Gangliosidosis, GM1
Gangliosidoses
Gangliosidoses, GM2
G(M2) Ganglioside
Sandhoff Disease
Hexosaminidase A
Tay-Sachs Disease
beta-N-Acetylhexosaminidases
Hexosaminidase B
Mucopolysaccharidosis IV
G(M1) Ganglioside
G(M2) Activator Protein
Hexosaminidases
beta-Galactosidase
Lipidoses
Galactosidases
Gangliosides
Sphingolipidoses
Sphingolipid Activator Proteins
Dog Diseases
1-Deoxynojirimycin
beta-Hexosaminidase beta Chain
Imino Sugars
Chromatography, Thin Layer
Cat Diseases
Carbohydrate Metabolism, Inborn Errors
Saposins
Leukodystrophy, Globoid Cell
Fucosidosis
Lysosomes
Fibroblasts
Molecular basis of GM1 gangliosidosis and Morquio disease, type B. Structure-function studies of lysosomal beta-galactosidase and the non-lysosomal beta-galactosidase-like protein. (1/69)
GM1 gangliosidosis and Morquio B disease are distinct disorders both clinically and biochemically yet they arise from the same beta-galactosidase enzyme deficiency. On the other hand, galactosialidosis and sialidosis share common clinical and biochemical features, yet they arise from two separate enzyme deficiencies, namely, protective protein/cathepsin A and neuraminidase, respectively. However distinct, in practice these disorders overlap both clinically and biochemically so that easy discrimination between them is sometimes difficult. The principle reason for this may be found in the fact that these three enzymes form a unique complex in lysosomes that is required for their stability and posttranslational processing. In this review, I focus mainly on the primary and secondary beta-galactosidase deficiency states and offer some hypotheses to account for differences between GM1 gangliosidosis and Morquio B disease. (+info)Processing of lysosomal beta-galactosidase. The C-terminal precursor fragment is an essential domain of the mature enzyme. (2/69)
Lysosomal beta-D-galactosidase (beta-gal), the enzyme deficient in the autosomal recessive disorders G(M1) gangliosidosis and Morquio B, is synthesized as an 85-kDa precursor that is C-terminally processed into a 64-66-kDa mature form. The released approximately 20-kDa proteolytic fragment was thought to be degraded. We now present evidence that it remains associated to the 64-kDa chain after partial proteolysis of the precursor. This polypeptide was found to copurify with beta-gal and protective protein/cathepsin A from mouse liver and Madin-Darby bovine kidney cells and was immunoprecipitated from human fibroblasts but not from fibroblasts of a G(M1) gangliosidosis and a galactosialidosis patient. Uptake of wild-type protective protein/cathepsin A by galactosialidosis fibroblasts resulted in a significant increase of mature and active beta-gal and its C-terminal fragment. Expression in COS-1 cells of mutant cDNAs encoding either the N-terminal or the C-terminal domain of beta-gal resulted in the synthesis of correctly sized polypeptides without catalytic activity. Only when co-expressed, the two subunits associate and become catalytically active. Our results suggest that the C terminus of beta-gal is an essential domain of the catalytically active enzyme and provide evidence that lysosomal beta-galactosidase is a two-subunit molecule. These data may give new significance to mutations in G(M1) gangliosidosis patients found in the C-terminal part of the molecule. (+info)Characterization of beta-galactosidase mutations Asp332-->Asn and Arg148-->Ser, and a polymorphism, Ser532-->Gly, in a case of GM1 gangliosidosis. (3/69)
We have identified and characterized three missense mutations in a patient with type 1 G(M1) gangliosidosis, namely a substitution of G for A at nucleotide position 1044 (G1044-->A; in exon 10) on one allele, which converts Asp(332) into asparagine, and both a mutation (C492-->A in exon 4, leading to the amino acid change of Arg(148)-->Ser) and a polymorphism (A1644-->G in exon 15, leading to a change of Ser(532)-->Gly) on the other allele. This patient had less than 1% residual beta-galactosidase activity and minimally detectable levels of immunoreactive beta-galactosidase protein in fibroblasts. To account for the above findings, a series of expression and immunolocalization studies were undertaken to assess the impact of each mutation. Transient overexpression in COS-1 cells of cDNAs encoding Asp(332)Asn, Arg(148)Ser and Ser(532)Gly mutant beta-galactosidases produced abundant amounts of precursor beta-galactosidase, with activities of 0, 84 and 81% compared with the cDNA clone for wild-type beta-galactosidase (GP8). Since the level of vector-driven expression is much less in Chinese hamster ovary (CHO) cells than in COS-1 cells, and we knew that exogenous beta-galactosidase undergoes lysosomal processing when expressed in these cells, transient expression studies were performed of Arg(148)Ser and Ser(532)Gly, which yielded active forms of the enzyme. In this case, the Arg(148)Ser and Ser(532)Gly products gave rise to 11% and 86% of the control activity respectively. These results were not unexpected, since the Arg(148)Ser mutation introduced a major conformational change into the protein, and we anticipated that it would be degraded in the endoplasmic reticulum (ER), whereas the polymorphism was expected to produce near-normal activity. To examine the effect of the Asp(332)Asn mutation on the catalytic activity, we isolated CHO clones permanently transfected with the Asp(332)Asn and Asp(332)Glu constructs, purified the enzymes by substrate-analogue-affinity chromatography, and determined their kinetic parameters. The V(max) values of both mutant recombinant enzymes were markedly reduced (less than 0.9% of the control), and the K(m) values were unchanged compared with the corresponding wild-type enzyme isolated at the same time. Both the Arg(148)Ser beta-galactosidase in CHO cells and Asp(332)Asn beta-galactosidases (in COS-1 and CHO cells) produced abundant immunoreaction in the perinuclear area, consistent with localization in the ER. A low amount was detected in lysosomes. Incubation of patient fibroblasts in the presence of leupeptin, which reduces the rate of degradation of lysosomal beta-galactosidase by thiol proteases, had no effect on residual enzyme activity, and immunostaining was again detected largely in the perinuclear area (localized to the ER) with much lower amounts in the lysosomes. In summary, the Arg(148)Ser mutation has no effect on catalytic activity, whereas the Asp(332)Asn mutation seriously reduces catalytic activity, suggesting that Asp(332) might play a role in the active site. Immunofluorescence studies indicate the expressed mutant proteins with Arg(148)Ser and Asp(332)Asn mutations are held up in the ER, where they are probably degraded, resulting in only minimum amounts of the enzyme becoming localized in the lysosomes. These results are completely consistent with findings in the cultured fibroblasts. Our results imply that most of the missense mutations described in G(M1) gangliosidosis to date have little effect on catalytic activity, but do affect protein conformation such that the resulting protein cannot be transported out of the ER and fails to arrive in the lysosome. This accounts for the minimal amounts of enzyme protein and activity seen in most G(M1) gangliosidosis patient fibroblasts. (+info)Impaired elastic-fiber assembly by fibroblasts from patients with either Morquio B disease or infantile GM1-gangliosidosis is linked to deficiency in the 67-kD spliced variant of beta-galactosidase. (4/69)
We have previously shown that intracellular trafficking and extracellular assembly of tropoelastin into elastic fibers is facilitated by the 67-kD elastin-binding protein identical to an enzymatically inactive, alternatively spliced variant of beta-galactosidase (S-Gal). In the present study, we investigated elastic-fiber assembly in cultures of dermal fibroblasts from patients with either Morquio B disease or GM1-gangliosidosis who bore different mutations of the beta-galactosidase gene. We found that fibroblasts taken from patients with an adult form of GM1-gangliosidosis and from patients with an infantile form, carrying a missense mutations in the beta-galactosidase gene-mutations that caused deficiency in lysosomal beta-galactosidase but not in S-Gal-assembled normal elastic fibers. In contrast, fibroblasts from two cases of infantile GM1-gangliosidosis that bear nonsense mutations of the beta-galactosidase gene, as well as fibroblasts from four patients with Morquio B who had mutations causing deficiency in both forms of beta-galactosidase, did not assemble elastic fibers. We also demonstrated that S-Gal-deficient fibroblasts from patients with either GM1-gangliosidosis or Morquio B can acquire the S-Gal protein, produced by coculturing of Chinese hamster ovary cells permanently transected with S-Gal cDNA, resulting in improved deposition of elastic fibers. The present study provides a novel and natural model validating functional roles of S-Gal in elastogenesis and elucidates an association between impaired elastogenesis and the development of connective-tissue disorders in patients with Morquio B disease and in patients with an infantile form of GM1-gangliosidosis. (+info)GM1-gangliosidosis in Alaskan huskies: clinical and pathologic findings. (5/69)
Three Alaskan Huskies, two females and one male, were diagnosed with GM1-gangliosidosis. Clinically, diseased animals exhibited proportional dwarfism and developed progressive neurologic impairment with signs of cerebellar dysfunction at the age of 5-7 months. Skeletal lesions characterized by retarded enchondral ossification of vertebral epiphyses were revealed by radiographs of the male dog at 5.5 months of age. Histologic examination of the central nervous system (CNS) revealed that most neurons were enlarged with a foamy to granular cytoplasm due to tightly packed vacuoles that displaced the Nissl substance. Vacuoles in paraffin-embedded sections stained positively with Luxol fast blue and Grocott's method, and in frozen sections vacuoles were periodic acid-Schiff positive. Foamy vacuolation also occurred within neurons of the autonomic ganglia. Extracerebral cells such as macrophages and peripheral lymphocytes also displayed foamy cytoplasm and vacuolation. In the CNS of diseased animals, a mild demyelination and axonal degeneration was accompanied by a significant astrogliosis (P < 0.05) in the gray matter as compared with age- and sex-matched control dogs. There was also a significant loss (P < 0.05) of oligodendrocytes in the gray and white matter of affected animals as compared with controls. Ultrastructurally, the neuronal storage material consisted of numerous circular to concentric whorls of lamellated membranes or stacks of membranes in parallel arrays. GM1-gangliosidosis in Alaskan Huskies resembles beta-galactosidase deficiency in other canine breeds, and these CNS disorders may be a consequence of neuronal storage and disturbed myelin processing. (+info)Signaling pathways transduced through the elastin receptor facilitate proliferation of arterial smooth muscle cells. (6/69)
In this report we demonstrate that soluble peptides, elastin degradation products stimulate proliferation of arterial smooth muscle cells. We show that these effects are due to generation of intracellular signals transduced through the cell surface elastin receptor, which consists of peripheral 67-kDa elastin-binding protein (EBP) (spliced variant of beta-galactosidase), immobilized to the transmembrane sialidase and the protective protein. We found that elastin receptor-transduced signaling triggers activation of G proteins, opening of l-type calcium channels, and a sequential activation of tyrosine kinases: FAK, c-Src, platelet-derived growth factor-receptor kinase and then Ras-Raf-MEK1/2-ERK1/2 phosphorylation cascade. This, in turn, causes an increase in expression of cyclins and cyclin-dependent kinases, and a consequent increase in cellular proliferation. The EBP-transduced signals also induce tyrosine kinase-dependent phosphorylation of beta-tubulin, LC3, microtubule-associated protein 1, and alpha-actin and troponin-T, which could be linked to reorganization of cytoskeleton. We have also disclosed that induction of these signals can be abolished by anti-EBP antibody or by galactosugars, which cause shedding of EBP from the cell surface. Moreover, elastin-derived peptides did not induce proliferation of EBP-deficient cells derived from patients bearing a nonsense mutation of the beta-galactosidase gene or sialidase-deficient cells from patients with congenital sialidosis. (+info)Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. (7/69)
Mouse models of the GM2 gangliosidoses [Tay-Sachs, late onset Tay-Sachs (LOTS), Sandhoff] and GM1 gangliosidosis have been studied to determine whether there is a common neuro-inflammatory component to these disorders. During the disease course, we have: (i) examined the expression of a number of inflammatory markers in the CNS, including MHC class II, CD68, CD11b (CR3), 7/4, F4/80, nitrotyrosine, CD4 and CD8; (ii) profiled cytokine production [tumour necrosis factor alpha (TNF alpha), transforming growth factor (TGF beta 1) and interleukin 1 beta (IL1 beta)]; and (iii) studied blood-brain barrier (BBB) integrity. The kinetics of apoptosis and the expression of Fas and TNF-R1 were also assessed. In all symptomatic mouse models, a progressive increase in local microglial activation/expansion and infiltration of inflammatory cells was noted. Altered BBB permeability was evident in Sandhoff and GM1 mice, but absent in LOTS mice. Progressive CNS inflammation coincided with the onset of clinical signs in these mouse models. Substrate reduction therapy in the Sandhoff mouse model slowed the rate of accumulation of glycosphingolipids in the CNS, thus delaying the onset of the inflammatory process and disease pathogenesis. These data suggest that inflammation may play an important role in the pathogenesis of the gangliosidoses. (+info)Brain ceramide hexosides in Tay-Sachs disease and generalized gangliosidosis (GM1-gangliosidosis). (8/69)
The carbohydrate composition was determined for ceramide hexosides isolated from brains of patients with Tay-Sachs disease and generalized gangliosidosis (hereby named GM1-gangliosidosis). Gray matter of patients with each disease showed a characteristic abnormal ceramide hexoside pattern. In Tay-Sachs gray matter, ceramide trihexoside is the major component, whereas ceramide tetrahexoside is barely detectable. In GM1-gangliosidosis, ceramide tetrahexoside is the major ceramide hexoside, while ceramide trihexoside is present only in small amount. These two major components have been characterized as the asialo derivatives of, respectively, the "Tay-Sachs ganglioside" (GM2-ganglioside) and the normal major monosialoganglioside (GM1-ganglioside). In both diseases, more than half the ceramide monohexoside of gray matter was glucocerebroside. Gray matter ceramide dihexoside, present in both diseases at higher than normal levels, was mostly ceramide lactoside, with possibly a small amount of ceramide digalactoside. Sulfatide contained only galactose. The abnormal ceramide hexoside pattern is limited to gray matter: white matter showed normal ceramide hexosides, i.e. a preponderance of monohexosides and sulfatide, with no detectable glucocerebroside. (+info)GM1 gangliosidosis is a rare inherited lysosomal storage disorder caused by the deficiency of an enzyme called β-galactosidase. This enzyme is responsible for breaking down certain complex fats (gangliosides) in the body. When this enzyme is lacking or not working properly, these gangliosides accumulate in various cells, particularly in nerve cells of the brain, leading to progressive neurological deterioration.
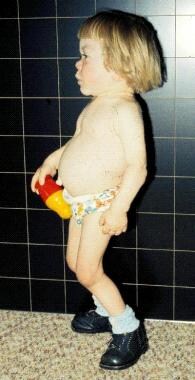
The condition can present at different ages and with varying severity, depending on the amount of functional β-galactosidase enzyme activity. The three main types of GM1 gangliosidosis are:
1. Early infantile (type I): This is the most severe form, with symptoms appearing within the first few months of life. Infants may appear normal at birth but then develop rapidly progressing neurological problems such as developmental delay, muscle weakness, seizures, and cherry-red spots in the eyes. Life expectancy is typically less than 2 years.
2. Late infantile/juvenile (type II): Symptoms begin between ages 1 and 3 years or later in childhood. Affected individuals may have developmental delay, motor difficulties, muscle weakness, and cognitive decline. Some individuals with this form may also develop corneal clouding and bone abnormalities.
3. Adult/chronic (type III): This is the least severe form of GM1 gangliosidosis, with symptoms appearing in late childhood, adolescence, or adulthood. Symptoms can include neurological problems such as muscle weakness, tremors, and difficulties with coordination and speech.
Currently, there is no cure for GM1 gangliosidosis, and treatment is primarily supportive to manage symptoms and improve quality of life.
Gangliosidoses are a group of inherited metabolic disorders caused by the accumulation of certain complex lipids called gangliosides in the brain and nervous system. This buildup is due to a deficiency of specific enzymes needed to break down these substances. The three main types of gangliosidoses are:
1. Type 1 - Infantile Neurovisceral or Tay-Sachs Disease: Characterized by the absence of the enzyme hexosaminidase A, leading to severe neurological symptoms such as muscle weakness, blindness, and developmental delay in early infancy, with rapid progression and death usually occurring before age 4.
2. Type 2 - Juvenile or Subacute GM1 Gangliosidosis: Caused by a deficiency of the enzyme beta-galactosidase, resulting in progressive neurological symptoms such as motor and cognitive decline, beginning between ages 6 months and 2 years. Affected individuals may survive into adolescence or early adulthood.
3. Type 3 - Adult or Chronic GM1 Gangliosidosis: Characterized by a deficiency of beta-galactosidase, leading to milder neurological symptoms that appear in late childhood, adolescence, or even adulthood. The progression is slower compared to the other types, and life expectancy varies widely.
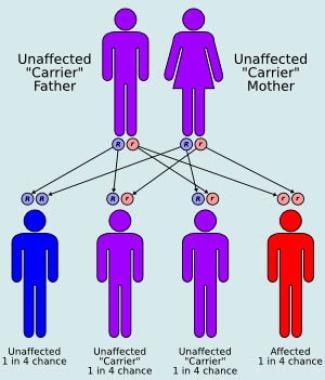
Gangliosidoses are autosomal recessive disorders, meaning an individual must inherit two copies of the defective gene (one from each parent) to develop the condition.
GM2 gangliosidoses are a group of inherited metabolic disorders caused by the accumulation of harmful amounts of GM2 gangliosides in the body's cells, particularly in the nerve cells of the brain. There are three main types of GM2 gangliosidoses: Tay-Sachs disease, Sandhoff disease, and AB variant of GM2 gangliosidosis. These conditions are characterized by progressive neurological degeneration, which can lead to severe physical and mental disabilities, and ultimately death in childhood or early adulthood.
The underlying cause of GM2 gangliosides is a deficiency in the enzyme hexosaminidase A (Tay-Sachs and AB variant) or both hexosaminidase A and B (Sandhoff disease), which are responsible for breaking down GM2 gangliosides. Without sufficient enzyme activity, GM2 gangliosides accumulate in the lysosomes of cells, leading to cell dysfunction and death.
Symptoms of GM2 gangliosidoses can vary depending on the specific type and severity of the disorder, but often include developmental delay, muscle weakness, loss of motor skills, seizures, blindness, and dementia. There is currently no cure for GM2 gangliosidoses, and treatment is focused on managing symptoms and improving quality of life.
Sandhoff disease is a rare inherited disorder that affects the nervous system. It's a type of GM2 gangliosidosis, which is a group of conditions characterized by the body's inability to break down certain fats (lipids) called gangliosides.
In Sandhoff disease, deficiencies in the enzymes hexosaminidase A and B lead to an accumulation of GM2 ganglioside in various cells, particularly in nerve cells of the brain. This accumulation results in progressive damage to the nervous system.
The symptoms of Sandhoff disease typically appear between 6 months and 2 years of age and can include developmental delay, seizures, an exaggerated startle response, muscle weakness, loss of motor skills, and vision and hearing loss. The condition is often fatal by around age 3. It's caused by mutations in the HEXB gene, and it's inherited in an autosomal recessive manner, meaning an individual must inherit two copies of the mutated gene (one from each parent) to develop the disease.
Hexosaminidase A is an enzyme that is responsible for breaking down certain complex molecules in the body, specifically gangliosides. This enzyme is composed of two subunits, alpha and beta, which are encoded by the genes HEXA and HEXB, respectively.
Deficiency or mutation in the HEXA gene can lead to a genetic disorder called Tay-Sachs disease, which is characterized by an accumulation of gangliosides in the nerve cells, leading to progressive neurological degeneration. The function of hexosaminidase A is to break down these gangliosides into simpler molecules that can be eliminated from the body. Without sufficient levels of this enzyme, the gangliosides build up and cause damage to the nervous system.
Tay-Sachs Disease is a rare, inherited autosomal recessive disorder that affects the nervous system's functioning. It results from the deficiency of an enzyme called hexosaminidase A (Hex-A), which is necessary for breaking down gangliosides, a type of fatty substance found in nerve cells. When Hex-A is absent or insufficient, gangliosides accumulate abnormally in the nerve cells, leading to their progressive destruction and severe neurological deterioration.
The classic infantile form of Tay-Sachs Disease manifests within the first six months of life with symptoms such as loss of motor skills, seizures, paralysis, dementia, blindness, and eventually death, usually by age four. Late-onset forms of the disease also exist, which may present in childhood or adulthood with milder symptoms.
Tay-Sachs Disease is more prevalent among individuals of Ashkenazi Jewish, French Canadian, and Cajun descent. Genetic counseling and prenatal testing are recommended for couples at risk of passing on the disease.
Beta-N-Acetylhexosaminidases are a group of enzymes that play a role in the breakdown and recycling of complex carbohydrates in the body. Specifically, they help to break down gangliosides, which are a type of molecule found in cell membranes.
There are several different isoforms of beta-N-Acetylhexosaminidases, including A, B, and S. These isoforms are formed by different combinations of subunits, which can affect their activity and substrate specificity.
Mutations in the genes that encode for these enzymes can lead to a variety of genetic disorders, including Tay-Sachs disease and Sandhoff disease. These conditions are characterized by an accumulation of gangliosides in the brain, which can cause progressive neurological deterioration and death.
Treatment for these conditions typically involves managing symptoms and providing supportive care, as there is currently no cure. Enzyme replacement therapy has been explored as a potential treatment option, but its effectiveness varies depending on the specific disorder and the age of the patient.
Hexosaminidase B is a type of enzyme that is involved in the breakdown of complex lipids called gangliosides in the body. These enzymes are found in lysosomes, which are structures inside cells that break down and recycle various materials.
Hexosaminidase B specifically helps to break down a particular type of ganglioside called GM2 ganglioside, which is abundant in the nervous system. Mutations in the gene that provides instructions for making this enzyme can lead to a condition called Tay-Sachs disease, which is characterized by the accumulation of GM2 gangliosides in the nerve cells, leading to progressive neurological deterioration.
In summary, Hexosaminidase B is an essential enzyme for breaking down certain types of lipids in the body, and its deficiency can lead to serious health consequences.
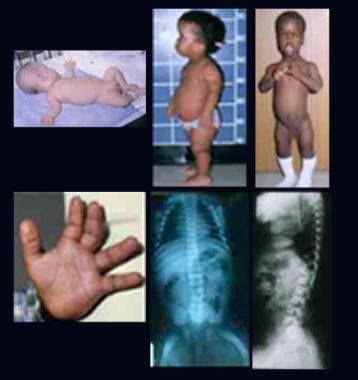
Mucopolysaccharidosis IV (MPS IV), also known as Morquio Syndrome, is a rare genetic disorder that belongs to the family of diseases called mucopolysaccharidoses. It is characterized by the accumulation of glycosaminoglycans (GAGs or mucopolysaccharides) in various tissues and organs due to deficiencies in specific enzymes needed to break down these complex carbohydrates.
There are two types of MPS IV: Type A and Type B, which are caused by deficiencies in different enzymes (GALNS and B3GALNT1, respectively). Both types result in similar symptoms but may vary in severity. The accumulation of GAGs primarily affects the bones, cartilage, eyes, ears, heart, and respiratory system.
Common features of MPS IV include:
* Dwarfism with short trunk and long limbs
* Progressive skeletal abnormalities such as kyphosis (hunchback), scoliosis (curvature of the spine), pectus carinatum (protruding breastbone), and joint laxity or stiffness
* Coarse facial features
* Corneal clouding
* Hearing loss
* Heart valve abnormalities
* Respiratory issues
* Hypermobile and dislocated joints
* Carpal tunnel syndrome
* Spinal cord compression
Treatment for MPS IV primarily focuses on managing symptoms, improving quality of life, and preventing complications. Enzyme replacement therapy (ERT) is available for Type B but not for Type A. Other treatments may include physical therapy, surgery, and medications to address specific symptoms.
Hexosaminidases are a group of enzymes that play a crucial role in the breakdown of complex carbohydrates, specifically glycoproteins and glycolipids, in the human body. These enzymes are responsible for cleaving the terminal N-acetyl-D-glucosamine (GlcNAc) residues from these molecules during the process of glycosidase digestion.
There are several types of hexosaminidases, including Hexosaminidase A and Hexosaminidase B, which are encoded by different genes and have distinct functions. Deficiencies in these enzymes can lead to serious genetic disorders, such as Tay-Sachs disease and Sandhoff disease, respectively. These conditions are characterized by the accumulation of undigested glycolipids and glycoproteins in various tissues, leading to progressive neurological deterioration and other symptoms.
Beta-galactosidase is an enzyme that catalyzes the hydrolysis of beta-galactosides into monosaccharides. It is found in various organisms, including bacteria, yeast, and mammals. In humans, it plays a role in the breakdown and absorption of certain complex carbohydrates, such as lactose, in the small intestine. Deficiency of this enzyme in humans can lead to a disorder called lactose intolerance. In scientific research, beta-galactosidase is often used as a marker for gene expression and protein localization studies.
Lipidoses are a group of genetic disorders characterized by abnormal accumulation of lipids (fats or fat-like substances) in various tissues and cells of the body due to defects in lipid metabolism. These disorders include conditions such as Gaucher's disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, and Wolman disease, among others. The accumulation of lipids can lead to progressive damage in multiple organs, resulting in a range of symptoms and health complications. Early diagnosis and management are essential for improving the quality of life and prognosis of affected individuals.
Galactosidases are a group of enzymes that catalyze the hydrolysis of galactose-containing sugars, specifically at the beta-glycosidic bond. There are several types of galactosidases, including:
1. Beta-galactosidase: This is the most well-known type of galactosidase and it catalyzes the hydrolysis of lactose into glucose and galactose. It has important roles in various biological processes, such as lactose metabolism in animals and cell wall biosynthesis in plants.
2. Alpha-galactosidase: This enzyme catalyzes the hydrolysis of alpha-galactosides, which are found in certain plant-derived foods like legumes. A deficiency in this enzyme can lead to a genetic disorder called Fabry disease.
3. N-acetyl-beta-glucosaminidase: This enzyme is also known as hexosaminidase and it catalyzes the hydrolysis of N-acetyl-beta-D-glucosamine residues from glycoproteins, glycolipids, and other complex carbohydrates.
Galactosidases are widely used in various industrial applications, such as food processing, biotechnology, and biofuel production. They also have potential therapeutic uses, such as in the treatment of lysosomal storage disorders like Fabry disease.
Gangliosides are a type of complex lipid molecule known as sialic acid-containing glycosphingolipids. They are predominantly found in the outer leaflet of the cell membrane, particularly in the nervous system. Gangliosides play crucial roles in various biological processes, including cell recognition, signal transduction, and cell adhesion. They are especially abundant in the ganglia (nerve cell clusters) of the peripheral and central nervous systems, hence their name.
Gangliosides consist of a hydrophobic ceramide portion and a hydrophilic oligosaccharide chain that contains one or more sialic acid residues. The composition and structure of these oligosaccharide chains can vary significantly among different gangliosides, leading to the classification of various subtypes, such as GM1, GD1a, GD1b, GT1b, and GQ1b.
Abnormalities in ganglioside metabolism or expression have been implicated in several neurological disorders, including Parkinson's disease, Alzheimer's disease, and various lysosomal storage diseases like Tay-Sachs and Gaucher's diseases. Additionally, certain bacterial toxins, such as botulinum neurotoxin and tetanus toxin, target gangliosides to gain entry into neuronal cells, causing their toxic effects.
Sphingolipidoses are a group of inherited metabolic disorders characterized by the accumulation of sphingolipids in various tissues and organs due to deficiencies in enzymes involved in sphingolipid metabolism. Sphingolipids are a type of lipid molecule that play important roles in cell membranes, signal transduction, and cell recognition.
Examples of sphingolipidoses include Gaucher's disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, and Krabbe disease, among others. These disorders can affect various organs and systems in the body, including the brain, liver, spleen, bones, and nervous system, leading to a range of symptoms such as developmental delay, seizures, movement disorders, enlarged organs, and skin abnormalities.
Treatment for sphingolipidoses typically involves managing symptoms and addressing complications, although some forms of these disorders may be amenable to enzyme replacement therapy or stem cell transplantation.
Sphingolipid activator proteins (SAPs), also known as saposins, are a group of small proteins that play a crucial role in the metabolism of sphingolipids, a class of lipids found in cell membranes. These proteins are produced by the cleavage of a precursor protein called prosaposin.
SAPs facilitate the hydrolysis of sphingolipids by activating specific lysosomal hydrolases, enzymes that break down these lipids into simpler molecules. Each SAP has a unique structure and function, and they are named SapA, SapB, SapC, and SapD.
SapA and SapB activate the enzyme glucocerebrosidase, which breaks down glucosylceramide into glucose and ceramide. SapC activates the enzyme galactocerebrosidase, which breaks down galactosylceramide into galactose and ceramide. SapD has multiple functions, including activating the enzyme acid sphingomyelinase, which breaks down sphingomyelin into ceramide and phosphorylcholine.
Deficiencies in SAPs can lead to lysosomal storage disorders, such as Gaucher disease (caused by a deficiency in glucocerebrosidase) and Krabbe disease (caused by a deficiency in galactocerebrosidase). These disorders are characterized by the accumulation of undigested sphingolipids in various tissues, leading to cell dysfunction and tissue damage.
There is no medical definition for "dog diseases" as it is too broad a term. However, dogs can suffer from various health conditions and illnesses that are specific to their species or similar to those found in humans. Some common categories of dog diseases include:
1. Infectious Diseases: These are caused by viruses, bacteria, fungi, or parasites. Examples include distemper, parvovirus, kennel cough, Lyme disease, and heartworms.
2. Hereditary/Genetic Disorders: Some dogs may inherit certain genetic disorders from their parents. Examples include hip dysplasia, elbow dysplasia, progressive retinal atrophy (PRA), and degenerative myelopathy.
3. Age-Related Diseases: As dogs age, they become more susceptible to various health issues. Common age-related diseases in dogs include arthritis, dental disease, cancer, and cognitive dysfunction syndrome (CDS).
4. Nutritional Disorders: Malnutrition or improper feeding can lead to various health problems in dogs. Examples include obesity, malnutrition, and vitamin deficiencies.
5. Environmental Diseases: These are caused by exposure to environmental factors such as toxins, allergens, or extreme temperatures. Examples include heatstroke, frostbite, and toxicities from ingesting harmful substances.
6. Neurological Disorders: Dogs can suffer from various neurological conditions that affect their nervous system. Examples include epilepsy, intervertebral disc disease (IVDD), and vestibular disease.
7. Behavioral Disorders: Some dogs may develop behavioral issues due to various factors such as anxiety, fear, or aggression. Examples include separation anxiety, noise phobias, and resource guarding.
It's important to note that regular veterinary care, proper nutrition, exercise, and preventative measures can help reduce the risk of many dog diseases.
1-Deoxynojirimycin (DNJ) is an antagonist of the enzyme alpha-glucosidase, which is involved in the digestion of carbohydrates. DNJ is a naturally occurring compound found in some plants, including mulberry leaves and the roots of the African plant Moringa oleifera. It works by binding to the active site of alpha-glucosidase and inhibiting its activity, which can help to slow down the digestion and absorption of carbohydrates in the small intestine. This can help to reduce postprandial glucose levels (the spike in blood sugar that occurs after a meal) and may have potential benefits for the management of diabetes and other metabolic disorders. DNJ is also being studied for its potential anti-cancer effects.
Beta-Hexosaminidase beta chain is a subunit of the beta-Hexosaminidase enzyme, which is responsible for breaking down complex lipids called gangliosides in the body. Specifically, it helps to break down a type of ganglioside called GM2 ganglioside into simpler components. Defects in this enzyme can lead to a group of genetic disorders known as the GM2 gangliosidoses, which include Tay-Sachs disease and Sandhoff disease. These conditions are characterized by the accumulation of GM2 gangliosides in various tissues, particularly in the nervous system, leading to progressive neurological deterioration.
Iminosugars are a class of naturally occurring compounds that are structural analogs of simple sugars (monosaccharides), in which the oxygen atom in the furan ring is replaced by a nitrogen atom. This small change in structure gives iminosugars unique biological properties, particularly their ability to inhibit carbohydrate-processing enzymes such as glycosidases and glycosyltransferases.
Iminosugars are found in various plants, animals, and microorganisms, and have been studied for their potential therapeutic applications in a variety of diseases, including diabetes, viral infections, and cancer. Some iminosugars have been shown to act as potent inhibitors of glycosidases involved in the replication of certain viruses, such as HIV and hepatitis C virus, making them promising candidates for antiviral therapy.
In addition, iminosugars have been investigated for their potential to modulate the immune system and reduce inflammation, which has led to interest in their use as therapeutic agents for autoimmune diseases and other inflammatory conditions. However, further research is needed to fully understand the mechanisms of action and safety profiles of iminosugars before they can be widely used in clinical settings.
Thin-layer chromatography (TLC) is a type of chromatography used to separate, identify, and quantify the components of a mixture. In TLC, the sample is applied as a small spot onto a thin layer of adsorbent material, such as silica gel or alumina, which is coated on a flat, rigid support like a glass plate. The plate is then placed in a developing chamber containing a mobile phase, typically a mixture of solvents.
As the mobile phase moves up the plate by capillary action, it interacts with the stationary phase and the components of the sample. Different components of the mixture travel at different rates due to their varying interactions with the stationary and mobile phases, resulting in distinct spots on the plate. The distance each component travels can be measured and compared to known standards to identify and quantify the components of the mixture.
TLC is a simple, rapid, and cost-effective technique that is widely used in various fields, including forensics, pharmaceuticals, and research laboratories. It allows for the separation and analysis of complex mixtures with high resolution and sensitivity, making it an essential tool in many analytical applications.
There are many diseases that can affect cats, and the specific medical definitions for these conditions can be quite detailed and complex. However, here are some common categories of feline diseases and examples of each:
1. Infectious diseases: These are caused by viruses, bacteria, fungi, or parasites. Examples include:
* Feline panleukopenia virus (FPV), also known as feline parvovirus, which can cause severe gastrointestinal symptoms and death in kittens.
* Feline calicivirus (FCV), which can cause upper respiratory symptoms such as sneezing and nasal discharge.
* Feline leukemia virus (FeLV), which can suppress the immune system and lead to a variety of secondary infections and diseases.
* Bacterial infections, such as those caused by Pasteurella multocida or Bartonella henselae, which can cause abscesses or other symptoms.
2. Neoplastic diseases: These are cancerous conditions that can affect various organs and tissues in cats. Examples include:
* Lymphoma, which is a common type of cancer in cats that can affect the lymph nodes, spleen, liver, and other organs.
* Fibrosarcoma, which is a type of soft tissue cancer that can arise from fibrous connective tissue.
* Squamous cell carcinoma, which is a type of skin cancer that can be caused by exposure to sunlight or tobacco smoke.
3. Degenerative diseases: These are conditions that result from the normal wear and tear of aging or other factors. Examples include:
* Osteoarthritis, which is a degenerative joint disease that can cause pain and stiffness in older cats.
* Dental disease, which is a common condition in cats that can lead to tooth loss, gum inflammation, and other problems.
* Heart disease, such as hypertrophic cardiomyopathy (HCM), which is a thickening of the heart muscle that can lead to congestive heart failure.
4. Hereditary diseases: These are conditions that are inherited from a cat's parents and are present at birth or develop early in life. Examples include:
* Polycystic kidney disease (PKD), which is a genetic disorder that causes cysts to form in the kidneys and can lead to kidney failure.
* Hypertrophic cardiomyopathy (HCM), which can be inherited as an autosomal dominant trait in some cats.
* Progressive retinal atrophy (PRA), which is a group of genetic disorders that cause degeneration of the retina and can lead to blindness.
Inborn errors of carbohydrate metabolism refer to genetic disorders that affect the body's ability to break down and process carbohydrates, which are sugars and starches that provide energy for the body. These disorders are caused by defects in enzymes or transport proteins that play a critical role in the metabolic pathways involved in carbohydrate metabolism.
There are several types of inborn errors of carbohydrate metabolism, including:
1. Galactosemia: This disorder affects the body's ability to metabolize the sugar galactose, which is found in milk and other dairy products. It is caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase.
2. Glycogen storage diseases: These disorders affect the body's ability to store and break down glycogen, which is a complex carbohydrate that serves as a source of energy for the body. There are several types of glycogen storage diseases, each caused by a deficiency in a different enzyme involved in glycogen metabolism.
3. Hereditary fructose intolerance: This disorder affects the body's ability to metabolize the sugar fructose, which is found in fruits and sweeteners. It is caused by a deficiency of the enzyme aldolase B.
4. Pentose phosphate pathway disorders: These disorders affect the body's ability to metabolize certain sugars and generate energy through the pentose phosphate pathway. They are caused by defects in enzymes involved in this pathway.
Symptoms of inborn errors of carbohydrate metabolism can vary widely depending on the specific disorder and its severity. Treatment typically involves dietary restrictions, supplementation with necessary enzymes or cofactors, and management of complications. In some cases, enzyme replacement therapy or even organ transplantation may be considered.
Saposins are a group of naturally occurring lipid-binding proteins that play an essential role in the metabolism of lipids within cells. They are named after a skin disease called "Niemann-Pick disease," where defects in saposin function lead to an accumulation of lipids in various tissues, including the brain.
There are four types of saposins (SapA, SapB, SapC, and SapD) that are produced by the cleavage of a larger precursor protein called prosaposin. These proteins help to facilitate the breakdown of lipids in lysosomes, which are specialized organelles within cells that break down and recycle various materials.
Saposins play an important role in activating certain enzymes that are involved in breaking down lipids, such as sphingolipids and gangliosides. They do this by binding to these enzymes and presenting them with their lipid substrates in a way that allows the enzymes to efficiently break them down.
Defects in saposin function can lead to a variety of diseases, including Niemann-Pick disease, Gaucher disease, and Krabbe disease, which are characterized by an accumulation of lipids in various tissues and neurological symptoms.
Globoid cell leukodystrophy, also known as Krabbe disease, is a rare inherited disorder that affects the nervous system. It is characterized by the accumulation of abnormal quantities of a protein called psychosine in the brain's nerve cells, leading to their destruction and progressive damage to the protective sheath (myelin) that covers the nerves.
The term "leukodystrophy" refers to a group of disorders that affect the white matter of the brain, which is primarily composed of myelin. In globoid cell leukodystrophy, the accumulation of psychosine in the brain's nerve cells, particularly in macrophages (which are then referred to as "globoid cells"), results in progressive demyelination and severe neurological symptoms.
Early-onset forms of Krabbe disease typically present within the first six months of life, with symptoms such as irritability, feeding difficulties, muscle weakness, and developmental delays. Late-onset forms may not become apparent until later in childhood or even adulthood, with symptoms that can include vision loss, hearing impairment, muscle stiffness, and difficulty coordinating movements. The progression of the disease is often rapid, leading to severe disability and a shortened lifespan.
There is currently no cure for globoid cell leukodystrophy, but various treatments, such as bone marrow transplantation and enzyme replacement therapy, are being investigated to help manage the symptoms and slow down the progression of the disease.
Fucosidosis is a rare inherited metabolic disorder caused by the deficiency of the enzyme alpha-L-fucosidase. This enzyme is responsible for breaking down complex sugars called glycoproteins and glycolipids in the body. Without sufficient levels of this enzyme, these substances accumulate in various tissues and organs, leading to progressive cellular damage and impaired function.
The condition is characterized by a wide range of symptoms, including coarse facial features, developmental delays, intellectual disability, seizures, vision and hearing loss, cardiac problems, and skeletal abnormalities. There are two main types of fucosidosis, type 1 and type 2, which differ in the age of onset and severity of symptoms.
Fucosidosis is an autosomal recessive disorder, meaning that an individual must inherit two copies of the defective gene, one from each parent, to develop the condition. It is typically diagnosed through enzyme assays and genetic testing. Currently, there is no cure for fucosidosis, and treatment is focused on managing symptoms and improving quality of life.
Imino furanoses are not a recognized medical term, but they may be referred to in the field of biochemistry and carbohydrate research. In this context, imino furanoses are a type of sugar ring structure that contains an imine group (-C=N-) instead of the usual oxygen atom in the furanose form of sugars. Imino furanoses can be formed under certain conditions during chemical reactions involving carbohydrates, but they are not typically found in biological systems.
Lysosomes are membrane-bound organelles found in the cytoplasm of eukaryotic cells. They are responsible for breaking down and recycling various materials, such as waste products, foreign substances, and damaged cellular components, through a process called autophagy or phagocytosis. Lysosomes contain hydrolytic enzymes that can break down biomolecules like proteins, nucleic acids, lipids, and carbohydrates into their basic building blocks, which can then be reused by the cell. They play a crucial role in maintaining cellular homeostasis and are often referred to as the "garbage disposal system" of the cell.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
Lysosomal storage diseases (LSDs) are a group of rare inherited metabolic disorders caused by defects in lysosomal function. Lysosomes are membrane-bound organelles within cells that contain enzymes responsible for breaking down and recycling various biomolecules, such as proteins, lipids, and carbohydrates. In LSDs, the absence or deficiency of specific lysosomal enzymes leads to the accumulation of undigested substrates within the lysosomes, resulting in cellular dysfunction and organ damage.
These disorders can affect various organs and systems in the body, including the brain, nervous system, bones, skin, and visceral organs. Symptoms may include developmental delays, neurological impairment, motor dysfunction, bone abnormalities, coarse facial features, hepatosplenomegaly (enlarged liver and spleen), and recurrent infections.
Examples of LSDs include Gaucher disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, Pompe disease, and mucopolysaccharidoses (MPS). Treatment options for LSDs may include enzyme replacement therapy, substrate reduction therapy, or bone marrow transplantation. Early diagnosis and intervention can help improve the prognosis and quality of life for affected individuals.
 GM1 gangliosidosis: MedlinePlus Genetics
GM1 gangliosidosis: MedlinePlus Genetics
 GM1 gangliosidoses - Wikipedia
GM1 gangliosidoses - Wikipedia
 GM1 Gangliosidosis: Background, Pathophysiology, Epidemiology
GM1 Gangliosidosis: Background, Pathophysiology, Epidemiology
 Myelin abnormalities in the optic and sciatic nerves in mice with GM1-gangliosidosis
Myelin abnormalities in the optic and sciatic nerves in mice with GM1-gangliosidosis
 Identification of Two Novel Pathogenic Mutations of GM1 Gangliosidosis in Japanese Domestic Shorthair Cats - WSAVA 2015...
Identification of Two Novel Pathogenic Mutations of GM1 Gangliosidosis in Japanese Domestic Shorthair Cats - WSAVA 2015...
Gene therapy for GM1 gangliosidosis: challenges of translational medicine. - Department of Experimental Psychology
GM1 Gangliosidosis: Background, Pathophysiology, Epidemiology
 GM1 Gangliosidosis - United Brain Association
GM1 Gangliosidosis - United Brain Association
 Hyperphosphatasemia in GM1 gangliosidosis.<...
Hyperphosphatasemia in GM1 gangliosidosis.<...
 GM1-Gangliosidosis typ I-II, differential diagnosis | Amedes Genetics
GM1-Gangliosidosis typ I-II, differential diagnosis | Amedes Genetics
GM1 Gangliosidosis Treatment Market trends - Article Wood - Bloggers Unite India
 Improvements in Developmental Milestones Observed With GM1 Gangliosidosis Gene Therapy
Improvements in Developmental Milestones Observed With GM1 Gangliosidosis Gene Therapy
 Passage Bio Reports Positive Interim Clinical Data from Study of First Eight Patients with GM1 Gangliosidosis - Global Genes
Passage Bio Reports Positive Interim Clinical Data from Study of First Eight Patients with GM1 Gangliosidosis - Global Genes
 ICGNMD Publications | International Centre for Genomic Medicine in Neuromuscular Diseases - UCL - University College London
ICGNMD Publications | International Centre for Genomic Medicine in Neuromuscular Diseases - UCL - University College London
 Lipid Storage Diseases | National Institute of Neurological Disorders and Stroke
Lipid Storage Diseases | National Institute of Neurological Disorders and Stroke
Genetic Brain Disorders | MedlinePlus
 Shop cat DNA tests | Wisdom Panel™ Complete for Cats
Shop cat DNA tests | Wisdom Panel™ Complete for Cats
 Azafaros Announces Enrollment of First Patient in Phase 2 RAINBOW Study Evaluating AZ-3102 in GM2 and NP-C Patients - DutchNews...
Azafaros Announces Enrollment of First Patient in Phase 2 RAINBOW Study Evaluating AZ-3102 in GM2 and NP-C Patients - DutchNews...
Index by author - October 01, 1998, 19 (9) | American Journal of Neuroradiology
A Complete List of Available Canine and Feline Genetic Tests - TUFTSBG2003 - VIN
 GLB1-Related Disorders - Norton & Elaine Sarnoff Center for Jewish Genetics
GLB1-Related Disorders - Norton & Elaine Sarnoff Center for Jewish Genetics
 WikiGenes
WikiGenes
 FIFes A&R regler Appendix I - Felis Danica
FIFes A&R regler Appendix I - Felis Danica
 Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US
Pantothenate kinase-associated neurodegeneration
 World Journal of Clinical Cases - Baishideng Publishing Group
World Journal of Clinical Cases - Baishideng Publishing Group
 Krabbe disease | Radiology Reference Article | Radiopaedia.org
Krabbe disease | Radiology Reference Article | Radiopaedia.org
Fucosidosis | Radiology Reference Article | Radiopaedia.org
 A Surfeit of Sphingolipids Sours Aβ Digestion | ALZFORUM
A Surfeit of Sphingolipids Sours Aβ Digestion | ALZFORUM
GLB110
- Variants (also called mutations) in the GLB1 gene cause GM1 gangliosidosis. (medlineplus.gov)
- The GLB1 gene variants produce versions of β-galactosidase that are not as effective at breaking down GM1 ganglioside as the normal version of the enzyme. (medlineplus.gov)
- The GM1 gangliosidoses, usually shortened to GM1, are gangliosidoses caused by mutation in the GLB1 gene resulting in a deficiency of beta-galactosidase. (wikipedia.org)
- GM1 Gangliosidoses disorders are caused by mutations in the GLB1 gene, which codes for lysosomal hydrolase, acid beta-galactosidase (β-gal). (wikipedia.org)
- GM1 gangliosidosis is a fatal, progressive, neurodegenerative lysosomal storage disease caused by mutations in the β-galactosidase ( GLB1 ) gene. (vin.com)
- GM1 gangliosidosis is caused by an abnormal variation (mutation) in the GLB1 gene, which plays a role in producing beta-galactosidase. (unitedbrainassociation.org)
- GM1 is a rare, fatal lysosomal storage disease in which mutations in the GLB1 gene result in very low activity of the enzyme beta-galactosidase (β-Gal). (globalgenes.org)
- GLB1-related conditions including GM1-gangliosidosis and mucopolysaccharidosis type IVB (MPSIVB) are two distinct lysosomal storage disorders caused by mutations the GLB1 gene. (jewishgenetics.org)
- Glb1 knockout mouse model shares natural history with type II GM1 gangliosidosis patients. (umassmed.edu)
- During the first half of 2020, the company plans to submit an IND for PBGM01, designed to treat GM1 gangliosidosis by targeting the lysosomal enzyme β-galactosidase (GLB1). (diwou.com)
Gangliosides9
- Low levels of β-gal cause an accumulation of GM1 gangliosides. (wikipedia.org)
- Reduced β-gal activity results in the accumulation of toxic levels of GM1 gangliosides in neurons throughout the brain, causing rapidly progressive neurodegeneration. (globalgenes.org)
- Furthermore, Dose 2 of PBGM01 has shown the ability to achieve healthy control levels of β-Gal activity and GM1 gangliosides in CSF, with durability up to 12 months after treatment. (globalgenes.org)
- The effects on CSF β-Gal activity and GM1 gangliosides were dose-dependent, with Dose 1 of PBGM01 exhibiting modest effects. (globalgenes.org)
- GM1 gangliosidosis and GM2 gangliosidosis (Tay-Sachs and Sandhoff diseases) are lysosomal storage disorders caused by the accumulation of GM1 or GM2 gangliosides, respectively, in the central nervous system (CNS), resulting in progressive and severe neurological impairment and early death. (dutchnews.nl)
- This leads to a build up of GM1 gangliosides which causes the destruction of certain cells in many organs, particularly those in the brain. (jewishgenetics.org)
- Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. (wikigenes.org)
- The gangliosidoses are progressive, fatal neurological diseases of cats where gangliosides. (labogen.com)
- The gangliosidoses are progressive, fatal neurological diseases of cats, humans and other animals where gangliosides accumulate principally in neuronal lysosomes. (labogen.com)
Sandhoff5
- GM1 gangliosidosis is similar to Tay-Sachs disease and Sandhoff disease, but the disorders are caused by different gene mutations and enzyme deficiencies. (unitedbrainassociation.org)
- In 2022, the compound received Fast Track Designation for GM1 and GM2 gangliosidoses as well as NP-C and Orphan Drug Designations (ODD) for GM2 gangliosidosis (Sandhoff and Tay-Sachs Diseases) and NP-C from the FDA. (dutchnews.nl)
- In 2018, Axovant Gene Therapies licensed exclusive worldwide rights from UMass Medical School for the development and commercialization of gene therapy programs for GM1 gangliosidosis and GM2 gangliosidosis, including Tay-Sachs and Sandhoff diseases. (umassmed.edu)
- Research into potential therapies for lysosomal storage diseases such as Tay-Sachs, Sandhoff disease and GM1 gangliosidosis at UMass Medical School and Auburn University has led to significant advances in the field. (umassmed.edu)
- National Tay-Sachs & Allied Diseases Association (NTSAD) , a nonprofit patient advocacy group, is leading the worldwide fight to treat and cure Tay-Sachs, Canavan, GM1 gangliosidosis, and Sandhoff diseases by driving and funding research, forging collaboration, fostering community, and supporting families. (passagebio.com)
Ganglioside12
- β-galactosidase helps break down several molecules, including a substance called GM1 ganglioside. (medlineplus.gov)
- GM1 ganglioside is important for normal functioning of neurons in the brain. (medlineplus.gov)
- Without enough functional β-galactosidase, GM1 ganglioside cannot be broken down when it is no longer needed. (medlineplus.gov)
- Damage caused by the buildup of GM1 ganglioside leads to the destruction of neurons, causing many of the signs and symptoms of GM1 gangliosidosis. (medlineplus.gov)
- These individuals are still able to break down some amount of GM1 ganglioside, and less of the enzyme builds up inside lysosomes. (medlineplus.gov)
- G M1 gangliosidosis is an autosomal recessive lysosomal storage disorder characterized by the generalized accumulation of G M1 ganglioside, oligosaccharides, and the mucopolysaccharide keratan sulfate (and their derivatives). (medscape.com)
- It occurs when a molecule called GM1 ganglioside accumulates in the brain and nerve cells, causing damage to the cells and eventually causing the cells to die. (unitedbrainassociation.org)
- When there is not enough beta-galactosidase in the cells, molecules called GM1 ganglioside accumulate and impair the nerve cells' function. (unitedbrainassociation.org)
- GM1 ganglioside is essential for normal cell function, but excess levels of the molecule have a toxic effect. (unitedbrainassociation.org)
- Dose 2 of PBGM01 also decreased CSF GM1 ganglioside levels and demonstrated the ability to achieve normal adult levels at one-year post-dose. (globalgenes.org)
- GM1 ganglioside levels continued to decline over time in all patients treated with Dose 2. (globalgenes.org)
- N B-DGJ significantly reduced total ganglioside and GM1 content in cerebrum-brainstem (C-BS) and in cerebellum of normal and beta-gal-/- mice. (wikigenes.org)
Accumulation2
- GM1-gangliosidosis is a glycosphingolipid lysosomal storage disease involving accumulation of GM1 and its asialo form (GA1) primarily in the brain. (nih.gov)
- GM1 accumulation also results in progressive damage to other tissues including the heart, liver, and bones and manifests with hypotonia (reduced muscle tone), progressive CNS dysfunction, seizures, and rapid developmental regression. (globalgenes.org)
Deficiency4
- Deficiency of the lysosomal hydrolase, acid β -galactosidase, causes G M1 gangliosidosis and Morquio disease type B (ie, mucopolysaccharidosis type IVB ). (medscape.com)
- Suzuki Y, Oshima A, Nanba E. B-Galactosidase deficiency (B-Galactosidosis): GM1 gangliosidosis and Morquio B disease. (medscape.com)
- GM1 gangliosidosis is caused by a deficiency of an enzyme called beta-galactosidase. (unitedbrainassociation.org)
- GM1 gangliosidosis is due to an inherited deficiency of the enzyme beta-galactosidase, whereas GM2 gangliosidosis is caused by a lack of the enzyme beta-hexosaminidase. (labogen.com)
Onset5
- GM1 has three forms classified by age of onset. (wikipedia.org)
- Onset of late infantile GM1 is typically between ages 1 and 3 years. (wikipedia.org)
- Onset of adult GM1 is typically in adolescence or adulthood and is the slowest progressing of the subtypes. (wikipedia.org)
- GM1 gangliosidosis is usually classified as one of three different types depending on the age of onset. (unitedbrainassociation.org)
- Type IIa (Late Onset) Infantile GM1 Gangliosidosis (GM1). (cgtlive.com)
Beta-galactosidase2
- GM1 storage in cerebral gray matter is 10-fold elevated (20-50-fold increased in viscera) Galactose-containing oligosacchariduria and moderate keratan sulfaturia Morquio disease Type B: Mutations with higher residual beta-galactosidase activity for the GM1 substrate than for keratan sulfate and other galactose-containing oligosaccharides have minimal neurologic involvement but severe dysostosis resembling Morquio disease type A (Mucopolysaccharidosis type 4). (wikipedia.org)
- Beta-galactosidase enzyme is deficient in the following conditions: GM1 gangliosidosis, Morquio syndrome B, and galactosialidosis. (mayocliniclabs.com)
Late infantile2
- GM1 gangliosidosis type II occurs in one of two forms: the late infantile or the juvenile forms. (medlineplus.gov)
- Imagine-1 is a phase 1/2, global, open-label, dose-escalation study of the AAVhu68 gene therapy PBGM01 delivered by intra-cisterna magna (ICM) injection in six cohorts of pediatric subjects with early and late infantile GM1 Gangliosidosis (GM1). (globalgenes.org)
Lysosomes1
- Conditions like GM1 gangliosidosis that cause molecules to build up inside lysosomes are called lysosomal storage disorders. (medlineplus.gov)
PBGM014
- New data from the ongoing Imagine-1 clinical trial (NCT04713475) demonstrate good safety and impressive improvements in clinically meaningful end points in patients with GM1 gangliosidosis treated with PBGM01, an investigational gene therapy from Passage Bio. (cgtlive.com)
- We are highly encouraged by the compelling data emerging from our Imagine-1 study, with results underscoring the potential of PBGM01 to be a transformative therapy for GM1 patients," said William Chou, president and CEO of Passage Bio. (globalgenes.org)
- Infantile GM1 patients receiving PBGM01 showed a survival benefit, with one hundred percent survival beyond 20 months of age. (globalgenes.org)
- In three of four children, Dose 2 of PBGM01 resulted in a 4.7-16.1x increase in CSF β-Gal activity at 30 days post-treatment relative to baseline, exceeding average levels seen in healthy adults and GM1 Natural History Study. (globalgenes.org)
Mutation1
- Recently, two different families of feline GM1 gangliosidosis caused by each different novel mutation were found in Japanese domestic shorthair cats. (vin.com)
Autosomal recessive1
- All 3 types of G M1 gangliosidosis are inherited as autosomal recessive traits and have equal sex distributions. (medscape.com)
Neurological2
- AZ-3102 is an orally available azasugar with a unique dual mode of action, developed as a potential treatment for rare lysosomal storage disorders with neurological involvement, including GM1 and GM2 gangliosidoses and Niemann-Pick disease type C (NP-C). (dutchnews.nl)
- The study aims to characterize prospectively longitudinal progression of neurological domains in GM1 and GM2 Gangliosidosis patients with high-quality standards (GCP compliant). (ucsf.edu)
Lysosomal storage d2
- GM1 gangliosidosis is one of a group of diseases called lysosomal storage disorders. (unitedbrainassociation.org)
- There are 2 major forms, GM1 and GM2, both of which may be involved in lysosomal storage disorders. (msdmanuals.com)
Disorders1
- Azafaros is developing the compound as a potentially disease-modifying treatment in severe metabolic disorders including GM1 and GM2 gangliosidoses and NP-C. Enrollment of the first patient in the study is an important milestone for Azafaros in its mission to bring new treatment options to these patients and their families. (dutchnews.nl)
Enzyme1
- Enzyme activity is markedly reduced in patients with G M1 gangliosidosis. (medscape.com)
Mutations2
- In this study, we successfully identified the two novel pathogenic mutations in feline GM1 gangliosidosis. (vin.com)
- There are 2 main types of GM2 gangliosidosis, each of which can be caused by numerous different mutations. (msdmanuals.com)
Morquio1
- Careful review of clinical findings will help distinguish between GM1 gangliosidosis and Morquio syndrome type B. (mayocliniclabs.com)
Nerve cells2
- GM1 gangliosidosis is an inherited disorder that destroys nerve cells (neurons) in the brain and spinal cord. (medlineplus.gov)
- For instance, in November 2021, Sio Gene Therapies reported positive interim data for gene therapy trial of Phase I/II of AXO-AAV-GM1 for the treatment of GM1 gangliosidosis, a genetic disorder that progressively destroys nerve cells in the brain and spinal cord. (medgadget.com)
Severe1
- The signs and symptoms of the most severe form of GM1 gangliosidosis, called type I or the infantile form, usually develop by the age of 6 months. (medlineplus.gov)
Gene therapy2
- There is no cure for GM1, although several gene therapy trials are underway. (wikipedia.org)
- Gene therapy for GM1 gangliosidosis: challenges of translational medicine. (ox.ac.uk)
Developmental regression1
- Individuals with GM1 gangliosidosis type II experience developmental regression but usually do not have cherry-red spots, coarse facial features, or enlarged organs. (medlineplus.gov)
Fatal1
- Although both types of gangliosidoses cause fatal progressive brain disease, they are caused by entirely different genetic errors of two different lysosomal enzymes. (labogen.com)
Galactosidase1
- In general, people with GM1 gangliosidosis have milder signs and symptoms if they have higher levels of functional β-galactosidase activity. (medlineplus.gov)
Infantile form1
- The infantile form of G M1 gangliosidosis (type 1) typically presents between birth and age 6 months with progressive organomegaly, dysostosis multiplex, facial coarsening, and rapid neurologic deterioration within the first year of life. (medscape.com)
Adult2
- GM1 gangliosidosis type III is the adult or chronic form of the condition, and this is the mildest form. (medlineplus.gov)
- A high frequency of G M1 gangliosidosis has been reported from Southern Brazil, and a large number of Japanese patients with the adult form have been reported. (medscape.com)
Type5
- The age at which symptoms first appear varies in people with GM1 gangliosidosis type III, although most affected individuals develop signs and symptoms in their teens. (medlineplus.gov)
- Life expectancy varies among people with GM1 gangliosidosis type III. (medlineplus.gov)
- Inheritance of GM1 gangliosidosis appears to be type-specific. (unitedbrainassociation.org)
- The clinical trial is being conducted in Brazil and the US and will evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics across two doses of its lead asset, AZ-3102, in patients with GM2 gangliosidosis and Niemann-Pick disease type C (NP-C). (dutchnews.nl)
- Abnormal peaks with slower migration times than the tetrasaccharide position were observed for fucosidosis, α-mannosidosis, GM1 gangliosidosis, GM2 gangliosidosis variant 0, Pompe disease, and glycogen storage disease type 3. (nih.gov)
Symptoms2
- However, the signs and symptoms of these three types can overlap, leading some researchers to believe that GM1 gangliosidosis occurs on a spectrum instead of as three distinct types. (medlineplus.gov)
- The early signs of GM1 gangliosidosis disease vary from case to case, and different forms of the disease have various initial symptoms. (unitedbrainassociation.org)
Genetic1
- Diagnosis of GM1 can be obtained by genetic and enzymatic testing. (wikipedia.org)
Disease3
- GM1 gangliosidosis is a progressive, degenerative brain and central nervous system disease. (unitedbrainassociation.org)
- We remain focused on execution across our ongoing clinical programs and are excited to have dosed additional patients in our Imagine-1 trial for GM1 gangliosidosis as well as the first patient in our GALax-C trial for infantile Krabbe disease," said Bruce Goldsmith, Ph.D., president and chief executive officer of Passage Bio. (biospace.com)
- This trial studies how the disease progresses in patients with GM1 and GM2 forms of Tay-Sachs Disease. (ucsf.edu)
Clinical1
- Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. (medscape.com)
Mice2
- The levels of GM1 and GA1 were significantly increased in both the optic nerve and sciatic nerve of the β-gal -/- mice. (nih.gov)
- The abnormalities in GM1 and myelin lipids in optic nerve of β-gal -/- mice correlated with a reduction in the relative amount of myelin and periodicity in fresh nerve. (nih.gov)
100,0003
- GM1 gangliosidosis is estimated to occur in 1 in 100,000 to 200,000 newborns. (medlineplus.gov)
- GM1 is a rare lysosomal storage disorder with a prevalence of 1 to every 100,000 to 200,000 live births worldwide, although rates are higher in some regions. (wikipedia.org)
- Life expectancy for infants with GM1 ranges from 2-10 years, and infantile GM1 represents approximately 60 percent of the global GM1 incidence of 0.5 to 1 in 100,000 live births. (globalgenes.org)
Incidence1
- G M1 gangliosidosis is a rare disorder, and data concerning incidence are not widely available. (medscape.com)
Early1
- This form of G M1 gangliosidosis most frequently presents in early infancy and may be evident at birth. (medscape.com)
Rare1
- GM1 gangliosidosis is a rare condition that causes the progressive degeneration of elements of the central nervous system. (unitedbrainassociation.org)
Treatment2
- citation needed] Treatment for GM1 is symptom-based and palliative. (wikipedia.org)
- Global GM1 Gangliosidosis Treatment market is poised to value over USD 447.8 million by 2028 end at a CAGR of over 36% during the forecast period 2021 to 2028. (articlewood.com)
Types1
- There are two types of gangliosidoses, GM1 and GM2 gangliosidosis. (labogen.com)
Research2
- Dive into the research topics of 'Hyperphosphatasemia in GM1 gangliosidosis. (vub.be)
- Cure GM1 Foundation, a nonprofit patient advocacy group, provides families caring for a child with GM1 gangliosidosis with support and resources, opportunities to meet others families who understand the challenges of GM1 gangliosidosis, and direct funding toward research for a cure. (passagebio.com)