11.2 deletion were diagnosed with the autosomal dominant form of Opitz G/BBB syndrome and Cayler cardiofacial syndrome. (medlineplus.gov)
- To avoid confusion, this condition is usually called 22q11.2 deletion syndrome, a description based on its underlying genetic cause. (medlineplus.gov)
- 22q11.2 deletion syndrome affects an estimated 1 in 4,000 people. (medlineplus.gov)
- Researchers are working to identify all of the genes that contribute to the features of 22q11.2 deletion syndrome. (medlineplus.gov)
- Chromosome 22q11.2 deletion syndrome is a neurogenetic disorder associated with high rates of schizophrenia and other psychiatric conditions. (nih.gov)
- The authors report what is to their knowledge the first large-scale collaborative study of rates and sex distributions of psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome. (nih.gov)
- The 1,402 participants with 22q11.2 deletion syndrome, ages 6-68 years, were assessed for psychiatric disorders with validated diagnostic instruments. (nih.gov)
- To the authors' knowledge, this is the largest study of psychiatric morbidity in 22q11.2 deletion syndrome. (nih.gov)
- These results highlight the need to monitor and reduce the long-term burden of psychopathology in 22q11.2 deletion syndrome. (nih.gov)
- Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. (bvsalud.org)
- Patients with 22q11.2 deletion syndrome have heterogeneous clinical presentations including immunodeficiency, cardiac anomalies, and hypocalcemia . (bvsalud.org)
- We characterized the microRNA expression patterns in the peripheral blood of patients with 22q11.2 deletion syndrome (n=31) compared to normal controls (n=22). (bvsalud.org)
- 22q11.2 deletion syndrome cohort exhibited microRNA expression hyper-variability and group dysregulation. (bvsalud.org)
- In summary, microRNA profiling of chromosome 22q11.2 deletion syndrome /DiGeorge patients revealed a signature microRNA expression pattern distinct from normal controls with clinical relevance . (bvsalud.org)
- Bleeding Severity and Phenotype in 22q11.2 Deletion Syndrome-A Cross-Sectional Investigation. (harvard.edu)
- Abnormalities in gray matter microstructure in young adults with 22q11.2 deletion syndrome. (harvard.edu)
- Failed Progenitor Specification Underlies the Cardiopharyngeal Phenotypes in a Zebrafish Model of 22q11.2 Deletion Syndrome. (harvard.edu)
- 22q11.2 deletion syndrome, also known commonly as DiGeorge syndrome, is a condition that varies greatly in severity among affected individuals (including family members). (childrensdayton.org)
- how rare is 22q11.2 deletion syndrome? (childrensdayton.org)
- are kids born with 22q11.2 deletion syndrome? (childrensdayton.org)
- what is the life expectancy of someone with 22q11.2 deletion syndrome? (childrensdayton.org)
- how do I know if my child will be born with 22q11.2 deletion syndrome? (childrensdayton.org)
- After birth, if 22q11.2 deletion syndrome is suspected, tests such as CT, MRI, heart ultrasound and other testing may be ordered. (childrensdayton.org)
- what are the genes responsible for 22q11.2 deletion syndrome? (childrensdayton.org)
- how do you treat 22q11.2 deletion syndrome? (childrensdayton.org)
- Patients with 22q11.2 Deletion Syndrome (22q11.2DS) have a 25-30% risk of developing schizophrenia, and also suffer frequent hearing loss. (biorxiv.org)
- In the Df1 /+ mouse model of human 22q11.2 Deletion Syndrome, we find that hearing loss shapes measures that are considered schizophrenia-relevant endophenotypes, such as central auditory gain and auditory sensorimotor gating. (biorxiv.org)
Microdeletion3
- DiGeorge syndrome represents a part of the highly variable clinical spectrum of microdeletion 22q11.2. (medicover-genetics.com)
- Noninvasive prenatal testing (NIPT), also known as microdeletion tests, were first created to test for Down syndrome. (aboutlawsuits.com)
- Doctors decide what to order and companies sell microdeletion testing as an option to add-on to the Down syndrome screening. (aboutlawsuits.com)
Angelo DiGeorge2
- The syndrome was first described in 1968 by American physician Angelo DiGeorge. (wikipedia.org)
- DiGeorge Syndrome is named after my father doctor Angelo DiGeorge who died three years ago. (scienceblogs.com)
Immunodeficiency syndromes2
Disorder9
- Take, for instance, a common group of birth defects - forms of a disorder called DiGeorge syndrome. (scienceblogs.com)
- Jasmine Jefferson was born with a disorder called DiGeorge syndrome that has caused her to be in strict quarantine since March 16th. (abc.com)
- ABSTRACT Sanjad Sakati syndrome is a rare autosomal recessive disorder that has been described in Arabs. (who.int)
- Sanjad Sakati syndrome or hypoparathyroidism-retardation-dysmorphism (HRD) is an autosomal recessive disorder that was first described in 1988 [1]. (who.int)
- Dr. Mathews added that although the findings need to be replicated, clinicians should monitor pregnancies for these risk factors if either parent has Tourette syndrome or if there is a family history of the disorder. (medscape.com)
- Tourette syndrome is a phenomenologically heterogeneous neuropsychiatric disorder comprising multiple motor and vocal tics that begin in early childhood and persist for at least a year. (medscape.com)
- The primary outcome measures were diagnoses of Tourette syndrome and either Tourette syndrome or chronic tic disorder (grouped together as Tourette syndrome/chronic tic disorder) based on criteria from the Diagnostic and Statistical Manual of Mental Disorder, Fourth Edition, Text Revision (DSM-IV-TR) when the children were 13 or 14 years old, as well as tic-related information from maternal questionnaires. (medscape.com)
- A total of 50 of the teens had Tourette syndrome, and 72 had chronic tic disorder. (medscape.com)
- P = .002 for Tourette syndrome/chronic tic disorder). (medscape.com)
Genes4
- DiGeorge syndrome is typically due to the deletion of 30 to 40 genes in the middle of chromosome 22 at a location known as 22q11.2. (wikipedia.org)
- This condition is described as a contiguous gene deletion syndrome because it results from the loss of many genes that are close together. (medlineplus.gov)
- The syndrome arises from hemizygous deletions of up to 3Mb on chromosome 22q11.2, a region that contains 60 genes and 4 microRNAs . (bvsalud.org)
- El síndrome DiGeorge es causado por la supresión de varios genes del cromosoma 22. (epnet.com)
22q11 deletion4
- 22q11 Deletion Syndrome" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (harvard.edu)
- This graph shows the total number of publications written about "22q11 Deletion Syndrome" by people in Harvard Catalyst Profiles by year, and whether "22q11 Deletion Syndrome" was a major or minor topic of these publication. (harvard.edu)
- Below are the most recent publications written about "22q11 Deletion Syndrome" by people in Profiles. (harvard.edu)
- Frontal Hypoactivation During a Working Memory Task in Children With 22q11 Deletion Syndrome. (harvard.edu)
Diagnosis1
- The availability of genetic testing enables accurate diagnosis of affected children, discovery of carriers and prospective counselling as well as prenatal diagnosis of Sanjad Sakati syndrome in high-risk families. (who.int)
Prader-Willi1
- Prader-Willi and Angelman syndromes can cause seizures and inability to control food consumption and offer a child little chance of living independently as an adult. (aboutlawsuits.com)
Anomaly1
- DiGeorge anomaly is part of a rare congenital abnormality that is the result of defects during early fetal developmental. (lu.se)
Cayler1
- Cayler cardiofacial syndrome. (childrensdayton.org)
Mental retardation1
- Down syndrome is the most common cause of mental retardation and malformation in a newborn. (healthofchildren.com)
Chromosome 223
- Around one in 4000 is born with this syndrome, which arises from a deletion of a short segment of chromosome 22. (scienceblogs.com)
- The small deletion on the long arm (q) of chromosome 22 causes a syndrome with a diverse collection of symptoms. (medicover-genetics.com)
- DiGeorge syndrome is a rare congenital disease that affects an infant's immune system and that is due to a large deletion from chromosome 22. (healthofchildren.com)
Genetic deletion1
- I enjoy following the tremendous research being done all over the world in connection with DiGeorge Syndrome, which is by all accounts the most common chromosomal genetic deletion syndrome. (scienceblogs.com)
Disorders4
- When the genetic basis of 22q11.2 syndrome was identified, it was discovered that several other conditions thought to be different disorders were all in fact part of the same condition. (medicover-genetics.com)
- Medical problems commonly associated with DiGeorge syndrome include heart defects, poor immune system function, a cleft palate, complications related to low levels of calcium in the blood and behavioral disorders. (weheal.org)
- Once the genetic basis for these disorders was identified, doctors determined that they were all part of a single syndrome with many possible signs and symptoms. (medlineplus.gov)
- Inadequate maternal weight gain, cannabis use during pregnancy, and birth order have been identified as 3 new potential risk factors for Tourette syndrome and chronic tic disorders in children, new research shows. (medscape.com)
Severity1
- The number and severity of problems associated with DiGeorge syndrome vary greatly. (weheal.org)
Symptoms2
- The reason for this is that the symptoms of DiGeorge syndrome vary considerably. (scienceblogs.com)
- INTRODUCTION - DiGeorge syndrome (DGS) is a constellation of signs and symptoms associated with defective development of the pharyngeal pouch system. (medilib.ir)
4,000 people1
- DiGeorge syndrome occurs in about 1 in 4,000 people. (wikipedia.org)
Fluorescence1
- In attempting to refine the shortest region of overlap for these syndromes we have employed fluorescence in situ hybridisation. (ox.ac.uk)
Deletion syndrome2
- I am also pleased that you correctly refer to it as "DiGeorge Syndrome" rather than as 22 q 11 deletion syndrome or by one of its many other monikers. (scienceblogs.com)
- Available at: http://www.stanfordchildrens.org/en/topic/default?id=22q112-deletion-syndrome-90-P01682. (epnet.com)
Thymic1
- Chandran M, Deftos LJ, Stuenkel CA, Haghighi P, Orloff LA. Thymic parathyroid carcinoma and postoperative hungry bone syndrome. (medscape.com)
Developmental1
- In addition, variable developmental problems and schizoid features are also associated with this syndrome. (harvard.edu)
Deletions1
- 2009 Feb 25;10:16) Not all deletions at 22q11 result in the 22q11deletion syndrome. (harvard.edu)
Hypocalcemia1
- DiGeorge syndrome may be first spotted when an affected newborn has heart defects or convulsions from hypocalcemia due to malfunctioning parathyroid glands and low levels of parathyroid hormone (parathormone). (wikipedia.org)
Commonly1
- 22q11.2 syndrome is caused by a genetic mutation change, more commonly now known as a genetic variant. (childrensdayton.org)
Facial1
- They found that knockouts of specific transcription factors that were not previously linked to Digeorge were tied to distinctive combinations of facial muscle and cardiac defects resemble the congenital defects in babies. (scienceblogs.com)
Features of this syndrome2
- The features of this syndrome vary widely, even among members of the same family, and affect many parts of the body. (wikipedia.org)
- However, due to the variable features of this syndrome, it is often underdiagnosed and researchers estimate it occurs more frequently. (childrensdayton.org)
Patients1
- This case series reports 8 patients with Sanjad Sakati syndrome from 7 families in Jordan, 6 of whom had genetic testing. (who.int)
Heart3
- And yes, they revealed that, at least for those with DiGeorge syndrome, a face can tell something about the heart. (scienceblogs.com)
- Available at: http://www.heart.org/HEARTORG/Conditions/CongenitalHeartDefects/DiGeorge-Syndrome_UCM_309017_Article.jsp. (epnet.com)
- For example, DiGeorge syndrome can cause heart defects. (aboutlawsuits.com)