Elliptocytosis, Hereditary
Spectrin
Erythrocyte Deformability
Spherocytosis, Hereditary
Erythrocyte Membrane
Erythrocytes, Abnormal
Collodion
Neuropeptides
Paper
Pedigree
Cytoskeletal Proteins
Erythrocytes
Membrane Proteins
Mutation
Electrophoresis, Polyacrylamide Gel
Trypsin
Molecular Sequence Data
Exons
Extraction of erythrocyte membrane proteins by sulfhydryl inhibitors. (1/138)
Human red cell membrane proteins were extracted by incubation of the ghost with hypotonic phosphate buffer (pH 7.4), N-ethylmaleimide and p-hydroxy-mercuribenzoate. In paroxysmal nocturnal hemoglobinuria (PNH), hereditary spherocytosis (HS) and hereditary elliptocytosis, the amount of proteins extracted by these procedures was significantly less than the amount extractable from the ghost of normal and aplastic anemia red cells. Polypeptide patterns of red cell membranes in these hematological disorders were essentially similar to those of normal ghosts. Analysis of the supernatant by SDS polyacrylamide gel electrophoresis revealed that this reduction was mainly due to the reduced amount of peripheral proteins extracted. The extraction of peripheral proteins by sulfhydryl reagents was accompanied by shape changes resulting in the formation of membrane vesicles, suggesting an important role of peripheral proteins in the maintenance of ghost shape. It is also suggested that qualitative abnormalities of peripheral proteins such as altered reactivity to sulfhydryl reagents and/or strong binding to the membrane are present in PNH, HS and hereditary elliptocytosis red cells. (+info)Hereditary spherocytosis and elliptocytosis erythrocytes show a normal transbilayer phospholipid distribution. (2/138)
Phosphatidylserine (PS) asymmetry was determined in red blood cells from patients with hereditary spherocytosis and elliptocytosis. No PS-exposing subpopulations were detected using the very sensitive method with fluorescently labeled annexin V. Treatment with N-ethylmaleimide or adenosine triphosphate (ATP) depletion to inactivate the flipase did not lead to formation of PS-exposing subpopulations in these cells, but elevated intracellular calcium levels did lead to extensive scrambling of the PS asymmetry. Although interactions of the membrane skeleton with the phospholipid bilayer have been suggested to stabilize the asymmetric distribution of PS across the bilayer, our data show that red blood cells with a severely damaged membrane skeleton are able to preserve asymmetry, even under conditions in which restoration of the asymmetric distribution is excluded. Moreover, the loss of membrane asymmetry in these cells requires active scrambling involving high levels of intracellular calcium as in normal cells. Our data show that the severe disorder of the membrane skeleton found in these cells does not affect the activity of flipase or scramblase, indicating that these proteins are not regulated by, nor coupled to the membrane skeleton assembly, and that possible thrombotic events in spherocytosis patients are not likely associated with altered PS topology of the red blood cells. (+info)Prevention of cerebral malaria in children in Papua New Guinea by southeast Asian ovalocytosis band 3. (3/138)
Southeast Asian ovalocytosis (SAO) occurs at high frequency in malarious regions of the western Pacific and may afford a survival advantage against malaria. It is caused by a deletion of the erythrocyte membrane band 3 gene and the band 3 protein mediates the cytoadherence of parasitized erythrocytes in vitro. The SAO band 3 variant may prevent cerebral malaria but it exacerbates malaria anemia and may also increase acidosis, a major determinant of mortality in malaria. We undertook a case-control study of children admitted to hospital in a malarious region of Papua New Guinea. The SAO band 3, detected by the polymerase chain reaction, was present in 0 of 68 children with cerebral malaria compared with six (8.8%) of 68 matched community controls (odds ratio = 0, 95% confidence interval = 0-0.85). Median hemoglobin levels were 1.2 g/dl lower in malaria cases with SAO than in controls (P = 0.035) but acidosis was not affected. The remarkable protection that SAO band 3 affords against cerebral malaria may offer a valuable approach to a better understanding of the mechanisms of adherence of parasitized erythrocytes to vascular endothelium, and thus of the pathogenesis of cerebral malaria. (+info)Autosomal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosis. (4/138)
BACKGROUND: A defect in the anion exchanger 1 (AE1) of the basolateral membrane of type A intercalated cells in the renal collecting duct may result in a failure to maintain a cell-to-lumen H+ gradient, leading to distal renal tubular acidosis (dRTA). Thus, dRTA may occur in Southeast Asian ovalocytosis (SAO), a common AE1 gene abnormality observed in Southeast Asia and Melanesia. Our study investigated whether or not this renal acidification defect exists in individuals with SAO. METHODS: Short and three-day NH4Cl loading tests were performed in 20 individuals with SAO and in two subjects, including their families, with both SAO and dRTA. Mutations of AE1 gene in individuals with SAO and members of the two families were also studied. RESULTS: Renal acidification in the 20 individuals with SAO and in the parents of the two families was normal. However, the two clinically affected individuals with SAO and dRTA had compound heterozygosity of 27 bp deletion in exon 11 and missense mutation G701D resulting from a CGG-->CAG substitution in exon 17 of the AE1 gene. Red cells of the two subjects with dRTA and SAO and the family members with SAO showed an approximate 40% reduction in sulfate influx with normal 4,4'-di-isothiocyanato-stilbene-2,2'-disulfonic acid sensitivity and pH dependence. CONCLUSION: These findings suggest that compound heterozygosity of abnormal AE1 genes causes autosomal recessive dRTA in SAO. (+info)Elliptocytosis in patients with C-terminal domain mutations of protein 4.1 correlates with encoded messenger RNA levels rather than with alterations in primary protein structure. (5/138)
Early biochemical studies defined 4 functional domains of the erythroid protein 4.1 (4.1R). From amino-terminal to carboxy-terminal, these are 30 kd, 16 kd, 10 kd, and 22/24 kd in size. Although the functional properties of both the 30-kd and the 10-kd domain have been demonstrated in red cells, no functional activities have been assigned to either the 16-kd or the 22/24-kd domain in these cells. We here describe new mutations in the sequence encoding the C-terminal 22/24-kd domain that are associated with hereditary elliptocytosis. An unusually mild phenotype observed in heterozygous and homozygous members of 1 family suggested heterogeneity in the pattern of expression of 4.1R deficiency. Using a variety of protein and messenger RNA (mRNA) quantification strategies, we showed that, regardless of the alteration in the C-terminal primary sequence, when the protein is produced, it assembles at the cell membrane. In addition, we found that alterations in red cell morphologic features and membrane function correlate with the amount of membrane-associated protein-and therefore with the amount of mRNA accumulated-rather than with the primary structure of the variant proteins. These data suggest that an intact sequence at exons 19 through 21 encoding part of the C-terminal 22/24-kd region is not required for proper protein 4.1R assembly in mature red cells. (Blood. 2000;95:1834-1841) (+info)Mild spherocytic hereditary elliptocytosis and altered levels of alpha- and gamma-adducins in beta-adducin-deficient mice. (6/138)
The membrane skeleton, a dynamic network of proteins associated with the plasma membrane, determines the shape and mechanical properties of erythrocytes. Deficiencies or defects in membrane skeletal proteins are associated with inherited disorders of erythrocyte morphology and function. Adducin is one of the proteins localized at the spectrin-actin junction of the membrane skeleton. In this work we show that deficiency of beta-adducin produces an 80% decrease of alpha-adducin and a fourfold up-regulation of gamma-adducin in erythrocytes. beta-Adducin or any other isoform generated by translation of abnormally spliced messenger RNAs could not be detected by our antibodies either in ghosts or in cytoplasm of -/- erythrocytes. Actin levels were diminished in mutant mice, suggesting alterations in the actin-spectrin junctional complexes due to the absence of adducin. Elliptocytes, ovalocytes, and occasionally spherocytes were found in the blood film of -/- mice. Hematological values showed an increase in reticulocyte counts and mean corpuscular hemoglobin concentration, decreased mean corpuscular volume and hematocrit, and normal erythrocyte counts that, associated to splenomegaly, indicate that the mice suffer from mild anemia with compensated hemolysis. These modifications are due to a loss of membrane surface and dehydration that result in an increase in the osmotic fragility of red blood cells. The marked alteration in osmotic fragility together with the predominant presence of elliptocytes is reminiscent of the human disorder called spherocytic hereditary elliptocytosis. Our results suggest that the amount of adducin remaining in the mutant animals (presumably alphagamma adducin) could be functional and might account for the mild phenotype. (Blood. 2000;95:3978-3985) (+info)Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. (7/138)
We describe three mutations of the red-cell anion exchangerband 3 (AE1, SLC4A1) gene associated with distalrenal tubular acidosis (dRTA) in families from Malaysia and Papua NewGuinea: Gly(701)-->Asp (G701D), Ala(858)-->Asp(A858D) and deletion of Val(850) (DeltaV850). The mutationsA858D and DeltaV850 are novel; all three mutations seem to berestricted to South-East Asian populations. South-East Asianovalocytosis (SAO), resulting from the band 3 deletion of residues400-408, occurred in many of the families but did not itselfresult in dRTA. Compound heterozygotes of each of the dRTA mutationswith SAO all had dRTA, evidence of haemolytic anaemia and abnormal red-cell properties. The A858D mutation showed dominant inheritance and therecessive DeltaV850 and G701D mutations showed a pseudo-dominantphenotype when the transport-inactive SAO allele was also present. Red-cell and Xenopus oocyte expression studies showed that theDeltaV850 and A858D mutant proteins have greatly decreased aniontransport when present as compound heterozygotes (DeltaV850/A858D,DeltaV850/SAO or A858D/SAO). Red cells with A858D/SAO had only 3% ofthe SO(4)(2-) efflux of normal cells, thelowest anion transport activity so far reported for human red cells. The results suggest dRTA might arise by a different mechanism for eachmutation. We confirm that the G701D mutant protein has an absoluterequirement for glycophorin A for movement to the cell surface. Wesuggest that the dominant A858D mutant protein is possibly mis-targetedto an inappropriate plasma membrane domain in the renal tubular cell,and that the recessive DeltaV850 mutation might give dRTA because ofits decreased anion transport activity. (+info)Defective spectrin integrity and neonatal thrombosis in the first mouse model for severe hereditary elliptocytosis. (8/138)
Mutations affecting the conversion of spectrin dimers to tetramers result in hereditary elliptocytosis (HE), whereas a deficiency of human erythroid alpha- or beta-spectrin results in hereditary spherocytosis (HS). All spontaneous mutant mice with cytoskeletal deficiencies of spectrin reported to date have HS. Here, the first spontaneous mouse mutant, sph(Dem)/ sph(Dem), with severe HE is described. The sph(Dem) mutation is the insertion of an intracisternal A particle element in intron 10 of the erythroid alpha-spectrin gene. This causes exon skipping, the in-frame deletion of 46 amino acids from repeat 5 of alpha-spectrin and alters spectrin dimer/tetramer stability and osmotic fragility. The disease is more severe in sph(Dem)/sph(Dem) neonates than in alpha-spectrin-deficient mice with HS. Thrombosis and infarction are not, as in the HS mice, limited to adults but occur soon after birth. Genetic background differences that exist between HE and HS mice are suspect, along with red blood cell morphology differences, as modifiers of thrombosis timing. sph(Dem)/sph(Dem) mice provide a unique model for analyzing spectrin dimer- to-tetramer conversion and identifying factors that influence thrombosis. (+info)Hereditary elliptocytosis is a genetic condition characterized by the presence of abnormally shaped red blood cells (RBCs), which are often oval or elliptical in shape instead of the typical biconcave disc shape. This condition is caused by mutations in genes that encode proteins responsible for maintaining the stability and flexibility of RBCs, such as spectrin and ankyrin.
There are several types of hereditary elliptocytosis, including:
1. Type 1 Hereditary Elliptocytosis (HE): This is the most common form and is usually a mild condition with few or no symptoms. It is caused by mutations in the spectrin gene.
2. Type 2 Hereditary Elliptocytosis (HE): This form is less common and can be more severe than type 1, with symptoms such as anemia, fatigue, and jaundice. It is caused by mutations in the gene that encodes the protein ankyrin.
3. Spherocytic Elliptocytosis (SE): This is a rare form of hereditary elliptocytosis that combines features of both hereditary elliptocytosis and hereditary spherocytosis, another genetic RBC disorder. SE is caused by mutations in genes that encode spectrin or ankyrin.
In general, people with hereditary elliptocytosis have few or no symptoms and do not require treatment. However, in some cases, severe hemolysis (breakdown of RBCs) can occur, leading to anemia, jaundice, gallstones, and other complications. In these cases, treatment may be necessary to manage the symptoms and prevent further complications.
Spectrin is a type of cytoskeletal protein that is responsible for providing structural support and maintaining the shape of red blood cells (erythrocytes). It is a key component of the erythrocyte membrane skeleton, which provides flexibility and resilience to these cells, allowing them to deform and change shape as they pass through narrow capillaries. Spectrin forms a network of fibers just beneath the cell membrane, along with other proteins such as actin, band 4.1, and band 3. Mutations in spectrin genes can lead to various blood disorders, including hereditary spherocytosis and hemolytic anemia.
Hemolytic anemia, congenital is a type of anemia that is present at birth and characterized by the abnormal breakdown (hemolysis) of red blood cells. This can occur due to various genetic defects that affect the structure or function of the red blood cells, making them more susceptible to damage and destruction.
There are several types of congenital hemolytic anemias, including:
1. Congenital spherocytosis: A condition caused by mutations in genes that affect the shape and stability of red blood cells, leading to the formation of abnormally shaped and fragile cells that are prone to hemolysis.
2. G6PD deficiency: A genetic disorder that affects the enzyme glucose-6-phosphate dehydrogenase (G6PD), which is essential for protecting red blood cells from damage. People with this condition have low levels of G6PD, making their red blood cells more susceptible to hemolysis when exposed to certain triggers such as infections or certain medications.
3. Hereditary elliptocytosis: A condition caused by mutations in genes that affect the structure and flexibility of red blood cells, leading to the formation of abnormally shaped and fragile cells that are prone to hemolysis.
4. Pyruvate kinase deficiency: A rare genetic disorder that affects an enzyme called pyruvate kinase, which is essential for the production of energy in red blood cells. People with this condition have low levels of pyruvate kinase, leading to the formation of fragile and abnormally shaped red blood cells that are prone to hemolysis.
Symptoms of congenital hemolytic anemia can vary depending on the severity of the condition but may include fatigue, weakness, pale skin, jaundice, dark urine, and an enlarged spleen. Treatment may involve blood transfusions, medications to manage symptoms, and in some cases, surgery to remove the spleen.
Erythrocyte deformability refers to the ability of red blood cells (erythrocytes) to change shape and bend without rupturing, which is crucial for their efficient movement through narrow blood vessels. This deformability is influenced by several factors including the cell membrane structure, hemoglobin concentration, and intracellular viscosity. A decrease in erythrocyte deformability can negatively impact blood flow and oxygen delivery to tissues, potentially contributing to various pathological conditions such as sickle cell disease, diabetes, and cardiovascular diseases.
Hereditary Spherocytosis is a genetic disorder that affects the red blood cells (RBCs) causing them to take on a spherical shape instead of their normal biconcave disc shape. This occurs due to mutations in the genes responsible for the proteins that maintain the structure and flexibility of RBCs, such as ankyrin, band 3, spectrin, and protein 4.2.
The abnormally shaped RBCs are fragile and prone to hemolysis (premature destruction), which can lead to anemia, jaundice, and gallstones. Symptoms can vary from mild to severe and may include fatigue, weakness, shortness of breath, and a yellowing of the skin and eyes (jaundice). Diagnosis is typically made through a combination of family history, physical examination, complete blood count (CBC), and specialized tests such as osmotic fragility test, eosin-5'-maleimide binding test, or direct antiglobulin test. Treatment may include monitoring, supplementation with folic acid, and in severe cases, splenectomy (surgical removal of the spleen) to reduce RBC destruction.
An erythrocyte, also known as a red blood cell, is a type of cell that circulates in the blood and is responsible for transporting oxygen throughout the body. The erythrocyte membrane refers to the thin, flexible barrier that surrounds the erythrocyte and helps to maintain its shape and stability.
The erythrocyte membrane is composed of a lipid bilayer, which contains various proteins and carbohydrates. These components help to regulate the movement of molecules into and out of the erythrocyte, as well as provide structural support and protection for the cell.
The main lipids found in the erythrocyte membrane are phospholipids and cholesterol, which are arranged in a bilayer structure with the hydrophilic (water-loving) heads facing outward and the hydrophobic (water-fearing) tails facing inward. This arrangement helps to maintain the integrity of the membrane and prevent the leakage of cellular components.
The proteins found in the erythrocyte membrane include integral proteins, which span the entire width of the membrane, and peripheral proteins, which are attached to the inner or outer surface of the membrane. These proteins play a variety of roles, such as transporting molecules across the membrane, maintaining the shape of the erythrocyte, and interacting with other cells and proteins in the body.
The carbohydrates found in the erythrocyte membrane are attached to the outer surface of the membrane and help to identify the cell as part of the body's own immune system. They also play a role in cell-cell recognition and adhesion.
Overall, the erythrocyte membrane is a complex and dynamic structure that plays a critical role in maintaining the function and integrity of red blood cells.
Abnormal erythrocytes refer to red blood cells that have an abnormal shape, size, or other characteristics. This can include various types of abnormalities such as:
1. Anisocytosis: Variation in the size of erythrocytes.
2. Poikilocytosis: Variation in the shape of erythrocytes, including but not limited to teardrop-shaped cells (dacrocytes), crescent-shaped cells (sickle cells), and spherical cells (spherocytes).
3. Anemia: A decrease in the total number of erythrocytes or a reduction in hemoglobin concentration, which can result from various underlying conditions such as iron deficiency, chronic disease, or blood loss.
4. Hemoglobinopathies: Abnormalities in the structure or function of hemoglobin, the protein responsible for carrying oxygen in erythrocytes, such as sickle cell anemia and thalassemia.
5. Inclusion bodies: Abnormal structures within erythrocytes, such as Heinz bodies (denatured hemoglobin) or Howell-Jolly bodies (nuclear remnants).
These abnormalities can be detected through a complete blood count (CBC) and peripheral blood smear examination. The presence of abnormal erythrocytes may indicate an underlying medical condition, and further evaluation is often necessary to determine the cause and appropriate treatment.
Collodion is a clear, colorless, viscous solution that is used in medicine and photography. Medically, collodion is often used as a temporary protective dressing for wounds, burns, or skin abrasions. When applied to the skin, it dries to form a flexible, waterproof film that helps to prevent infection and promote healing. Collodion is typically made from a mixture of nitrocellulose, alcohol, and ether.
In photography, collodion was historically used as a medium for wet plate photography, which was popular in the mid-19th century. The photographer would coat a glass plate with a thin layer of collodion, then sensitize it with silver salts before exposing and developing the image while the collodion was still wet. This process required the photographer to carry a portable darkroom and develop the plates immediately after exposure. Despite its challenges, the wet plate collodion process was able to produce highly detailed images, making it a popular technique for portrait photography during its time.
Neuropeptides are small protein-like molecules that are used by neurons to communicate with each other and with other cells in the body. They are produced in the cell body of a neuron, processed from larger precursor proteins, and then transported to the nerve terminal where they are stored in secretory vesicles. When the neuron is stimulated, the vesicles fuse with the cell membrane and release their contents into the extracellular space.
Neuropeptides can act as neurotransmitters or neuromodulators, depending on their target receptors and the duration of their effects. They play important roles in a variety of physiological processes, including pain perception, appetite regulation, stress response, and social behavior. Some neuropeptides also have hormonal functions, such as oxytocin and vasopressin, which are produced in the hypothalamus and released into the bloodstream to regulate reproductive and cardiovascular function, respectively.
There are hundreds of different neuropeptides that have been identified in the nervous system, and many of them have multiple functions and interact with other signaling molecules to modulate neural activity. Dysregulation of neuropeptide systems has been implicated in various neurological and psychiatric disorders, such as chronic pain, addiction, depression, and anxiety.
I'm sorry for any confusion, but "paper" is not a medical term. In general, it refers to a thin material made from pulp derived from wood, rags, or other fibrous substances, which is used for writing, printing, drawing, or packaging. If you have a question about a specific medical concept or condition, I'd be happy to help if I can!
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Cytoskeletal proteins are a type of structural proteins that form the cytoskeleton, which is the internal framework of cells. The cytoskeleton provides shape, support, and structure to the cell, and plays important roles in cell division, intracellular transport, and maintenance of cell shape and integrity.
There are three main types of cytoskeletal proteins: actin filaments, intermediate filaments, and microtubules. Actin filaments are thin, rod-like structures that are involved in muscle contraction, cell motility, and cell division. Intermediate filaments are thicker than actin filaments and provide structural support to the cell. Microtubules are hollow tubes that are involved in intracellular transport, cell division, and maintenance of cell shape.
Cytoskeletal proteins are composed of different subunits that polymerize to form filamentous structures. These proteins can be dynamically assembled and disassembled, allowing cells to change their shape and move. Mutations in cytoskeletal proteins have been linked to various human diseases, including cancer, neurological disorders, and muscular dystrophies.
Erythrocytes, also known as red blood cells (RBCs), are the most common type of blood cell in circulating blood in mammals. They are responsible for transporting oxygen from the lungs to the body's tissues and carbon dioxide from the tissues to the lungs.
Erythrocytes are formed in the bone marrow and have a biconcave shape, which allows them to fold and bend easily as they pass through narrow blood vessels. They do not have a nucleus or mitochondria, which makes them more flexible but also limits their ability to reproduce or repair themselves.
In humans, erythrocytes are typically disc-shaped and measure about 7 micrometers in diameter. They contain the protein hemoglobin, which binds to oxygen and gives blood its red color. The lifespan of an erythrocyte is approximately 120 days, after which it is broken down in the liver and spleen.
Abnormalities in erythrocyte count or function can lead to various medical conditions, such as anemia, polycythemia, and sickle cell disease.
Heterozygote detection is a method used in genetics to identify individuals who carry one normal and one mutated copy of a gene. These individuals are known as heterozygotes and they do not typically show symptoms of the genetic disorder associated with the mutation, but they can pass the mutated gene on to their offspring, who may then be affected.
Heterozygote detection is often used in genetic counseling and screening programs for recessive disorders such as cystic fibrosis or sickle cell anemia. By identifying heterozygotes, individuals can be informed of their carrier status and the potential risks to their offspring. This information can help them make informed decisions about family planning and reproductive options.
Various methods can be used for heterozygote detection, including polymerase chain reaction (PCR) based tests, DNA sequencing, and genetic linkage analysis. The choice of method depends on the specific gene or mutation being tested, as well as the availability and cost of the testing technology.
Membrane proteins are a type of protein that are embedded in the lipid bilayer of biological membranes, such as the plasma membrane of cells or the inner membrane of mitochondria. These proteins play crucial roles in various cellular processes, including:
1. Cell-cell recognition and signaling
2. Transport of molecules across the membrane (selective permeability)
3. Enzymatic reactions at the membrane surface
4. Energy transduction and conversion
5. Mechanosensation and signal transduction
Membrane proteins can be classified into two main categories: integral membrane proteins, which are permanently associated with the lipid bilayer, and peripheral membrane proteins, which are temporarily or loosely attached to the membrane surface. Integral membrane proteins can further be divided into three subcategories based on their topology:
1. Transmembrane proteins, which span the entire width of the lipid bilayer with one or more alpha-helices or beta-barrels.
2. Lipid-anchored proteins, which are covalently attached to lipids in the membrane via a glycosylphosphatidylinositol (GPI) anchor or other lipid modifications.
3. Monotopic proteins, which are partially embedded in the membrane and have one or more domains exposed to either side of the bilayer.
Membrane proteins are essential for maintaining cellular homeostasis and are targets for various therapeutic interventions, including drug development and gene therapy. However, their structural complexity and hydrophobicity make them challenging to study using traditional biochemical methods, requiring specialized techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single-particle cryo-electron microscopy (cryo-EM).
Genetic variation refers to the differences in DNA sequences among individuals and populations. These variations can result from mutations, genetic recombination, or gene flow between populations. Genetic variation is essential for evolution by providing the raw material upon which natural selection acts. It can occur within a single gene, between different genes, or at larger scales, such as differences in the number of chromosomes or entire sets of chromosomes. The study of genetic variation is crucial in understanding the genetic basis of diseases and traits, as well as the evolutionary history and relationships among species.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Electrophoresis, polyacrylamide gel (EPG) is a laboratory technique used to separate and analyze complex mixtures of proteins or nucleic acids (DNA or RNA) based on their size and electrical charge. This technique utilizes a matrix made of cross-linked polyacrylamide, a type of gel, which provides a stable and uniform environment for the separation of molecules.
In this process:
1. The polyacrylamide gel is prepared by mixing acrylamide monomers with a cross-linking agent (bis-acrylamide) and a catalyst (ammonium persulfate) in the presence of a buffer solution.
2. The gel is then poured into a mold and allowed to polymerize, forming a solid matrix with uniform pore sizes that depend on the concentration of acrylamide used. Higher concentrations result in smaller pores, providing better resolution for separating smaller molecules.
3. Once the gel has set, it is placed in an electrophoresis apparatus containing a buffer solution. Samples containing the mixture of proteins or nucleic acids are loaded into wells on the top of the gel.
4. An electric field is applied across the gel, causing the negatively charged molecules to migrate towards the positive electrode (anode) while positively charged molecules move toward the negative electrode (cathode). The rate of migration depends on the size, charge, and shape of the molecules.
5. Smaller molecules move faster through the gel matrix and will migrate farther from the origin compared to larger molecules, resulting in separation based on size. Proteins and nucleic acids can be selectively stained after electrophoresis to visualize the separated bands.
EPG is widely used in various research fields, including molecular biology, genetics, proteomics, and forensic science, for applications such as protein characterization, DNA fragment analysis, cloning, mutation detection, and quality control of nucleic acid or protein samples.
Trypsin is a proteolytic enzyme, specifically a serine protease, that is secreted by the pancreas as an inactive precursor, trypsinogen. Trypsinogen is converted into its active form, trypsin, in the small intestine by enterokinase, which is produced by the intestinal mucosa.
Trypsin plays a crucial role in digestion by cleaving proteins into smaller peptides at specific arginine and lysine residues. This enzyme helps to break down dietary proteins into amino acids, allowing for their absorption and utilization by the body. Additionally, trypsin can activate other zymogenic pancreatic enzymes, such as chymotrypsinogen and procarboxypeptidases, thereby contributing to overall protein digestion.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
Exons are the coding regions of DNA that remain in the mature, processed mRNA after the removal of non-coding intronic sequences during RNA splicing. These exons contain the information necessary to encode proteins, as they specify the sequence of amino acids within a polypeptide chain. The arrangement and order of exons can vary between different genes and even between different versions of the same gene (alternative splicing), allowing for the generation of multiple protein isoforms from a single gene. This complexity in exon structure and usage significantly contributes to the diversity and functionality of the proteome.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
 Hereditary elliptocytosis
Hereditary elliptocytosis
List of hematologic conditions
Elliptocyte
HAGH
Hereditary pyropoikilocytosis
Anemia
PRPF31
Glycophorin C
Protein 4.1
Southeast Asian ovalocytosis
SPTB
Human genetic resistance to malaria
Protein 4.2
Spectrin, alpha 1
Aldolase A deficiency
Hemolytic anemia
Red blood cell
Spectrin
Congenital hemolytic anemia
Hemolysis
Normocytic anemia
Asymptomatic
Neonatal jaundice
List of diseases (H)
List of MeSH codes (C15)
Hematologic disease
List of MeSH codes (C16)
List of OMIM disorder codes
Microcytic anemia
Hereditary elliptocytosis - Wikipedia
 Hereditary Elliptocytosis: Practice Essentials, Pathophysiology, Etiology
Hereditary Elliptocytosis: Practice Essentials, Pathophysiology, Etiology
 Elliptocytosis, Hereditary; Ovalocytosis, Hereditary
Elliptocytosis, Hereditary; Ovalocytosis, Hereditary
Hereditary Elliptocytosis: Background, Pathophysiology, Epidemiology
 Hereditary Spherocytosis and Hereditary Elliptocytosis - Hematology and Oncology - MSD Manual Professional Edition
Hereditary Spherocytosis and Hereditary Elliptocytosis - Hematology and Oncology - MSD Manual Professional Edition
 Types of Hemolytic Anemia | Hematology-Oncology Associates of CNY
Types of Hemolytic Anemia | Hematology-Oncology Associates of CNY
 Blood smear: MedlinePlus Medical Encyclopedia
Blood smear: MedlinePlus Medical Encyclopedia
 hemolytic anemia
hemolytic anemia
Splenectomy - Health Library
 Dr. William Mitchell, MD, Pediatric Hematology-Oncology Specialist - Bronx, NY | Sharecare
Dr. William Mitchell, MD, Pediatric Hematology-Oncology Specialist - Bronx, NY | Sharecare
 Jon Morrow, PhD, MD | Directory of Faculty Research Interests
Jon Morrow, PhD, MD | Directory of Faculty Research Interests
 Samuel E. Lux IV, MD - Dana-Farber Cancer Institute | Boston, MA
Samuel E. Lux IV, MD - Dana-Farber Cancer Institute | Boston, MA
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency - Hematology and Oncology - Merck Manuals Professional Edition
 Corinna L. Schultz, MD
Corinna L. Schultz, MD
Kernicterus: Background, Pathophysiology, Etiology
 Diagnostic Pathology Nonneoplastic Pediatrics, 2nd edition - Angelica R. Putnam - 1020 - ELSEVIER HEALTH SCIENCES -...
Diagnostic Pathology Nonneoplastic Pediatrics, 2nd edition - Angelica R. Putnam - 1020 - ELSEVIER HEALTH SCIENCES -...
2012 ICD-9-CM Diagnosis Code 283.0 : Autoimmune hemolytic anemias
 Abhd10 Mouse Gene Details | abhydrolase domain containing 10 | International Mouse Phenotyping Consortium
Abhd10 Mouse Gene Details | abhydrolase domain containing 10 | International Mouse Phenotyping Consortium
 Highlighting Biophysics Research During Sickle Cell Awareness Month
Highlighting Biophysics Research During Sickle Cell Awareness Month
Alport syndrome and mental retardation: clinical and genetic dissection of the contiguous gene deletion syndrome in Xq22.3 (ATS...
 Labrador Retriever Facts - Wisdom Panel™ Dog Breeds
Labrador Retriever Facts - Wisdom Panel™ Dog Breeds
 MyDogDNA™
MyDogDNA™
 Hydrops Fetalis Precision Panel - IGENOMIX Latino América
Hydrops Fetalis Precision Panel - IGENOMIX Latino América
Specific PHGKB|Rare Diseases PHGKB|PHGKB
 ANAEMIA - Dr SHAH's Homeopathy
ANAEMIA - Dr SHAH's Homeopathy
Anemia Facts, Anemia Types and Anemia Kinds
 Haematology MCQs with Answer and Explnation | Chapter 1 Questions, Answers and Explanations
Haematology MCQs with Answer and Explnation | Chapter 1 Questions, Answers and Explanations
Dr DEEPAN P SHAH - MD(HOMOEOPATHY)
 Hemolytic anemia
Hemolytic anemia
Ovalocytosis4
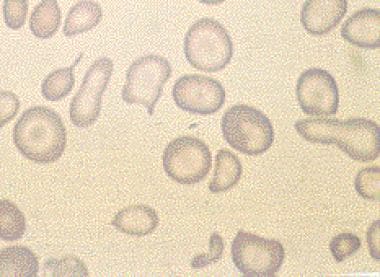
- Hereditary elliptocytosis, also known as ovalocytosis, is an inherited blood disorder in which an abnormally large number of the person's red blood cells are elliptical rather than the typical biconcave disc shape. (wikipedia.org)
- Hereditary elliptocytosis (ovalocytosis) is a rare autosomal dominant disorder in which RBCs are oval or elliptical. (msdmanuals.com)
- Presence of RBCs with an oval shape may be a sign of hereditary elliptocytosis or hereditary ovalocytosis . (medlineplus.gov)
- A11 - Torlontano G, FIORITONI G: Hereditary Ovalocytosis with Dyserythropoiesis. (giuseppefioritoni.it)
Spherocytosis and hereditary elliptocytosis2
- Hereditary spherocytosis and hereditary elliptocytosis are congenital red blood cell (RBC) membrane disorders that can cause a mild hemolytic anemia. (msdmanuals.com)
- Hereditary spherocytosis and hereditary elliptocytosis are examples of inherited hemolytic anemias. (ghcgenetics.com)
Pyropoikilocytosis3
- The mode of inheritance is autosomal dominant, except for hereditary pyropoikilocytosis (HPP) , which is autosomal recessive. (medscape.com)
- Hemolysis is usually absent or slight, with little or no anemia except in some homozygous patients (hereditary pyropoikilocytosis). (msdmanuals.com)
- Defects in the "horizontal" interactions that hold the membrane skeleton together cause hereditary elliptocytosis or its severe variant, hereditary pyropoikilocytosis. (basicmedicalkey.com)
Congenital4
- Hereditary spherocytosis is the most common congenital hemolytic anemia among Caucasians with an estimated prevalence ranging from 1:2,000 to 1:5,000. (ghcgenetics.com)
- Congenital hemolytic anemia (or hereditary hemolytic anemia ) refers to hemolytic anemia which is primarily due to congenital disorders . (en-academic.com)
- Congenital hemolytic jaundice - Known also as hereditary spherocytosis (HS), this is a genetic disorder of the red blood cell membrane clinically characterized by anemia, jaundice (yellowing) and splenomegaly (enlargement of the spleen). (en-academic.com)
- Congenital and hereditary forms of fibrinogen deficiency are described as early as 1920. (treatment-diabetes-info.com)
Cytoskeleton2
- Normally, this deformation lasts only as long as a cell is present in a capillary, but in hereditary elliptocytosis the instability of the cytoskeleton means that erythrocytes deformed by passing through a capillary are forever rendered elliptical. (wikipedia.org)
- In hereditary elliptocytosis, genetic mutations result in weakness of the cytoskeleton of the cell, leading to deformation of the cell. (msdmanuals.com)
Deficiency3
- It is thought that elliptocytosis in glycophorin C deficiency is actually the consequence of a band 4.1 deficit, as glycophorin C deficient individuals also have reduced intracellular band 4.1 (probably due to the reduced number of binding sites for band 4.1 in the absence of glycoprotein C).[citation needed] Inheritance of multiple mutations tends to infer more serious disease. (wikipedia.org)
- Hereditary elliptocytosis and severe iron deficiency. (thebloodproject.com)
- In a disease such as hereditary spherocytosis, erythrocytes have a smaller ratio of surface area to volume and are thus more susceptible to osmotic stress, as opposed to the increased resistance characteristic of thalassemia, iron deficiency anemia, or any other condition that would cause an increased surface area-to-volume ratio. (medscape.com)
Thalassemia1
- The osmotic fragility test is mainly used in helping with the diagnosis of hereditary spherocytosis but it is also used in some countries as a method to screen for beta thalassemia, especially where laboratory resources are limited. (medscape.com)
Genetic1
- EL2 and EL3: The most common genetic defects (present in two-thirds of all cases of hereditary elliptocytosis) are in genes for the polypeptides α-spectrin or β-spectrin. (wikipedia.org)
RBCs3
- Hereditary elliptocytosis (HE) encompasses inherited disorders of erythrocytes that have the common feature of elliptical RBCs on morphologic examination and shortened RBC survival. (medscape.com)
- Hereditary spherocytosis is characterized by hemolysis of spheroidal RBCs and anemia. (msdmanuals.com)
- In hereditary spherocytosis, because RBCs are spheroidal and the mean corpuscular volume (MCV) is normal, the mean corpuscular diameter is below normal, and RBCs resemble spherocytes. (msdmanuals.com)
Diagnosis3
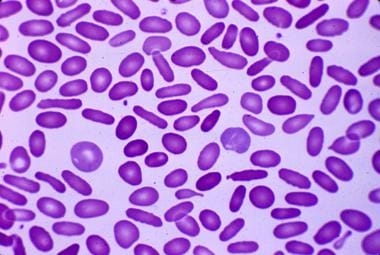
- The diagnosis of hereditary elliptocytosis is usually made by coupling a family history of the condition with an appropriate clinical presentation and confirmation on a blood smear. (wikipedia.org)
- Only in the complete absence of any clinical and laboratory signs of these syndromes and the diseases that cause them, the diagnosis of a hereditary violation of fibrinogen synthesis becomes eligible. (treatment-diabetes-info.com)
- The diagnosis of such hereditary forms is confirmed by stable, life-long insufficiency of factor I, as well as hypofibrinogenemia in the parents and other blood relatives of the patient. (treatment-diabetes-info.com)
Defect1
- For instance, the most common genotype responsible for HPP occurs when the affected individual inherits an α-spectrin mutation from one parent (i.e. one parent has hereditary elliptocytosis) and the other parent passes on an as-yet-undefined defect that causes the affected individual's cells to preferentially produce the defective α-spectrin rather than normal α-spectrin. (wikipedia.org)
Defects in membrane2
- Hereditary defects in membrane skeleton proteins. (dana-farber.org)
- Nonimmune anemias Hereditary red cell membrane disorders are accounted for by nonimmune hereditary hemolytic anemias caused by defects in membrane proteins located in distinct layers of the red cell membrane. (bredagenetics.com)
Erythrocytes2
- These elliptical cells are taken up by the spleen and removed from circulation when they are younger than they would normally be, meaning that the erythrocytes of people with hereditary elliptocytosis have a shorter than average life-span (a normal person's erythrocytes average 120 days or more). (wikipedia.org)
- Hereditary elliptocytosis: Peripheral blood smear reveals cigar-shaped erythrocytes (elliptocytes). (medscape.com)
Diseases1
- The Red Blood Cell Membrane and Its Disorders: Hereditary Spherocytosis, Elliptocytosis, and Related Diseases. (medscape.com)
Elliptical1
- As discussed in the following sections, molecular defects in vertical interactions cause hereditary spherocytosis, whereas defects in horizontal interactions result in the formation of elliptical red cells. (basicmedicalkey.com)
Hemolytic anemia among1
- Hereditary spherocytosis (SFER-o-si-to-sis) is the most common cause of hemolytic anemia among people of Northern European descent. (hoacny.com)
Caused by defects1
- During the past decade our laboratory and others have shown that hereditary spherocytosis is caused by defects in the connections that attach the membrane skeleton to the overlying lipid bilayer. (dana-farber.org)
Clinical features1
- The pathogenesis and clinical features of hereditary spherocytosis. (basicmedicalkey.com)
Genes2
- Individuals with a single mutation in one of the spectrin genes are usually asymptomatic, but those who are homozygotes or are compound heterozygotes (i.e. they are heterozygous for two different elliptocytosis-causing mutations) have sufficient cell membrane instability to have a clinically significant haemolytic anaemia. (wikipedia.org)
- Proporciona un análisis completo de los genes involucrados en esta enfermedad utilizando secuenciación de próxima generación (NGS) para comprender completamente el espectro de genes relevantes involucrados. (igenomix.com)
Symptoms1
- In hereditary spherocytosis, symptoms and signs are usually mild. (msdmanuals.com)
Anemias1
- Spectrin Oligomerization is Cooperatively Coupled to Membrane Assembly: A Linkage Targeted by Many Hereditary Hemolytic Anemias? (yale.edu)
Skeleton1
- Defects in the "vertical" connections between the membrane skeleton and band 3 result in hereditary spherocytosis. (basicmedicalkey.com)
Cell2
- In hereditary spherocytosis, the cell membrane surface area is decreased disproportionately to the intracellular content due to loss of proteins associated with the cell membrane. (msdmanuals.com)
- Like hereditary spherocytosis, this condition also involves a problem with the cell membrane. (hoacny.com)
Normal1
- Although pathological in humans, elliptocytosis is normal in camelids. (wikipedia.org)
Patients2
- Hereditary spherocytosis or hereditary elliptocytosis is suspected in patients with unexplained hemolysis (as suggested by the presence of anemia and reticulocytosis), particularly if splenomegaly, a family history of similar manifestations, or suggestive RBC indices are present. (msdmanuals.com)
- In patients with hereditary spherocytosis (HS), the presence of non-deformable spherocytes causes hemolysis of varying severity. (basicmedicalkey.com)
Disease1
- Orivet is also included within the World Small Animal Veterinary Association hereditary disease checking out database. (aussievetproducts.com.au)
Present1
- hemolytic anemia that is present from birth and in which the lifespan of red blood cells is diminished, such as occurs in hereditary spherocytosis. (en-academic.com)
Type1
- X linked Alport syndrome (ATS, OMIM 301050) is a hereditary glomerulonephritis resulting from either point mutations or intragenic deletions of the COL4A5 gene encoding the α5 chain of type IV collagen. (bmj.com)