Double Outlet Right Ventricle
Heart Septal Defects, Ventricular
Heart Defects, Congenital
Ventricular Septum
Transposition of Great Vessels
Heart Septal Defects
Pulmonary Valve Stenosis
Tetralogy of Fallot
Heart Ventricles
Evolution of risk factors influencing early mortality of the arterial switch operation. (1/36)
OBJECTIVES: The present study was undertaken to determine the independent risk factors for early mortality in the current era after arterial switch operation (ASO). BACKGROUND: Prior reports on factors affecting outcome of the ASO demonstrated that abnormal coronary arterial patterns were associated with increased risk of early mortality. As diagnostic, surgical and perioperative management techniques continue to evolve, the risk factors for the ASO may have changed. METHODS: All patients who underwent the ASO at Children's Hospital, Boston between January 1, 1992 and December 31, 1996 were included. Hospital charts, echocardiographic and cardiac catheterization data and operative reports of all patients were reviewed. Demographics and preoperative, intraoperative and postoperative variables were recorded. RESULTS: Of the 223 patients included in the study (median age at ASO = 6 days and median weight = 3.5 kg), 26 patients had aortic arch obstruction or interruption, 12 had Taussig-Bing anomaly, 12 had multiple ventricular septal defects, 8 had right ventricular hypoplasia and 6 were premature. There were 16 early deaths (7%), with 3 deaths in the 109 patients considered "low risk" (2.7%). Coronary artery pattern was not associated with an increased risk of death. Compared with usual coronary anatomy pattern, however, inverted coronary patterns and single right coronary patterns were associated with increased incidence of delayed sternal closure (p = 0.003) and longer duration of mechanical ventilation (p = 0.008). In a multivariate logistic regression model using only preoperative variables, aortic arch repair at a separate procedure before ASO and smaller birth weight were independent predictors of early mortality. In a second model that included both pre- and intraoperative variables, circulatory arrest time and right ventricular hypoplasia were independent predictors of early death. CONCLUSIONS: The ASO can be performed in the current era without excess early mortality related to uncommon coronary artery patterns. Aortic arch repair before ASO, right ventricular hypoplasia, lower birth weight and longer intraoperative support continue to be independent risk factors for early mortality after the ASO. (+info)Double-outlet right ventricle: an antenatal diagnostic dilemma. (2/36)
OBJECTIVE: The purpose of this study was to describe the antenatal ultrasonographic findings of fetuses with double-outlet right ventricle (DORV). DESIGN: The records were reviewed of all fetuses scanned in our ultrasound unit which were suspected of having DORV during a 6-year period ending in April 1996. A medical record search for all infants with a postnatal diagnosis of DORV was also undertaken to identify cases that were not detected antenatally. Records were examined to determine the accuracy of antenatal diagnosis and the reasons for diagnostic errors. Fetuses without follow-up were excluded. RESULTS: There were 20 fetuses with antenatally detected conotruncal defects that had DORV included in the differential diagnosis. Three fetuses were excluded, seven did not have DORV and ten were confirmed postnatally as having DORV. Two additional infants were found to have DORV from the medical record search, producing a total of 12 cases. Antenatal sonographic cardiac findings included malpositioned (overriding or transposed) great arteries (n = 11), ventricular septal defect (n = 11) and small pulmonary artery suggesting stenosis (n = 4). Confirmed postnatal cardiac findings that were missed antenatally included aortic coarctation (n = 2), right-sided aortic arch (n = 2) and pulmonary stenosis (n = 1). Seven of the 12 fetuses had extracardiac findings. Nine of the 12 fetuses tested had a normal karyotype. Eleven of the 12 infants were liveborn. Nine of these 11 survived the neonatal period and underwent surgical repair within the first year of life; two subsequently died. In total, seven fetuses survived and five did not. CONCLUSIONS: Most fetuses with DORV can be identified antenatally as having an abnormal heart. However, it is very difficult to distinguish this particular defect from other conotruncal abnormalities. (+info)Double outlet right ventricle with anterior and left-sided aorta and subpulmonary ventricular septal defect. (3/36)
Double outlet right ventricle (DORV) is a heterogeneous group of abnormal ventriculoarterial connections where, by definition, both great arteries (pulmonary artery and aorta) arise primarily from the morphologically right ventricle. This condition affects 1-1.5% of the patients with congenital heart diseases, with a frequency of 1 in each 10,000 live births. We report the case of an 18-day-old infant with DORV and extremely rare anatomical features, such as anterior and left-sided aorta and subpulmonary ventricular septal defect (VSD). In addition to the anatomic features, the role of the echocardiogram for guiding the diagnosis and the surgical therapy of this congenital heart disease are discussed. (+info)Antitachycardia burst pacing for pleomorphic reentrant ventricular tachycardias associated with non-coronary artery diseases: a morphology specific programming for ventricular tachycardias. (4/36)
To study the role of antitachycardia burst pacing in patients with reentrant pleomorphic ventricular tachycardia (VT) associated with non-coronary artery diseases, the efficacy of antitachycardia pacing and appropriate antitachycardia pacing cycle length were evaluated in each pleomorphic VT morphology of seven patients. Seven patients were included in this study. Clinically documented pleomorphic VTs were reproduced in an electrophysiologic study. For each VT, rapid ventricular pacing was attempted from the apex of the right ventricle at a cycle length which was 20 ms shorter than that of VT and repeated after a decrement of the cycle length in steps of 10 ms until the VT was terminated or accelerated. All 16 VTs could be entrained by the rapid pacing, and 13 of the 16 VTs (81%) were terminated, whereas pacing-induced acceleration was observed in the other 3 VTs of the 3 patients. VT cycle length (VTCL), block cycle length (BCL) which was defined as the longest VT interrupting paced cycle length, %BCL/VTCL and entrainment zone which was defined as VTCL minus BCL, varied in each VT morphology of each patient. In two patients, antitachycardia pacing was effective in all VT morphologies and the maximum difference of the %BCL/VTCL among the pleomorphic VTs was less than 10%. Thus, antitachycardia pacing seemed to be beneficial for these patients. In the other 5 patients, a difference of more than 10% in %BCL/VTCL was observed among the pleomorphic VT morphologies and/or at least one VT morphology showed pacing-induced acceleration. Compared to the 13 terminated VTs, three accelerated VTs had a wide entrainment zone [160 +/- 44 vs 90 +/- 48 ms, p < 0.04] and small %BCL/VTCL [61 +/- 6 vs 77 +/- 11%,p<0.03]. In pleomorphic VTs associated with non-coronary artery diseases, responses to rapid pacing was not uniform; VT might be terminable or accelerated even in the same patient. We need to pay close attention when programming antitachycardia pacing in patients with pleomorphic VT. (+info)Cardiovascular defects associated with abnormalities in midline development in the Loop-tail mouse mutant. (5/36)
Loop-tail (Lp) is a naturally occurring mouse mutant that develops severe neural tube defects. In this study, we describe complex cardiovascular defects in Lp homozygotes, which include double-outlet right ventricle, with obligatory perimembranous ventricular septal defects, and double-sided aortic arch, with associated abnormalities in the aortic arch arteries. Outflow tract and aortic arch defects are often related to abnormalities in the cardiac neural crest, but using molecular and anatomic markers, we show that neural crest migration is normal in Lp/Lp embryos. On the other hand, the heart fails to loop normally in Lp/Lp embryos, in association with incomplete axial rotation and reduced cervical flexion. As a consequence, the ventricular loop is shifted posteromedially relative to its position in wild-type embryos. This suggests that the observed cardiac alignment defects in the Lp mutant may be secondary to failure of neural tube closure and incomplete axial rotation. Double-sided aortic arch is a rare finding among mouse models. In humans, it is usually an isolated malformation, only rarely occurring in combination with other cardiac defects. We suggest that the double-sided arch arises as a primary defect in the Lp mutant, unrelated to the alignment defects, perhaps reflecting a role for the (as-yet-unknown) Lp gene in maintenance/regression of the aortic arch system. (+info)Echocardiographic characteristics and outcome of straddling mitral valve. (6/36)
OBJECTIVES: This study sought to characterize the echocardiographic features of straddling mitral valve (SMV) and to determine its surgical implications and midterm outcome in a large clinical cohort. BACKGROUND: Despite a relatively large body of literature on the postmortem anatomy of SMV, there is a paucity of information regarding its echocardiographic features, surgical implications and preoperative predictors of outcome. METHODS: A retrospective review identified 46 patients with SMV between 1982 and 1999 who underwent echocardiography and surgery and had follow-up data. A detailed review of the echocardiograms, surgical reports and all pertinent records was undertaken. RESULTS: Review of the echocardiograms revealed a widely varying anatomy among the study patients. However, four distinct groups with relatively uniform morphologic features could be distinguished on the basis of segmental analysis. Cardiac malposition associated with right ventricular hypoplasia, superior-inferior ventricles and criss-cross atrioventricular relations were common among patients with [S,D,L] (S = visceroatrial situs solitus, D = D-ventricular loop, L = L-malposition of the great arteries) (n = 6) and [S,L,D] (n = 5) segmental combinations but were rare among patients with [S,D,D] (n = 26) and [S,L,L] (n = 9) combinations. Surgical management consisted of a functional single-ventricle palliation in 38 patients (83%) and biventricular repair in 8 patients (17%). Overall mortality was 22%, but none of the seven patients who were operated on during the cohort's last five years (1994 to 1999) has died. By multivariate analysis, noncommitted ventricular septal defect was the strongest independent predictor of death (relative risk = 10.2), followed by multiple ventricular septal defects (relative risk = 4.7). CONCLUSIONS: This study demonstrates that echocardiography provides detailed noninvasive imaging of the complex anatomic features of SMV and its associated anomalies. Anatomic classification based on segmental analysis allows the distinction of four groups with more uniform morphologic features. Although a biventricular approach is feasible in selected patients, a functional univentricular palliation is indicated in those with major straddling and markedly hypoplastic ventricles. (+info)CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. (7/36)
Recent investigations identified heterozygous CFC1 mutations in subjects with heterotaxy syndrome, all of whom had congenital cardiac malformations, including malposition of the great arteries. We hypothesized that a subset of patients with similar types of congenital heart disease---namely, transposition of the great arteries and double-outlet right ventricle, in the absence of laterality defects---would also have CFC1 mutations. Our analysis of the CFC1 gene in patients with these cardiac disorders identified two disease-related mutations in 86 patients. The present study identifies the first autosomal single-gene defect for these cardiac malformations and indicates that some cases of transposition of the great arteries and double-outlet right ventricle can share a common genetic etiology with heterotaxy syndrome. In addition, these results demonstrate that the molecular pathway involving CFC1 plays a critical role in normal and abnormal cardiovascular development. (+info)Long-term predictors of aortic root dilation and aortic regurgitation after arterial switch operation. (8/36)
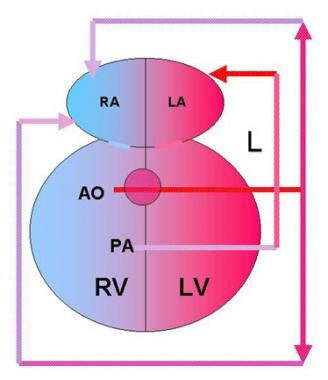
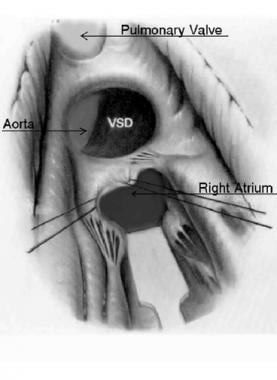
BACKGROUND: Neo-aortic root dilation (ARD) and neo-aortic regurgitation (AR) may be progressive after arterial switch operation (ASO) for d-loop transposition of the great arteries (dTGA). We sought to identify predictors of ARD and AR after ASO. METHODS AND RESULTS: 335 patients were identified who underwent ASO for dTGA with intact ventricular septum or ventricular septal defect (VSD), including double-outlet right ventricle (DORV), before 2001 with at least 1 postoperative echocardiogram at our institution, at least 1 year after ASO, and no previous atrial switch procedure (median follow-up of 5.0 years). Probability of freedom from ARD was 97%, 92%, 82%, and 51%, from at least moderate AR was 98%, 97%, 96%, and 93%, and from neo-aortic valve or root surgery was 100%, 100%, 99%, and 95%, at 1, 2, 5, and 10 years, respectively. For patients in whom ARD developed, progressive dilation was not observed during late follow-up. By Kaplan-Meier method, independent predictors of ARD, with neo-aortic root z-score of > or =3.0, were previous pulmonary artery band (PAB) (P=0.002, hazard ratio [HR]=2.4) and later time period when ASO was performed (P<0.002, HR=19.0). Risk factor for at least moderate AR was age > or =1 year at ASO (P=0.002, HR=5.8), which was closely related to VSD repair at ASO (P<0.001) and previous PAB. CONCLUSIONS: Significant ARD and AR continue to develop over time after ASO, but ARD does not tend to be progressive during late follow-up. Previous PAB was a significant risk factor for ARD. Older age at time of ASO, presence of VSD, and previous PAB were risk factors for AR. (+info)Double outlet right ventricle (DORV) is a congenital heart defect in which both great vessels (the aorta and the pulmonary artery) arise from the right ventricle. In a normal heart, the aorta arises from the left ventricle and the pulmonary artery arises from the right ventricle.
In DORV, there is a communication between the two ventricles (a ventricular septal defect), which allows oxygen-rich blood to mix with oxygen-poor blood. The location of this ventricular septal defect and the relationship of the great vessels to each other determine the physiology and the clinical manifestations of DORV.
DORV is a complex congenital heart defect that can range from mild to severe, and it often requires surgical intervention to improve blood flow and oxygenation. The prognosis for individuals with DORV depends on various factors, including the specific type of DORV, associated cardiac anomalies, and the timing and success of treatment.
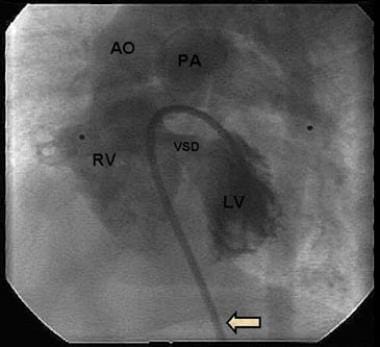
A ventricular septal defect (VSD) is a type of congenital heart defect that involves a hole in the wall separating the two lower chambers of the heart, the ventricles. This defect allows oxygenated blood from the left ventricle to mix with deoxygenated blood in the right ventricle, leading to inefficient oxygenation of the body's tissues. The size and location of the hole can vary, and symptoms may range from none to severe, depending on the size of the defect and the amount of blood that is able to shunt between the ventricles. Small VSDs may close on their own over time, while larger defects usually require medical intervention, such as medication or surgery, to prevent complications like pulmonary hypertension and heart failure.
Angiocardiography is a medical procedure used to examine the heart and blood vessels, particularly the chambers of the heart and the valves between them. It involves injecting a contrast agent into the bloodstream and taking X-ray images as the agent flows through the heart. This allows doctors to visualize any abnormalities such as blockages, narrowing, or leakage in the heart valves or blood vessels.
There are different types of angiocardiography, including:
* Left heart catheterization (LHC): A thin tube called a catheter is inserted into a vein in the arm or groin and threaded through to the left side of the heart to measure pressure and oxygen levels.
* Right heart catheterization (RHC): Similar to LHC, but the catheter is threaded through to the right side of the heart to measure pressure and oxygen levels there.
* Selective angiocardiography: A catheter is used to inject the contrast agent into specific blood vessels or chambers of the heart to get a more detailed view.
Angiocardiography can help diagnose and evaluate various heart conditions, including congenital heart defects, coronary artery disease, cardiomyopathy, and valvular heart disease. It is an invasive procedure that carries some risks, such as bleeding, infection, and damage to blood vessels or heart tissue. However, it can provide valuable information for diagnosing and treating heart conditions.
Congenital heart defects (CHDs) are structural abnormalities in the heart that are present at birth. They can affect any part of the heart's structure, including the walls of the heart, the valves inside the heart, and the major blood vessels that lead to and from the heart.
Congenital heart defects can range from mild to severe and can cause various symptoms depending on the type and severity of the defect. Some common symptoms of CHDs include cyanosis (a bluish tint to the skin, lips, and fingernails), shortness of breath, fatigue, poor feeding, and slow growth in infants and children.
There are many different types of congenital heart defects, including:
1. Septal defects: These are holes in the walls that separate the four chambers of the heart. The two most common septal defects are atrial septal defect (ASD) and ventricular septal defect (VSD).
2. Valve abnormalities: These include narrowed or leaky valves, which can affect blood flow through the heart.
3. Obstruction defects: These occur when blood flow is blocked or restricted due to narrowing or absence of a part of the heart's structure. Examples include pulmonary stenosis and coarctation of the aorta.
4. Cyanotic heart defects: These cause a lack of oxygen in the blood, leading to cyanosis. Examples include tetralogy of Fallot and transposition of the great arteries.
The causes of congenital heart defects are not fully understood, but genetic factors and environmental influences during pregnancy may play a role. Some CHDs can be detected before birth through prenatal testing, while others may not be diagnosed until after birth or later in childhood. Treatment for CHDs may include medication, surgery, or other interventions to improve blood flow and oxygenation of the body's tissues.
The ventricular septum is the thick, muscular wall that separates the left and right ventricles, which are the lower chambers of the heart. Its main function is to prevent the oxygen-rich blood in the left ventricle from mixing with the oxygen-poor blood in the right ventricle.
A congenital heart defect called a ventricular septal defect (VSD) can occur when there is an abnormal opening or hole in the ventricular septum, allowing blood to flow between the two ventricles. This can result in various symptoms and complications, depending on the size of the defect and the amount of blood that passes through it. VSDs are typically diagnosed and treated by pediatric cardiologists or cardiac surgeons.
Transposition of the Great Vessels is a congenital heart defect in which the two main vessels that carry blood from the heart to the rest of the body are switched in position. Normally, the aorta arises from the left ventricle and carries oxygenated blood to the body, while the pulmonary artery arises from the right ventricle and carries deoxygenated blood to the lungs. In transposition of the great vessels, the aorta arises from the right ventricle and the pulmonary artery arises from the left ventricle. This results in oxygen-poor blood being pumped to the body and oxygen-rich blood being recirculated back to the lungs, which can lead to serious health problems and is often fatal if not corrected through surgery soon after birth.
A heart septal defect is a type of congenital heart defect, which means it is present at birth. It involves an abnormal opening in the septum, the wall that separates the two sides of the heart. This opening allows oxygen-rich blood to leak into the oxygen-poor blood chambers in the heart.
There are several types of heart septal defects, including:
1. Atrial Septal Defect (ASD): A hole in the atrial septum, the wall between the two upper chambers of the heart (the right and left atria).
2. Ventricular Septal Defect (VSD): A hole in the ventricular septum, the wall between the two lower chambers of the heart (the right and left ventricles).
3. Atrioventricular Septal Defect (AVSD): A combination of an ASD and a VSD, often accompanied by malformation of the mitral and/or tricuspid valves.
The severity of a heart septal defect depends on the size of the opening and its location in the septum. Small defects may cause no symptoms and may close on their own over time. Larger defects can lead to complications, such as heart failure, pulmonary hypertension, or infective endocarditis, and may require medical or surgical intervention.
Pulmonary Valve Stenosis is a cardiac condition where the pulmonary valve, located between the right ventricle and the pulmonary artery, has a narrowed opening. This stenosis (narrowing) can cause obstruction of blood flow from the right ventricle to the lungs. The narrowing can be caused by a fusion of the valve leaflets, thickened or calcified valve leaflets, or rarely, a dysplastic valve.
The severity of Pulmonary Valve Stenosis is classified based on the gradient pressure across the valve, which is measured during an echocardiogram. A mild stenosis has a gradient of less than 30 mmHg, moderate stenosis has a gradient between 30-59 mmHg, and severe stenosis has a gradient of 60 mmHg or higher.
Mild Pulmonary Valve Stenosis may not require treatment, while more severe cases may need to be treated with balloon valvuloplasty or surgical valve replacement. If left untreated, Pulmonary Valve Stenosis can lead to right ventricular hypertrophy, heart failure, and other complications.
Tetralogy of Fallot is a congenital heart defect that consists of four components: ventricular septal defect (a hole between the lower chambers of the heart), pulmonary stenosis (narrowing of the pulmonary valve and outflow tract), overriding aorta (the aorta lies directly over the ventricular septal defect), and right ventricular hypertrophy (thickening of the right ventricular muscle). This condition results in insufficient oxygenation of the blood, leading to cyanosis (bluish discoloration of the skin and mucous membranes) and other symptoms such as shortness of breath, fatigue, and poor growth. Treatment typically involves surgical repair, which is usually performed during infancy or early childhood.
The heart ventricles are the two lower chambers of the heart that receive blood from the atria and pump it to the lungs or the rest of the body. The right ventricle pumps deoxygenated blood to the lungs, while the left ventricle pumps oxygenated blood to the rest of the body. Both ventricles have thick, muscular walls to generate the pressure necessary to pump blood through the circulatory system.
In medical terms, the heart is a muscular organ located in the thoracic cavity that functions as a pump to circulate blood throughout the body. It's responsible for delivering oxygen and nutrients to the tissues and removing carbon dioxide and other wastes. The human heart is divided into four chambers: two atria on the top and two ventricles on the bottom. The right side of the heart receives deoxygenated blood from the body and pumps it to the lungs, while the left side receives oxygenated blood from the lungs and pumps it out to the rest of the body. The heart's rhythmic contractions and relaxations are regulated by a complex electrical conduction system.
A newborn infant is a baby who is within the first 28 days of life. This period is also referred to as the neonatal period. Newborns require specialized care and attention due to their immature bodily systems and increased vulnerability to various health issues. They are closely monitored for signs of well-being, growth, and development during this critical time.
Gastric outlet obstruction (GOO) is a medical condition that refers to the blockage of the passage from the stomach to the small intestine, also known as the pylorus. This blockage can be caused by various factors, including tumors, scar tissue, or gallstones. As a result, food and digestive enzymes cannot pass through the pylorus into the small intestine, leading to symptoms such as vomiting, abdominal pain, bloating, and weight loss. In severe cases, GOO can lead to malnutrition, dehydration, and other complications if left untreated. Treatment options for GOO depend on the underlying cause of the obstruction and may include medication, endoscopic procedures, or surgery.
 Double outlet right ventricle
Double outlet right ventricle Double Outlet Right Ventricle With Transposition: Background, Pathophysiology, Etiology
Double Outlet Right Ventricle With Transposition: Background, Pathophysiology, Etiology Double-outlet right ventricle | Sparrow
Double-outlet right ventricle | Sparrow LHM Kidz - Double Outlet Right Ventricle - Little Hearts Matter
LHM Kidz - Double Outlet Right Ventricle - Little Hearts Matter Double Outlet Right Ventricle
Double Outlet Right Ventricle Double outlet right ventricle with pulmonary stenosis - ULTRASOUNDPAEDIA
Double outlet right ventricle with pulmonary stenosis - ULTRASOUNDPAEDIA double outlet right ventricle
double outlet right ventricle
 Double Outlet Right Ventricle - Pediatrics - MSD Manual Professional Edition
Double Outlet Right Ventricle - Pediatrics - MSD Manual Professional Edition What is Double Outlet Right Ventricle ? Find Treatment Now | Anavara
What is Double Outlet Right Ventricle ? Find Treatment Now | Anavara ESC 365 - Brain Abscess in an Adult with Unrepaired Double-Outlet Right Ventricle with Transposition of Great Arteries and...
ESC 365 - Brain Abscess in an Adult with Unrepaired Double-Outlet Right Ventricle with Transposition of Great Arteries and... Inpatient Hospitalization Costs Associated with Birth Defects Among Persons Aged 65 Years - United States, 2019 | MMWR
Inpatient Hospitalization Costs Associated with Birth Defects Among Persons Aged 65 Years - United States, 2019 | MMWR Double Outlet Right Ventricle | Fetology: Diagnosis and Management of the Fetal Patient, 2e | AccessObGyn | McGraw Hill Medical
Double Outlet Right Ventricle | Fetology: Diagnosis and Management of the Fetal Patient, 2e | AccessObGyn | McGraw Hill Medical Double outlet right ventricle with subpulmonary ventricular septal defect without pulmonary stenosis (Concept Id: C1956413)
-...
Double outlet right ventricle with subpulmonary ventricular septal defect without pulmonary stenosis (Concept Id: C1956413)
-... Congenital Heart Disease | Congenital Heart Defects | MedlinePlus
Congenital Heart Disease | Congenital Heart Defects | MedlinePlus Surgical treatment of subaortic stenosis after biventricular repair of double-outlet right ventricle Emre Belli, MD, Alain...
Surgical treatment of subaortic stenosis after biventricular repair of double-outlet right ventricle Emre Belli, MD, Alain... Two-dimensional echocardiographic diagnosis of double outlet left ventricle with subaortic ventricular septal defect, pulmonary...
Two-dimensional echocardiographic diagnosis of double outlet left ventricle with subaortic ventricular septal defect, pulmonary... The Fetal Care Center of Southern California - Fetal Care & Diagnostic Center
The Fetal Care Center of Southern California - Fetal Care & Diagnostic Center Diagnosis of severe congenital heart defects in Norway 2016 | Tidsskrift for Den norske legeforening
Diagnosis of severe congenital heart defects in Norway 2016 | Tidsskrift for Den norske legeforening Dr. Luis Scott, MD, Clinical Cardiac Electrophysiologist - Phoenix, AZ | Sharecare
Dr. Luis Scott, MD, Clinical Cardiac Electrophysiologist - Phoenix, AZ | Sharecare A Helping Hand for Children with Congenital Heart Disease | Maxim Healthcare Services
A Helping Hand for Children with Congenital Heart Disease | Maxim Healthcare Services Find a Location
Find a Location Infections
Infections