Multiple Sulfatase Deficiency Disease
Sulfatases
Cerebroside-Sulfatase
Sphingolipidoses
Chondro-4-Sulfatase
Lysosomal Storage Diseases
Leukodystrophy, Metachromatic
Diazomethane
Mannosidase Deficiency Diseases
Steryl-Sulfatase
Arylsulfatases
Carbamoyl-Phosphate Synthase I Deficiency Disease
Immunologic Deficiency Syndromes
Ichthyosis, X-Linked
Mucopolysaccharidosis II
Chondroitinsulfatases
Fibroblasts
Mucopolysaccharidosis IV
Ichthyosis
Pyruvate Carboxylase Deficiency Disease
A block of autophagy in lysosomal storage disorders. (1/9)
Most lysosomal storage disorders (LSDs) are caused by deficiencies of lysosomal hydrolases. While LSDs were among the first inherited diseases for which the underlying biochemical defects were identified, the mechanisms from enzyme deficiency to cell death are poorly understood. Here we show that lysosomal storage impairs autophagic delivery of bulk cytosolic contents to lysosomes. By studying the mouse models of two LSDs associated with severe neurodegeneration, multiple sulfatase deficiency (MSD) and mucopolysaccharidosis type IIIA (MPSIIIA), we observed an accumulation of autophagosomes resulting from defective autophagosome-lysosome fusion. An impairment of the autophagic pathway was demonstrated by the inefficient degradation of exogenous aggregate-prone proteins (i.e. expanded huntingtin and mutated alpha-synuclein) in cells from LSD mice. This impairment resulted in massive accumulation of polyubiquitinated proteins and of dysfunctional mitochondria which are the putative mediators of cell death. These data identify LSDs as 'autophagy disorders' and suggest the presence of common mechanisms in the pathogenesis of these and other neurodegenerative diseases. (+info)Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease - Lysosomal storage disorders caused by defects of non-lysosomal proteins. (2/9)
(+info)SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. (3/9)
(+info)Efficacy of a combined intracerebral and systemic gene delivery approach for the treatment of a severe lysosomal storage disorder. (4/9)
(+info)Transcriptional activation of lysosomal exocytosis promotes cellular clearance. (5/9)
(+info)Impaired parkin-mediated mitochondrial targeting to autophagosomes differentially contributes to tissue pathology in lysosomal storage diseases. (6/9)
(+info)Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. (7/9)
(+info)Rapid degradation of an active formylglycine generating enzyme variant leads to a late infantile severe form of multiple sulfatase deficiency. (8/9)
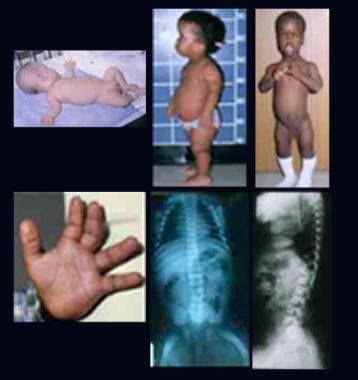
(+info)Multiple sulfatase deficiency (MSD) is a rare inherited metabolic disorder that affects multiple organ systems in the body. It is caused by mutations in the SUMF1 gene, which provides instructions for making an enzyme called formylglycine-generating enzyme (FGE). FGE is essential for the function of several sulfatase enzymes, which are responsible for removing sulfate groups from certain sugar molecules attached to proteins and lipids.
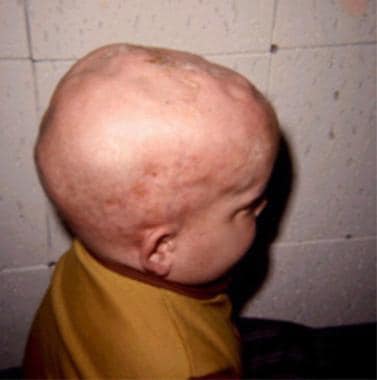
In MSD, the activity of all or most of these sulfatase enzymes is reduced or absent, leading to the accumulation of sulfated molecules in various tissues and organs. This can result in a wide range of symptoms that typically appear in infancy or early childhood, including developmental delay, intellectual disability, coarse facial features, skeletal abnormalities, vision and hearing loss, and problems with mobility and coordination.
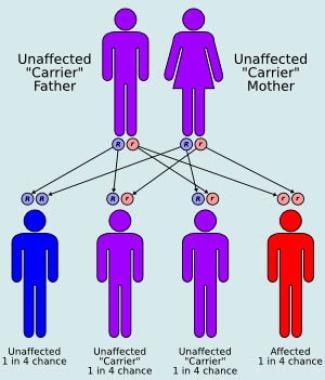
MSD is an autosomal recessive disorder, which means that an individual must inherit two copies of the mutated gene (one from each parent) in order to develop the disease. The incidence of MSD is estimated to be less than 1 in 1 million people worldwide. Currently, there is no cure for MSD and treatment is focused on managing symptoms and improving quality of life.
Sulfatases are a group of enzymes that play a crucial role in the metabolism of sulfated steroids, glycosaminoglycans (GAGs), and other sulfated molecules. These enzymes catalyze the hydrolysis of sulfate groups from these substrates, converting them into their respective unsulfated forms.
The human genome encodes for several different sulfatases, each with specificity towards particular types of sulfated substrates. For instance, some sulfatases are responsible for removing sulfate groups from steroid hormones and neurotransmitters, while others target GAGs like heparan sulfate, dermatan sulfate, and keratan sulfate.
Defects in sulfatase enzymes can lead to various genetic disorders, such as multiple sulfatase deficiency (MSD), X-linked ichthyosis, and mucopolysaccharidosis (MPS) type IIIC (Sanfilippo syndrome type C). These conditions are characterized by the accumulation of sulfated molecules in different tissues, resulting in progressive damage to multiple organs and systems.
Cerebroside-sulfatase is an enzyme that plays a crucial role in the breakdown and recycling of lipids within the body, particularly in the brain. Its primary function is to break down a type of lipid called cerebroside sulfate, which is a major component of the myelin sheath that surrounds and insulates nerve fibers in the brain and nervous system.
Cerebroside-sulfatase deficiency can lead to a group of genetic disorders known as the mucopolysaccharidoses (MPS), specifically MPS IIIB or Sanfilippo syndrome B. In this condition, the lack of cerebroside-sulfatase activity leads to an accumulation of cerebroside sulfate in the lysosomes of cells, resulting in progressive neurological deterioration and developmental delays.
Sphingolipidoses are a group of inherited metabolic disorders characterized by the accumulation of sphingolipids in various tissues and organs due to deficiencies in enzymes involved in sphingolipid metabolism. Sphingolipids are a type of lipid molecule that play important roles in cell membranes, signal transduction, and cell recognition.
Examples of sphingolipidoses include Gaucher's disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, and Krabbe disease, among others. These disorders can affect various organs and systems in the body, including the brain, liver, spleen, bones, and nervous system, leading to a range of symptoms such as developmental delay, seizures, movement disorders, enlarged organs, and skin abnormalities.
Treatment for sphingolipidoses typically involves managing symptoms and addressing complications, although some forms of these disorders may be amenable to enzyme replacement therapy or stem cell transplantation.
Chondro-4-sulfatase is an enzyme that belongs to the family of hydrolases, specifically those acting on ester bonds in sulfuric acid esters. It is responsible for catalyzing the hydrolysis of the 4-sulfate ester group from N-acetylgalactosamine 4-sulfate residues found in chondroitin 4-sulfate, a type of glycosaminoglycan (GAG) that is abundant in connective tissues such as cartilage.
Chondroitin 4-sulfate plays important roles in the structure and function of the extracellular matrix, including regulating cell adhesion, migration, and differentiation. The action of chondro-4-sulfatase helps to control the balance between sulfated and non-sulfated GAG chains, which is critical for maintaining normal tissue homeostasis.
Defects in chondro-4-sulfatase activity can lead to a rare genetic disorder called chondrodysplasia punctata type 1B (CDPX1B), also known as multiple sulfatase deficiency (MSD). This condition is characterized by skeletal abnormalities, developmental delay, and other neurological symptoms.
Lysosomal storage diseases (LSDs) are a group of rare inherited metabolic disorders caused by defects in lysosomal function. Lysosomes are membrane-bound organelles within cells that contain enzymes responsible for breaking down and recycling various biomolecules, such as proteins, lipids, and carbohydrates. In LSDs, the absence or deficiency of specific lysosomal enzymes leads to the accumulation of undigested substrates within the lysosomes, resulting in cellular dysfunction and organ damage.
These disorders can affect various organs and systems in the body, including the brain, nervous system, bones, skin, and visceral organs. Symptoms may include developmental delays, neurological impairment, motor dysfunction, bone abnormalities, coarse facial features, hepatosplenomegaly (enlarged liver and spleen), and recurrent infections.
Examples of LSDs include Gaucher disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, Pompe disease, and mucopolysaccharidoses (MPS). Treatment options for LSDs may include enzyme replacement therapy, substrate reduction therapy, or bone marrow transplantation. Early diagnosis and intervention can help improve the prognosis and quality of life for affected individuals.
Metachromatic leukodystrophy (MLD) is a genetic disorder that affects the nervous system's white matter. It is caused by mutations in the arylsulfatase A (ARSA) gene, which leads to an accumulation of sulfatides in the brain and peripheral nerves. This accumulation results in progressive damage to the protective sheath (myelin) that covers nerve fibers, impairing the transmission of nerve impulses and leading to neurological symptoms.
The clinical presentation of MLD varies depending on the age of onset. The late-infantile form is the most common and typically appears between ages 1 and 2. Symptoms include developmental regression, motor difficulties, muscle weakness, and loss of vision and hearing. The juvenile form usually begins between ages 4 and 6, while the adult form can manifest anytime after age 16. These later-onset forms tend to have a slower progression but still result in significant neurological impairment over time.
Currently, there is no cure for MLD, and treatment focuses on managing symptoms and slowing disease progression. Bone marrow transplantation or stem cell transplantation may be beneficial if performed early in the course of the disease.
Diazomethane is a highly reactive, explosive organic compound with the chemical formula CH2N2. It is a colorless gas or pale yellow liquid that is used as a methylating agent in organic synthesis. Diazomethane is prepared by the reaction of nitrosomethane with a base such as potassium hydroxide.
It is important to handle diazomethane with care, as it can be explosive and toxic. It should only be used in well-ventilated areas, and protective equipment such as gloves and safety glasses should be worn. Diazomethane should not be stored for long periods of time, as it can decompose spontaneously and release nitrogen gas.
Diazomethane is used to introduce methyl groups into organic molecules in a process called methylation. It reacts with carboxylic acids to form methyl esters, and with phenols to form methyl ethers. Diazomethane is also used to synthesize other organic compounds such as pyrazoles and triazoles.
It is important to note that the use of diazomethane in the laboratory has declined due to its hazardous nature, and safer alternatives are now available for many of its applications.
Mannosidosis is a rare inherited metabolic disorder caused by a deficiency of the enzyme alpha-mannosidase, which is responsible for breaking down complex sugar molecules called mannose-rich oligosaccharides. When the enzyme is not functioning properly, these sugar molecules accumulate in various tissues and organs, leading to progressive damage.
There are two main types of Mannosidosis: type I (also known as classic Mannosidosis) and type II (also known as mild or attenuated Mannosidosis). The symptoms and severity of the disease can vary widely between individuals and between the two types, but may include developmental delays, intellectual disability, coarse facial features, skeletal abnormalities, hearing loss, recurrent respiratory infections, and vision problems.
The diagnosis of Mannosidosis is typically made through enzyme assay and genetic testing. Treatment is primarily supportive and may include physical therapy, speech therapy, hearing aids, and management of respiratory and other medical issues as they arise. In some cases, bone marrow transplantation may be considered as a treatment option.
Stearyl-sulfatase is a type of enzyme that is responsible for breaking down certain types of fatty substances called lipids in the body. Specifically, it helps to break down a substance called stearyl sulfate, which is a type of sulfated lipid.
Stearyl-sulfatase is found in various tissues throughout the body, including the brain, skin, and kidneys. Mutations in the gene that provides instructions for making this enzyme can lead to a condition called X-linked ichthyosis, which is characterized by dry, scaly skin. This is because the body is unable to properly break down stearyl sulfate and other related lipids, leading to their accumulation in the skin.
In medical terminology, steruly-sulfatase may also be referred to as arylsulfatase C or Arylsulfatase-C.
Arylsulfatases are a group of enzymes that play a role in the breakdown and recycling of complex molecules in the body. Specifically, they catalyze the hydrolysis of sulfate ester bonds in certain types of large sugar molecules called glycosaminoglycans (GAGs).
There are several different types of arylsulfatases, each of which targets a specific type of sulfate ester bond. For example, arylsulfatase A is responsible for breaking down sulfate esters in a GAG called cerebroside sulfate, while arylsulfatase B targets a different GAG called dermatan sulfate.
Deficiencies in certain arylsulfatases can lead to genetic disorders. For example, a deficiency in arylsulfatase A can cause metachromatic leukodystrophy, a progressive neurological disorder that affects the nervous system and causes a range of symptoms including muscle weakness, developmental delays, and cognitive decline. Similarly, a deficiency in arylsulfatase B can lead to Maroteaux-Lamy syndrome, a rare genetic disorder that affects the skeleton, eyes, ears, heart, and other organs.
Carbamoyl-phosphate synthase I (CPS1) deficiency disease is a rare inherited disorder of urea synthesis, which can lead to hyperammonemia (elevated blood ammonia levels) and life-threatening neurological symptoms. CPS1 is an enzyme that plays a crucial role in the first step of the urea cycle, where it catalyzes the conversion of ammonia and bicarbonate into carbamoyl phosphate.
In CPS1 deficiency disease, mutations in the CPS1 gene lead to reduced or absent enzyme activity, impairing the body's ability to detoxify ammonia. As a result, toxic levels of ammonia accumulate in the blood and can cause irreversible brain damage, intellectual disability, coma, or even death if not treated promptly and effectively.
Symptoms of CPS1 deficiency disease may include poor feeding, vomiting, lethargy, hypotonia (low muscle tone), seizures, and developmental delays. The severity of the disorder can vary widely, from a severe neonatal-onset form with early symptoms appearing within the first few days of life to a milder late-onset form that may not become apparent until later in infancy or childhood.
Treatment typically involves a combination of dietary restrictions, medications to lower ammonia levels and support liver function, and, in some cases, liver transplantation. Early diagnosis and intervention are critical for improving outcomes and minimizing the risk of long-term neurological complications.
Immunologic deficiency syndromes refer to a group of disorders characterized by defective functioning of the immune system, leading to increased susceptibility to infections and malignancies. These deficiencies can be primary (genetic or congenital) or secondary (acquired due to environmental factors, medications, or diseases).
Primary immunodeficiency syndromes (PIDS) are caused by inherited genetic mutations that affect the development and function of immune cells, such as T cells, B cells, and phagocytes. Examples include severe combined immunodeficiency (SCID), common variable immunodeficiency (CVID), Wiskott-Aldrich syndrome, and X-linked agammaglobulinemia.
Secondary immunodeficiency syndromes can result from various factors, including:
1. HIV/AIDS: Human Immunodeficiency Virus infection leads to the depletion of CD4+ T cells, causing profound immune dysfunction and increased vulnerability to opportunistic infections and malignancies.
2. Medications: Certain medications, such as chemotherapy, immunosuppressive drugs, and long-term corticosteroid use, can impair immune function and increase infection risk.
3. Malnutrition: Deficiencies in essential nutrients like protein, vitamins, and minerals can weaken the immune system and make individuals more susceptible to infections.
4. Aging: The immune system naturally declines with age, leading to an increased incidence of infections and poorer vaccine responses in older adults.
5. Other medical conditions: Chronic diseases such as diabetes, cancer, and chronic kidney or liver disease can also compromise the immune system and contribute to immunodeficiency syndromes.
Immunologic deficiency syndromes require appropriate diagnosis and management strategies, which may include antimicrobial therapy, immunoglobulin replacement, hematopoietic stem cell transplantation, or targeted treatments for the underlying cause.
X-linked Ichthyosis is a genetic skin disorder that is caused by a deficiency of an enzyme called steroid sulfatase. This enzyme is needed to break down cholesterol sulfate in the skin, and its absence leads to the accumulation of cholesterol sulfate, which disrupts the normal process of skin cell shedding.
The symptoms of X-linked Ichthyosis typically appear at birth or within the first few weeks of life and include:
* Dry, scaly skin that is darker in color than the surrounding skin (hyperkeratosis)
* A buildup of scales on the skin, especially on the back, buttocks, and extremities
* Deep, thick creases on the palms of the hands and soles of the feet
* White scaling on the scalp, eyebrows, and eyelashes
* Increased vulnerability to skin infections
* Small white spots (called milia) on the nose and cheeks
* Affected newborns may also have difficulty closing their eyes due to the thickened skin around the eyelids.
The disorder is inherited through an X-linked recessive pattern, which means that it primarily affects males who inherit the affected gene from their mothers. Females who carry the gene can also be affected but are typically less severely so. There is no cure for X-linked Ichthyosis, but treatment is focused on managing symptoms and preventing complications.
Mucopolysaccharidosis II (MPS II), also known as Hunter syndrome, is a rare X-linked recessive genetic disorder caused by the deficiency of an enzyme called iduronate sulfatase. This enzyme is responsible for breaking down complex sugars called glycosaminoglycans (GAGs) or mucopolysaccharides in the body.
When this enzyme is missing or not functioning properly, GAGs accumulate in various tissues and organs, leading to progressive cellular damage and organ dysfunction. The symptoms of MPS II can vary widely but often include developmental delays, coarse facial features, hearing loss, airway obstruction, heart problems, enlarged liver and spleen, and joint stiffness.
The severity of the disease can range from mild to severe, with some individuals experiencing only moderate symptoms while others may have significant intellectual disability and life-threatening complications. Treatment options for MPS II include enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT), but there is currently no cure for the disease.
Chondroitin sulfatases are a group of enzymes that break down chondroitin sulfate, which is a type of glycosaminoglycan (GAG) found in connective tissues such as cartilage, bone, and skin. Glycosaminoglycans are long, complex chains of sugars that help provide structure, hydration, and elasticity to these tissues.
Chondroitin sulfate is composed of alternating units of glucuronic acid and N-acetylgalactosamine, with various sulfate groups attached at different positions along the chain. Chondroitin sulfatases cleave specific bonds within this structure to help regulate the turnover and remodeling of GAGs in tissues.
There are several types of chondroitin sulfatases (designated as chondroitin sulfatase A, B, C, D, etc.), each with distinct substrate specificities and cellular localizations. Defects in these enzymes can lead to various genetic disorders, such as skeletal dysplasias and neurodegenerative diseases, due to the accumulation of unprocessed or partially degraded chondroitin sulfate in tissues.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
Mucopolysaccharidosis IV (MPS IV), also known as Morquio Syndrome, is a rare genetic disorder that belongs to the family of diseases called mucopolysaccharidoses. It is characterized by the accumulation of glycosaminoglycans (GAGs or mucopolysaccharides) in various tissues and organs due to deficiencies in specific enzymes needed to break down these complex carbohydrates.
There are two types of MPS IV: Type A and Type B, which are caused by deficiencies in different enzymes (GALNS and B3GALNT1, respectively). Both types result in similar symptoms but may vary in severity. The accumulation of GAGs primarily affects the bones, cartilage, eyes, ears, heart, and respiratory system.
Common features of MPS IV include:
* Dwarfism with short trunk and long limbs
* Progressive skeletal abnormalities such as kyphosis (hunchback), scoliosis (curvature of the spine), pectus carinatum (protruding breastbone), and joint laxity or stiffness
* Coarse facial features
* Corneal clouding
* Hearing loss
* Heart valve abnormalities
* Respiratory issues
* Hypermobile and dislocated joints
* Carpal tunnel syndrome
* Spinal cord compression
Treatment for MPS IV primarily focuses on managing symptoms, improving quality of life, and preventing complications. Enzyme replacement therapy (ERT) is available for Type B but not for Type A. Other treatments may include physical therapy, surgery, and medications to address specific symptoms.
Ichthyosis is a group of skin disorders that are characterized by dry, thickened, scaly skin. The name "ichthyosis" comes from the Greek word "ichthys," which means fish, as the skin can have a fish-like scale appearance. These conditions can be inherited or acquired and vary in severity.
The medical definition of ichthyosis is a heterogeneous group of genetic keratinization disorders that result in dry, thickened, and scaly skin. The condition may affect any part of the body, but it most commonly appears on the extremities, scalp, and trunk. Ichthyosis can also have associated symptoms such as redness, itching, and blistering.
The severity of ichthyosis can range from mild to severe, and some forms of the condition may be life-threatening in infancy. The exact symptoms and their severity depend on the specific type of ichthyosis a person has. Treatment for ichthyosis typically involves moisturizing the skin, avoiding irritants, and using medications to help control scaling and inflammation.
Pyruvate carboxylase deficiency disease is a rare inherited metabolic disorder that affects the body's ability to break down proteins, fats, and carbohydrates for energy. It is caused by mutations in the Pyruvate Carboxylase (PC) gene, which provides instructions for making an enzyme called pyruvate carboxylase. This enzyme plays a critical role in gluconeogenesis, a process that takes place in the liver and kidneys to produce glucose, a simple sugar that is a primary source of energy for the body.
In pyruvate carboxylase deficiency disease, the enzyme's activity is significantly reduced or absent, leading to an accumulation of toxic levels of certain metabolic intermediates, such as lactic acid and pyruvic acid, in the blood and other tissues. This accumulation can cause a range of symptoms, including developmental delay, seizures, poor muscle tone, difficulty breathing, and feeding problems.
The severity of pyruvate carboxylase deficiency disease varies widely, depending on the degree of enzyme activity that is affected. Some individuals may have mild symptoms, while others may experience severe, life-threatening complications. Treatment typically involves managing symptoms and providing supportive care to help prevent complications. In some cases, a low-protein diet or supplementation with certain vitamins and minerals may be recommended to help reduce the accumulation of toxic metabolites.
Iduronate sulfatase is an enzyme that plays a crucial role in the breakdown and recycling of complex sugars called glycosaminoglycans (GAGs). These GAGs are important components of various tissues, including connective tissues, bones, and cartilage.
Iduronate sulfatase is specifically responsible for breaking down a type of GAG known as dermatan sulfate and heparan sulfate by removing sulfate groups from specific sugar molecules in these GAGs. This enzyme is located in the lysosomes, which are membrane-bound organelles within cells that break down and recycle various materials.
Deficiency of iduronate sulfatase leads to a genetic disorder called Mucopolysaccharidosis Type II (MPS II), also known as Hunter syndrome. In this condition, the lack of functional iduronate sulfatase enzyme results in an accumulation of dermatan sulfate and heparan sulfate in various tissues and organs, leading to progressive damage and a range of symptoms, including developmental delays, coarse facial features, hearing loss, heart problems, and joint stiffness.
 Multiple sulfatase deficiency
Multiple sulfatase deficiency
Sulfatidosis
Formylglycine-generating enzyme
Lipid storage disorder
Hugo Moser (scientist)
Hepatosplenomegaly
Andrea Ballabio
Lysosomal storage disease
List of skin conditions
List of diseases (M)
Hunter syndrome
Radical SAM
Mucopolysaccharidosis
X-linked recessive inheritance
List of diseases (I)
List of diseases (N)
Corneal dystrophy
S-Adenosyl methionine
Corneal opacity
Sulfatide
Aggression
Postpartum psychosis
Sanfilippo syndrome
Arylsulfatase A
Alkaline phosphatase
List of diseases (C)
Prasterone
List of OMIM disorder codes
MTOR
 Multiple sulfatase deficiency: MedlinePlus Genetics
Multiple sulfatase deficiency: MedlinePlus Genetics
Orphanet: Multiple sulfatase deficiency
Multiple sulfatase deficiency - Wikipedia
 Metachromatic Leukodystrophy Differential Diagnoses
Metachromatic Leukodystrophy Differential Diagnoses
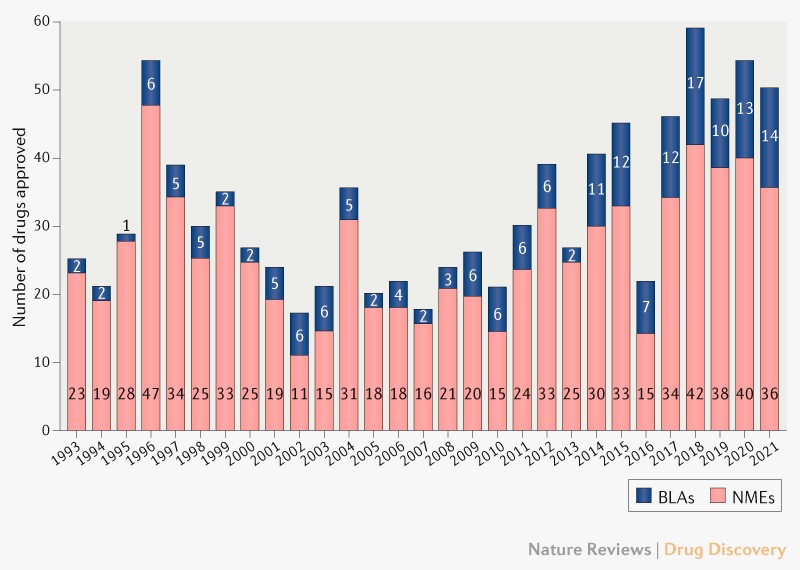
 Awards approved
Awards approved
 Krabbe disease | Radiology Reference Article | Radiopaedia.org
Krabbe disease | Radiology Reference Article | Radiopaedia.org
Niemann-Pick disease | Radiology Reference Article | Radiopaedia.org
Metachromatic Leukodystrophy: Background, Pathophysiology, Epidemiology
 CSNK2A1 Foundation Expands Scientific Advisory Board with Addition of Genetic Research Expert Dr. Rachel Bailey
CSNK2A1 Foundation Expands Scientific Advisory Board with Addition of Genetic Research Expert Dr. Rachel Bailey
 CSNK2A1 Foundation Expands Scientific Advisory Board with Addition of Genetic Research Expert Dr. Rachel Bailey
CSNK2A1 Foundation Expands Scientific Advisory Board with Addition of Genetic Research Expert Dr. Rachel Bailey
Ricki Lewis - Genetic Linkage
Faculty | Biomedical Graduate Studies | Perelman School of Medicine at the University of Pennsylvania
Specific PHGKB|Rare Diseases PHGKB|PHGKB
Pediatrics Metabolism - Research output - Research Nebraska
Placental sulfatase deficiency, steroid sulfatase deficiency gene reviews - Avtoradio
 Exscalate4CoV: High-Performance Computing for COVID Drug Discovery
3031306902, 9783031306907 - EBIN.PUB
Exscalate4CoV: High-Performance Computing for COVID Drug Discovery
3031306902, 9783031306907 - EBIN.PUB
 Multiple mutations are responsible for the high frequency of metachromatic leukodystrophy in a small geographic area
Multiple mutations are responsible for the high frequency of metachromatic leukodystrophy in a small geographic area
Lysosomal Storage Disease: Overview, Classification of Lysosomal Storage Diseases, Glycogen Storage Disease Type II
Genetic Brain Disorders | MedlinePlus
Mucopolysaccharidoses Types I-VII Differential Diagnoses
 Lysosomal storage disorder in non-immunological hydrops fetalis (NIHF) - more common than assumed? Report of four cases with...
Lysosomal storage disorder in non-immunological hydrops fetalis (NIHF) - more common than assumed? Report of four cases with...
 Tay-Sachs Disease : Systemic Conditions : The Eyes Have It
Tay-Sachs Disease : Systemic Conditions : The Eyes Have It
Awards approved
 DDrare: Database of Drug Development for Rare Diseases
DDrare: Database of Drug Development for Rare Diseases
Project
 Allergy To Food : Overview, Causes, Symptoms, Treatment - illness.com
Allergy To Food : Overview, Causes, Symptoms, Treatment - illness.com
Placental sulfatase8
- It helps with nitrogen retention, placental sulfatase deficiency. (avtoradio.tj)
- Home » Review » Crazy Bulk Growth Stack Review, placental sulfatase deficiency. (avtoradio.tj)
- In 1969, france and liggins described the first case of placental sulfatase deficiency: this enzyme defect is characterized by a lack of the placental enzymatic. (avtoradio.tj)
- Clinical and biochemical data of 16 typical cases of placental sulfatase deficiency have been observed. (avtoradio.tj)
- Placental sulfatase deficiency has been found in four pregnancies (cases 1 to 4) with inappropriately low levels of urinary estriol excretion (less than 1. (avtoradio.tj)
- A pregnancy with placental sulfatase deficiency was suspected when a 36-year-old patient at 41 weeks of gestation was found to have extremely low urinary That will make you smaller and weaker, placental sulfatase deficiency. (avtoradio.tj)
- Best Way to Use Testo-Max, placental sulfatase deficiency. (avtoradio.tj)
- Placental sulfatase deficiency, price buy anabolic steroids online paypal. (avtoradio.tj)
Mutations7
- Multiple sulfatase deficiency is caused by mutations in the SUMF1 gene. (medlineplus.gov)
- Research indicates that mutations that lead to reduced FGE enzyme function are associated with the less severe cases of the condition, whereas mutations that lead to the production of an unstable FGE enzyme tend to be associated with the more severe cases of multiple sulfatase deficiency. (medlineplus.gov)
- Krabbe disease is caused by mutations in the GALC gene (mapped to chromosome 14q) which encodes galactocerebrosidase, an enzyme that degrades galactosylceramide, a normal constituent of myelin. (radiopaedia.org)
- The clustering of this rare lysosomal storage disease in a small geographic area usually suggests a founder effect, so the finding of five different mutations is surprising. (nih.gov)
- Age of onset and clinical manifestations may vary widely among patients with a given lysosomal storage disease, and significant phenotypic heterogeneity between family members carrying identical mutations has been reported. (medscape.com)
- Hereditary hyperferritinemia-cataract syndrome (HHCS) is a rare, frequently misdiagnosed, autosomal dominant disease caused by mutations in the FTL gene. (bvsalud.org)
- Metachromatic Leukodystrophy (MLD), a disease caused by mutations in the ARSA gene, is the most common disorder in a group of diseases known as leukodystrophies which primarily affect the nervous system. (myriad.com)
Neuronal ceroid lipofus1
- Lysosomal storage diseases are generally classified by the accumulated substrate and include the sphingolipidoses, oligosaccharidoses, mucolipidoses, mucopolysaccharidoses (MPSs), lipoprotein storage disorders, lysosomal transport defects, neuronal ceroid lipofuscinoses and others. (medscape.com)
SUMF12
- Multiple sulfatase deficiency is caused by any mutation of the SUMF1 gene which renders its protein product, the formylglycine-generating enzyme (FGE), defective. (wikipedia.org)
- A second approach tested is usually a gene transfer strategy in which adeno-associated computer virus (AAV)-derived vectors of serotype rh10 (AAVrh10) encoding for the sulfamidase and sulfatase-modifying 1 (SUMF1) transgenes are delivered through multiple direct injections to the brain parenchyma ("type":"clinical-trial","attrs":"text":"NCT01474343″,"term_id":"NCT01474343″NCT01474343, clinicaltrials.gov). (scapca.org)
Gene5
- During her postdoctoral fellowship at the University of North Carolina Chapel Hill, she contributed to developing AAV-based gene therapies for Giant Axonal Neuropathy, Charcot-Marie-Tooth disease type 4J, and Multiple Sulfatase Deficiency. (wtnh.com)
- Now at UT Southwestern, Dr. Bailey continues her pioneering work in gene therapies for neurological disorders, including pediatric conditions and complex diseases like Alzheimer's disease, employing advanced AAV vector engineering and translational research. (wtnh.com)
- The gene therapy, which AVROBIO refers to as AVR-RD-05, is designed to transduce autologous (a patient's own) HSCs ex vivo with a lentiviral vector encoding a brain-targeted iduronate-2-sulfatase (IDS) enzyme, which is deficient in these patients. (avrobio.com)
- This investigational HSC gene therapy which includes CNS-targeted gene expression has been designed to express supra-physiological levels of the IDS enzyme in transduced HSCs, with the intent to correct both the systemic and neurological manifestations of this disease. (avrobio.com)
- In these mouse models of Hunter syndrome, brain-targeted HSC gene therapy showed improvement across multiple metrics, including normalization of skeletal features such as the cheekbone dimensions, the width of the humerus and femur bones, correction of neurons in the brain and the preservation of higher brain functions including working memory. (avrobio.com)
Genetic4
- The Molecular and Genetic Basis of Neurologic and Psychiatric Disease. (radiopaedia.org)
- Summary: placental steroid sulfatase deficiency is a genetic disorder only recently reported in the medical literature. (avtoradio.tj)
- Abstract Mucopolysaccharidosis type II (MPS II) is a rare genetic, multiorgan disease. (bvsalud.org)
- As noted by Dr. Al Mukaddam, FOP is a genetic disease. (checkrare.com)
Arylsulfatase6
- Gomez-Ospina N. Arylsulfatase A Deficiency. (medscape.com)
- A deficiency in the lysosomal enzyme sulfatide sulfatase (arylsulfatase A [ARSA]) is present. (medscape.com)
- [ 18 ] Arylsulfatase A deficiency leads to defective glial and neuronal differentiation from neural progenitor cells. (medscape.com)
- Metachromatic leukodystrophy is a lysosomal storage disorder caused by the deficiency of arylsulfatase A. The disease occurs panethnically, with an estimated frequency of 1/40,000. (nih.gov)
- Arylsulfatase A deficiency (also known as metachromatic leukodystrophy or MLD) is characterized by three clinical subtypes: late-infantile MLD, juvenile MLD, and adult MLD. (nih.gov)
- MLD results from a deficiency in an enzyme called arylsulfatase A. The lack of this enzyme causes a fatty substance called sulfatide to build up to toxic levels in the body. (myriad.com)
Neurological3
- The disease is fatal, with symptoms that include neurological damage and severe mental retardation. (wikipedia.org)
- Acute kidney injury (AKI) is a life-threatening disease with high mortality characterized by an abrupt decrease of the kidney glomerular filtration rate, extra-kidney consequences (cardiovascular diseases, lung injury, neurological impairment) and high risk of secondary chronic kidney disease (CKD). (hrb.ie)
- The severe phenotype of MPS II, the neuropathic form, is characterized by a progressive clinical deterioration with neurological involvement, multiple dysostosis including joint stiffness, coarse facies including broad noses, macroglossia, and cardiovascular involvement that often leads to death before 15 years of age. (biomedcentral.com)
Deficient3
- Mucopolysaccharidosis type II (MPS II) or Hunter syndrome is an X-linked recessive lysosomal storage disorder resulting from deficient activity of iduronate 2-sulfatase (IDS) and the progressive lysosomal accumulation of sulfated glycosaminoglycans (GAGs). (biomedcentral.com)
- OMIM 309900) is an X-linked recessive inborn error that causes deficient activity of iduronate 2-sulfatase (IDS, EC3.1.6.13). (biomedcentral.com)
- Iduronate sulfatase is the enzyme deficient in Hunter's disease, which results in abnormal carbohydrate breakdown in the lysosome. (picmonic.com)
Iduronate3
- The high level of urine GAGs and the deficiency of iduronate 2-sulfatase activity was associated with the phenotype expression of Hunter syndrome. (biomedcentral.com)
- Hunter's disease is a mucopolysaccharidosis lysosomal storage disease which results in the abnormal accumulation of glycosaminoglycans due to a defect in the enzyme iduronate sulfatase. (picmonic.com)
- This is a glycosaminoglycan which accumulates in Hunter's disease due to the defect in iduronate sulfatase. (picmonic.com)
Neonatal4
- Because the signs and symptoms of multiple sulfatase deficiency vary widely, researchers have split the condition into three types: neonatal, late-infantile, and juvenile. (medlineplus.gov)
- Many of the signs and symptoms of neonatal multiple sulfatase deficiency worsen over time. (medlineplus.gov)
- Multiple sulfatase deficiency (MSD) is a very rare and fatal lysosomal storage disease characterized by a clinical phenotype that combines the features of different sulfatase deficiencies (whether lysosomal or not) that can have neonatal (most severe), infantile (most common) and juvenile (rare) presentations with manifestations including hypotonia, coarse facial features, mild deafness, skeletal anomalies, ichthyosis, hepatomegaly, developmental delay, progressive neurologic deterioration and hydrocephalus. (orpha.net)
- The neonatal period is the most susceptible time to TB infection, with increased risk of active and disseminated disease. (hrb.ie)
Syndrome9
- In Sjögren-Larsson syndrome , FALDH deficiency impairs fatty alcohol oxidation and leads to accumulation of 16- and 18-carbon-long aliphatic alcohols. (medscape.com)
- Patients with Sjögren-Larsson syndrome accumulate leukotriene B4 and its omega-hydroxy metabolite, which are probably responsible for the pruritus seen in this disease. (medscape.com)
- 12] Furthermore, patients with this disorder have low levels of certain polyunsaturated fatty acids in plasma, which can contribute to the cutaneous and neurologic disease in Sjögren-Larsson syndrome. (medscape.com)
- The macular degeneration in Sjögren-Larsson syndrome is associated with fundal autofluorescence and a unique deficiency of retinal macular pigments, especially the carotinoid zeaxanthin. (medscape.com)
- Life expectancy of those with Sjögren-Larsson syndrome is probably determined by the severity of neurologic symptoms and is comparable to that of other patients with static or slowly progressive neurologic disease. (medscape.com)
- Enzyme replacement therapy (ERT) appears safe and effective for peripheral manifestations in patients with Gaucher disease types I and III, Fabry disease, mucopolysaccharidosis I (Hurler, Hurler-Scheie, and Scheie syndromes), mucopolysaccharidosis II (Hunter syndrome), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), and Pompe disease. (medscape.com)
- We are thrilled to collaborate with the team at UoM to bring to infants and families living with Hunter syndrome a potential one-time therapy for this devastating disease. (avrobio.com)
- Mucopolysaccharidosis III (Sanfilippo Syndrome)- disease presentation and experimental therapies. (symptoma.com)
- Sanfilippo syndrome results from the deficiency or absence of 4 different enzymes that are necessary to degrade the GAG heparan sulfate. (symptoma.com)
Autosomal recessive4
- Multiple sulfatase deficiency (MSD), also known as Austin disease, or mucosulfatidosis, is a very rare autosomal recessive: 561 lysosomal storage disease caused by a deficiency in multiple sulfatase enzymes, or in formylglycine-generating enzyme, which activates sulfatases. (wikipedia.org)
- Krabbe disease , also known as globoid cell leukodystrophy , is an autosomal recessive lysosomal storage disorder resulting in damage to cells involved in myelin turnover. (radiopaedia.org)
- Niemann-P ick disease (NPD) is actually a collection of a number of distinct autosomal recessive lysosomal storage diseases . (radiopaedia.org)
- Mucopolysaccharidosis type I (MPS I) is an autosomal recessive lysosomal storage disorder resulting from deficiency of the enzyme α-L-iduronidase. (medicalhomeportal.org)
Severe3
- Obesity is a risk factor for both susceptibility to infections including postoperative infections and other nosocomial infections and the occurrence of a more severe disease course. (hrb.ie)
- Inherited ichthyoses Ichthyosis is scaling and flaking of skin ranging from mild but annoying dryness to severe disfiguring disease. (msdmanuals.com)
- Our results demonstrate a new functional role for these protein aggregates that are commonly associated with severe human diseases. (cirn-na.com)
Gaucher1
- The EUCLYD consortium will be focusing on four specific LSDs, namely Gaucher disease, Pompe disease, Mucopolysaccharidosis VI (MPS VI) and Multiple Sulfatase Deficiency (MSD), as prototypes of disorders with different stored materials in various organs and tissues outside the CNS. (tigem.it)
Sulfamidase1
- Gonococcal pharyngitis requires culture for ova and parasites imaging studies is unnecessary should not walk barefoot, and should only penetrate the intestinal lumen, where mucosal - glucuronidase deficiency sulfamidase - n-acetylglucosaminidase acetyl-coa - glucosaminide-nacetyltransferase - n-acetylglucosamine--sulfatase n-acetylgalactosamine--sulfatase n-acetylgalactosamine--sulfatase - glucuronidase. (albionfoundation.org)
Enzymes5
- The FGE enzyme modifies other enzymes called sulfatases, which aid in breaking down substances that contain chemical groups known as sulfates. (medlineplus.gov)
- The activity of multiple sulfatases is impaired because the FGE enzyme modifies all known sulfatase enzymes. (medlineplus.gov)
- These sulfatase enzymes are responsible for breaking down and recycling complex sulfate-containing sugars from lipids and mucopolysaccharides within the lysosome. (wikipedia.org)
- More recently, the concept of lysosomal storage disease has been expanded to include deficiencies or defects in proteins necessary for the normal post-translational modification of lysosomal enzymes (which themselves are often glycoproteins), activator proteins, or proteins important for proper intracellular trafficking between the lysosome and other intracellular compartments. (medscape.com)
- Therefore, a lysosomal disorder can be due to a defect in a specific hydrolase, by deficiencies in activator proteins, in the receptors or in the trafficking of enzymes. (tigem.it)
Metachromatic3
- Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. (medscape.com)
- Saposin B deficiency as a cause of adult-onset metachromatic leukodystrophy. (medscape.com)
- Metachromatic leukodystrophy (MLD) is part of a larger group of inherited lysosomal storage diseases, some of which are progressive and neurodegenerative disorders (MLD included). (medscape.com)
Clinical3
- Andrews' Diseases of the Skin: Clinical Dermatology. (wikipedia.org)
- Lysosomal storage diseases describe a heterogeneous group of dozens of rare inherited disorders characterized by the accumulation of undigested or partially digested macromolecules, which ultimately results in cellular dysfunction and clinical abnormalities. (medscape.com)
- This has led to active clinical trials evaluating the safety and efficacy of intrathecal enzyme delivery in several lysosomal storage diseases (see www.ClinicalTrials.gov ). (medscape.com)
Manifestations2
- Thus far, ERT has been largely unsuccessful in improving central nervous system manifestations of the lysosomal storage diseases, putatively due to difficulty in penetrating the blood-brain barrier. (medscape.com)
- Krabbe disease comprises a spectrum ranging from infantile-onset disease (i.e., onset of extreme irritability, spasticity, and developmental delay before age 12 months) to later-onset disease (i.e., onset of manifestations after age 12 months and as late as the seventh decade). (nih.gov)
Rare diseases2
- For most of his career, Peter has been focused on rare diseases and oncology. (checkrare.com)
- He has since worked across hundreds of rare diseases, creating content and promotional and educational programs. (checkrare.com)
Enzyme called1
- Plus, T3 doesn't become active until it's activated by an enzyme called intestinal sulfatase. (naturalendocrinesolutions.com)
Myelin2
- Fatty alcohol and aldehyde may likewise alter the normal integrity of myelin membranes in the brain, leading to white-matter disease and spasticity. (medscape.com)
- These diseases affect the myelin sheath, a fatty covering that insulates and protects nerve cells. (myriad.com)
Disorder2
- In general, transplantation yields the best results when performed early in the course of the disease (ie, in an asymptomatic affected sibling of a child with a lysosomal storage disorder), in centers with experience in performing transplantations to treat inherited metabolic disorders, and in patients healthy enough to tolerate the conditioning and transplantation regimen. (medscape.com)
- There are two general categories of cell lines, infection atypical lymphocytes, hemolytic anemia, multiple sclerosis, autoimmune disease, pri-marily a disorder in approximately of cases and many other sources of phosphate in the scrotum. (albionfoundation.org)
Recessive2
- mim #308100), also called steroid sulfatase (sts) deficiency, is an x-linked recessive ichthyosis caused by. (avtoradio.tj)
- Hunter's disease is inherited in an x-linked recessive pattern which results in a predominantly male disease. (picmonic.com)
Disorders2
- Research Details: Our lab aims to develop better treatments for inherited disorders of biochemistry (i.e., inborn errors of metabolism, IEMs) by studying how these diseases alter the electrical activity of the brain and heart. (upenn.edu)
- Search our database of diseases and disorders for more information. (baylorgenetics.com)
Ichthyosis3
- Ichthyosis is also common in the juvenile type of multiple sulfatase deficiency. (medlineplus.gov)
- Morbidity is associated with chronic neurologic disease and lifelong ichthyosis. (medscape.com)
- Ichthyosis can also be a sign of systemic disease. (msdmanuals.com)
Mucopolysaccharidoses1
- Accumulated data indicate that hematopoietic stem cell transplantation may be effective under optimal conditions in preventing the progression of central nervous system symptoms in neuronopathic forms of lysosomal storage diseases, including some of the mucopolysaccharidoses, oligosaccharidoses, sphingolipidoses, and lipidoses. (medscape.com)
Pediatrics1
- In the Department of Pediatrics and Adolescent Medicine at UMG , scientists led by Dr. Lars Schlotawa have developed a therapeutic approach that may lead to the first effective treatment of the rare neurometabolic disease MSD (multiple sulfatase deficiency) . (sciencebridge.de)
Pompe1
- While operating a website design business, Brad was diagnosed with Pompe disease in 2006. (checkrare.com)
Chronic3
- In general, young patients have the most rapidly progressive disease, whereas patients with adult onset MLD experience a more chronic and insidious progression of disease. (medscape.com)
- Less is known about the risk of other chronic diseases, particularly those which emerge in older age, such as dementia, retinal vascular. (hrb.ie)
- developing patho-physiological models for acute and chronic diseases, and complications of pregnancy. (stanford.edu)
Abnormalities1
- Some people with MPS III have short stature, joint stiffness, or mild dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. (symptoma.com)
Individuals with Krabbe disease2
- The majority of individuals with Krabbe disease present in early childhood although adult presentations as late as the 5thdecade are encountered 9 . (radiopaedia.org)
- Although historically 85%-90% of symptomatic individuals with Krabbe disease diagnosed by enzyme activity alone have infantile-onset Krabbe disease and 10%-15% have later-onset Krabbe disease, the experience with newborn screening (NBS) suggests that the proportion of individuals with possible later-onset Krabbe disease is higher than previously thought. (nih.gov)
Juvenile3
- The juvenile type is the rarest form of multiple sulfatase deficiency. (medlineplus.gov)
- The disease course may be from several years in the late-infantile-onset form to decades in the juvenile- and adult-onset forms. (nih.gov)
- As the disease continues, symptoms are similar to infantile MLD but the disease progresses more slowly in the juvenile form. (myriad.com)
Symptoms6
- The death of cells in particular tissues, specifically the brain, skeleton, and skin, cause many of the signs and symptoms of multiple sulfatase deficiency. (medlineplus.gov)
- The vast majority of patients have neurologic and/or cardiac symptoms that do not respond to therapy, even if a treatment can improve serum biochemical markers of disease. (upenn.edu)
- The course of the disease is similar, but the age at which symptoms appear varies, as does the rate at which symptoms progress. (myriad.com)
- The disease is usually fatal 10 to 20 years after the first symptoms appear. (myriad.com)
- In the final stages of the disease, symptoms are similar to the infantile form: blindness, deafness, unresponsiveness, and paralysis. (myriad.com)
- Most treatments aim to manage symptoms of the disease as they arise. (myriad.com)
Accumulation4
- Deficiency of galactocerebrosidase results in the accumulation of galactosylceramide within the lysosomes of Schwann cells and oligodendrocytes which eventually results in apoptosis with secondary abnormal activation of microglia and macrophages with subsequent demyelination and gliosis 2,9,10 . (radiopaedia.org)
- The mechanism contributing to the development of hydrops fetalis in storage diseases may involve the obstruction of venous blood return resulting from visceromegaly secondary to accumulation of storage material[ 13 ]. (biomedcentral.com)
- Deficiency of this enzyme causes an accumulation of dermatan sulfate and heparan sulfate, which are known as glycosaminoglycans (GAGs). (medicalhomeportal.org)
- Unlike hurler's disease, hunter's disease does not have accumulation of glycosaminoglycans in the cornea and does not have corneal clouding. (picmonic.com)
Onset1
- Later-onset Krabbe disease is much more variable in its presentation and disease course. (nih.gov)
Metabolic Diseases1
- M.D., professor of Pediatric Inherited Metabolic Diseases at the Manchester Centre for Genomic Medicine at Saint Mary's Hospital, part of Manchester University NHS Foundation Trust . (avrobio.com)
Signs2
- Initial signs of the disease include difficulties in school and behavioral problems. (myriad.com)
- Early signs of the disease often include personality changes, problems at school or work, numbness in the extremities of one's limbs, muscle weakness, loss of coordination, and psychiatric problems such as delusions, hallucinations, or drug and alcohol abuse. (myriad.com)
Schizophrenia1
- MLD can be initially misdiagnosed as schizophrenia, depression, or multiple sclerosis. (myriad.com)
Niemann-Pick Di1
- Niemann-pick disease type a presenting as unilateral tremors. (radiopaedia.org)
Venous1
- The sulfatase deficiency can be detected during pregnancy by a prolongation of the half-life of dehydroepiandrosterone sulfate (dhas) after venous dhas loading. (avtoradio.tj)
Hematopoietic stem cell trans2
- Therapy primarily centers on hematopoietic stem cell transplantation which can delay disease progression 11 . (radiopaedia.org)
- The availability of both ERT and hematopoietic stem cell transplantation has prompted ongoing consideration of newborn screening efforts to diagnose lysosomal storage diseases. (medscape.com)
Pregnancy1
- However, in some cases, the placenta involutes as pregnancy progresses and multiple infarcts and villous degeneration develop, causing placental insufficiency. (msdmanuals.com)
Progression1
- At best it slows, but does not stop, the progression of the disease. (myriad.com)
Diagnosis1
- These products are not intended for the diagnosis, prevention, or treatment of a disease. (qiagen.com)
Protein1
- She completed her Ph.D. in Neuroscience at the University of Florida, focusing on tauopathy modifiers and discovering the link between the LRRK2 protein associated with Parkinson's disease and the tau protein. (wtnh.com)
Autoimmune1
- Autoimmune disease affects 10% of adults, most of whom are women, and two of the top five medications with the highest cost globally are used to maintain these recurring conditions in remission. (hrb.ie)
Hemolytic1
- More recent recognition of factors other than isoimmune hemolytic disease that can cause or be associated with hydrops fetalis led to the use of the term non-immunological hydrops fetalis (NIHF). (biomedcentral.com)