Farber Lipogranulomatosis
Acid Ceramidase
Galactosylgalactosylglucosylceramidase
In vivo delivery of human acid ceramidase via cord blood transplantation and direct injection of lentivirus as novel treatment approaches for Farber disease. (1/7)
(+info)Disseminated lipogranulomatosis. (2/7)
Farber disease or disseminated lipogranulomatosis is a rare inherited disorder of lipid metabolism resulting from a defect in ceramide degradation. Because of the feature of nodular swellings around various joints, this may sometimes be confused with juvenile idiopathic arthritis. We report a 4-year-old boy with Farber disease who presented with nodular swellings around the joint, angle of the mouth and conjunctiva, and was subsequently diagnosed to be a case of Farber Disease. (+info)A simple fluorogenic method for determination of acid ceramidase activity and diagnosis of Farber disease. (3/7)
(+info)Autologous transplantation of lentivector/acid ceramidase-transduced hematopoietic cells in nonhuman primates. (4/7)
(+info)Regulation of CC ligand 5/RANTES by acid sphingomyelinase and acid ceramidase. (5/7)
(+info)Novel biochemical abnormalities and genotype in Farber disease. (6/7)
Farber disease caused by acid ceramidase deficiency is characterised by a triad of painful and swollen joints, subcutaneous nodules, and laryngeal involvement. A one year old female with overlapping features of the classical and type 5 variants is reported. Sialuria and elevated plasma chitotriosidase were unusual findings. A novel mutation of the ASAH 1 gene was detected from DNA extracted from the umbilical stump. (+info)Systemic ceramide accumulation leads to severe and varied pathological consequences. (7/7)
(+info)Farber Lipogranulomatosis is a rare genetic disorder characterized by the accumulation of fatty substances (lipids) in various tissues and organs of the body. This condition is also known as Farber's disease or Infantile Ceramidase Deficiency. It is caused by mutations in the ASAH1 gene, which leads to a deficiency in the enzyme acid ceramidase. As a result, lipids accumulate in cells, causing inflammation and the formation of granulomas.
The symptoms of Farber Lipogranulomatosis typically appear within the first few months of life and can include:
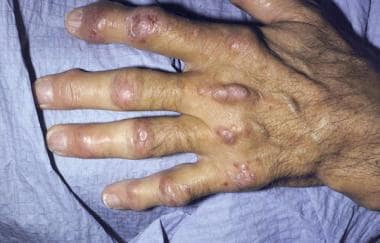
* Subcutaneous nodules (small lumps under the skin) that are painful and tender to the touch
* Hoarseness or weakened cry due to inflammation in the voice box (larynx)
* Joint stiffness and pain due to lipid accumulation in the synovial fluid of joints
* Difficulty swallowing and feeding due to lipid accumulation in the muscles used for swallowing
* Slow growth and development
* Respiratory problems, including recurrent pneumonia and bronchitis
There is no cure for Farber Lipogranulomatosis, but treatment can help manage the symptoms. This may include physical therapy to improve joint mobility, medications to reduce inflammation and pain, and feeding tubes to ensure adequate nutrition. In some cases, bone marrow transplantation may be considered as a potential treatment option.
Acid ceramidase is an enzyme that plays a role in the metabolism of ceramides, which are lipid molecules found in cell membranes. Specifically, acid ceramidase helps to break down ceramides into sphingosine and free fatty acids. This enzyme is active at an acidic pH and is located in the lysosomes, which are organelles within cells that help to break down and recycle various materials.
Defects in the gene that provides instructions for making acid ceramidase can lead to a condition called Farber disease, which is characterized by the accumulation of ceramides in various tissues and organs. This can cause a range of symptoms, including joint pain, muscle weakness, and developmental delays.
Galactosylgalactosylglucosylceramidase is a type of enzyme that is involved in the breakdown and recycling of complex lipids called glycosphingolipids in the body. More specifically, it helps to break down a particular type of glycosphingolipid known as globotriaosylceramide (Gb3 or CD77) into simpler components.
This enzyme is critical for maintaining the health and function of various tissues in the body, including the nervous system. Deficiencies in galactosylgalactosylglucosylceramidase have been linked to a number of serious genetic disorders, such as Tay-Sachs disease and Sandhoff disease, which are characterized by the accumulation of Gb3 and other glycosphingolipids in various tissues, leading to progressive neurological deterioration and other symptoms.
Ceramidases are a group of enzymes that catalyze the hydrolysis of ceramide into sphingosine and free fatty acids. Ceramides are important components of cell membranes, and their metabolism is tightly regulated in cells. The hydrolysis of ceramide by ceramidases produces sphingosine, which can be further phosphorylated to form sphingosine-1-phosphate (S1P), a signaling molecule involved in various cellular processes such as proliferation, differentiation, and survival.
There are several types of ceramidases that have been identified, including acid ceramidase, neutral ceramidase, and alkaline ceramidase. These enzymes differ in their subcellular localization, substrate specificity, and physiological functions. Dysregulation of ceramidase activity has been implicated in various diseases, including cancer, neurodegenerative disorders, and inflammatory conditions. Therefore, ceramidases are considered as potential therapeutic targets for the treatment of these diseases.
Ceramidase2
- Alves MQ, Le Trionnaire E, Ribeiro I, Carpentier S, Harzer K, Levade T, Ribeiro MG. Molecular basis of acid ceramidase deficiency in a neonatal form of Farber disease: identification of the first large deletion in ASAH1 gene. (medlineplus.gov)
- Inherited deficiency of acid ceramidase activity results in FARBER LIPOGRANULOMATOSIS. (nih.gov)
Farber's2
ASAH12
- Variants (also known as mutations) in the ASAH1 gene cause Farber lipogranulomatosis. (medlineplus.gov)
- The spectrum of ASAH1 -related disorders ranges from Farber disease (FD) to spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME). (nih.gov)
Disorder1
- Farber lipogranulomatosis is a rare disorder. (medlineplus.gov)
Features1
- Researchers had previously categorized Farber lipogranulomatosis into subtypes based on characteristic features, but the condition is now thought to be a spectrum of overlapping signs of symptoms. (medlineplus.gov)