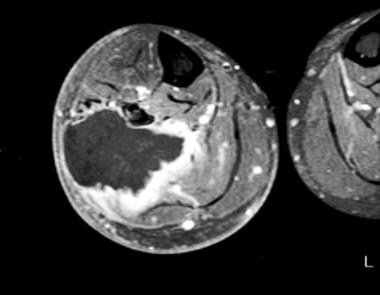
Neurofibroma
Neurofibroma, Plexiform
Neurofibromatosis 1
Neurofibromin 1
Neurofibrosarcoma
Nerve Sheath Neoplasms
Neurofibromatoses
Peripheral Nervous System Neoplasms
Genes, Neurofibromatosis 1
Schwann Cells
Cafe-au-Lait Spots
Neurilemmoma
Myelography
Vagus Nerve Diseases
Gingival Neoplasms
Scalp
Spinal Cord Neoplasms
Lipoma
S100 Proteins
Neurofibromatosis 2
Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. (1/54)
Neurofibromatosis type 1 (NF1) is one of the most common inherited disorders in humans and is caused by mutations in the NF1 gene. To date, the majority of the reported NF1 mutations are predicted to result in protein truncation, but very few studies have correlated the causative NF1 mutation with its effect at the mRNA level. We have applied a whole NF1 cDNA screening methodology to the study of 80 unrelated NF1 patients and have identified 44 different mutations, 32 being novel, in 52 of these patients. Mutations were detected in 87% of the familial cases, but in 51% of the sporadic ones. At least 15 of the 80 NF1 patients (19%) had recurrent mutations. The study shows that in 50% of the patients in whom the mutations were identified, these resulted in splicing alterations. Most of the splicing mutations did not involve the conserved AG/GT dinucleotides of the splice sites. One frameshift, two nonsense and two missense mutations were also responsible for alterations in mRNA splicing. The location and type of mutation within the NF1 gene, and its putative effect at the protein level, do not indicate any relationship to any specific clinical feature of NF1. The high proportion of aberrant spliced transcripts detected in NF1 patients stresses the importance of studying mutations at both the genomic and RNA level. It is possible that part of the clinical variability in NF1 could be due to mutations affecting mRNA splicing, which is the most common molecular defect in NF1. (+info)NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and malignant peripheral nerve sheath tumors. (2/54)
Although plexiform neurofibroma (PN) is thought to represent a benign neoplasm with the potential for malignant transformation (malignant peripheral nerve sheath tumor; MPNST), its neoplastic nature has been difficult to prove due to cellular heterogeneity, which hampers standard molecular genetic analysis. Its mixed composition typically includes Schwann cells, fibroblasts, perineurial-like cells, and mast cells. Although NF1 loss of heterozygosity has been reported in subsets of PNs, it remains uncertain which cell type(s) harbor these alterations. Using a dual-color fluorescence in situ hybridization and immunohistochemistry technique, we studied NF1 gene status in S-100 protein-positive and -negative cell subpopulations in archival paraffin-embedded specimens from seven PNs, two atypical PNs, one cellular/atypical PN, and eight MPNSTs derived from 13 patients, seven of which had neurofibromatosis type 1 (NF1). NF1 loss was detected in four of seven PNs and one atypical PN, with deletions entirely restricted to S-100 protein-immunoreactive Schwann cells. In contrast, all eight MPNSTs harbored NF1 deletions, regardless of S-100 protein expression or NF1 clinical status. Our results suggest that the Schwann cell is the primary neoplastic component in PNs and that S-100 protein-negative cells in MPNST represent dedifferentiated Schwann cells, which harbor NF1 deletions in both NF1-associated and sporadic tumors. (+info)Malignant schwannoma of the sciatic nerve originating in a spinal plexiform neurofibroma associated with neurofibromatosis type 1--case report. (3/54)
A 26-year-old man with neurofibromatosis type 1 (NF1) presented with a giant malignant schwannoma of the sciatic nerve. The differential diagnosis of malignant peripheral nerve sheath tumor (MPNST) was based on clinical, radiological, and histological evidence. The tumor apparently originated in a spinal plexiform neurofibroma. The lesion was resected totally without neural damage to the sciatic nerve. However, the tumor recurred within 2 months. The patient died of unknown factors probably associated with the spinal involvement. MPNST associated with NF1 has a poor prognosis due to recurrence or metastasis despite complete macroscopic removal. (+info)Orbit deformities in craniofacial neurofibromatosis type 1. (4/54)
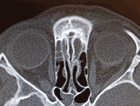
BACKGROUND AND PURPOSE: The possible relationship of orbit deformities in neurofibromatosis type 1 (NF1) to plexiform neurofibromas (PNFs) have not been fully elucidated. Our purpose was to review orbital changes in patients with craniofacial NF1. METHODS: We retrospectively reviewed CT and MR imaging abnormalities of the orbit in 31 patients (18 male, 13 female; mean age, 14 years; age range 1-40 years) with craniofacial NF1. RESULTS: Orbital abnormalities were documented in 24 patients. Six had optic nerve gliomas with enlarged optic canals. Twenty had PNFs in the orbit or contiguous to the anterior skull. The posterior orbit was distorted by encroachment from an expanded middle cranial fossa in 13 patients, and 18 had enlargement of the orbital rim. Other changes included focal decalcification or remodeling of orbital walls adjacent to PNFs in 18 patients and enlargement of cranial foramina resulting from tumor infiltration of sensory nerves in 16. These orbital deformities were sometimes progressive and always associated with orbital infiltration by PNFs. CONCLUSION: In our patients with craniofacial neurofibromatosis, bony orbital deformity occurred frequently and always with an optic nerve glioma or orbital PNF. PNFs were associated with orbital-bone changes in four patterns: expansion of the middle cranial fossa into the posterior orbit, enlargement of the orbital rim, bone erosion and decalcification by contiguous tumor, and enlargement of the cranial foramina. Orbital changes support the concept of secondary dysplasia, in which interaction of PNFs with the developing skull is a major component of the multifaceted craniofacial changes possible with NF1. (+info)Clinics in diagnostic imaging (96). Plexiform neurofibromatosis. (5/54)
A 10-year-old boy presented with a mass on the left side of his face for many years, left ear deafness, and limited vision in the left eye. Enhanced CT of the face and neck showed a multilobulated low attenuation mass in the left parotid space, with nodularity and involvement of branches of the left facial nerve. There was a mass in the left orbital apex that extended into the left cavernous sinus, exopthalmos of the left eye, and erosion of the medial portion of the greater wing of the left sphenoid. Partial removal of the mass in the left parotid space was performed. Histopathological examination revealed plexiform neurofibromas. CT and MR imaging findings in neurofibromatosis type 1 patients with craniofacial abnormalities are discussed. (+info)Plexiform neurofibroma of the cheek mucosa. A case report. (6/54)
The case reported deals with a solitary plexiform neurofibroma affecting the cheek submucosa. Neurofibroma is an uncommon tumor which rarely appears in oral cavity but it represents the most common neurogenic tumor. Furthermore, plexiform variety is less frequent. Clinically, oral neurofibromas usually appears as anodyne and asintomatic lesions. Sometimes, they produce nervous compression. In this case, tumor is big but asintomatic. There is no definitive radiologic image. It has association with polyglandular syndromes and phacomatosis. The treatment of choice is excision. There are doubts of the surgical results so that some authors are looking for new non-surgical treatments. The clinical characteristics, epidemiology, diagnosis and treatment are described as soon as a bibliographic revision. (+info)Molecular profiles of neurofibromatosis type 1-associated plexiform neurofibromas: identification of a gene expression signature of poor prognosis. (7/54)
PURPOSE: Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder with a complex variety of clinical symptoms. The hallmark of NF1 is the development of heterogeneous benign neurofibromas, which may appear as dermal neurofibromas or plexiform neurofibromas. NF1 patients with plexiform neurofibromas are at risk of developing malignant peripheral nerve sheath tumors. EXPERIMENTAL DESIGN: To obtain additional insight into the molecular pathogenesis of plexiform neurofibromas, we used real-time quantitative reverse transcription-PCR assays to quantify the mRNA expression of 349 selected genes in plexiform neurofibromas in comparison with dermal neurofibromas and patient-matched malignant peripheral nerve sheath tumors. RESULTS: Thirty genes were significantly up-regulated in plexiform neurofibromas compared with dermal neurofibromas. None were down-regulated. The up-regulated genes mainly encoded transcription factors and growth factors and secreted proteins, cytokines, and their receptors, pointing to a role of paracrine and autocrine signaling defects in the genesis of plexiform neurofibromas. We also identified a gene expression profile, based on MMP9, FLT4/VEGFR3, TNFRSF10B/TRAILR2, SHH, and GLI1, which discriminated those plexiform neurofibromas most likely to undergo malignant transformation. CONCLUSION: Our study has identified a limited number of signaling pathways that could be involved, when altered, in plexiform neurofibroma development. Some of the up-regulated genes could be useful diagnostic or prognostic markers or form the basis of novel therapeutic strategies. (+info)Molecular profiling of malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1, based on large-scale real-time RT-PCR. (8/54)
BACKGROUND: Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder with a complex range of clinical symptoms. The hallmark of NF1 is the onset of heterogeneous (dermal or plexiform) benign neurofibromas. Plexiform neurofibromas can give rise to malignant peripheral nerve sheath tumors (MPNSTs), and the underlying molecular mechanisms are largely unknown. RESULTS: To obtain further insight into the molecular pathogenesis of MPNSTs, we used real-time quantitative RT-PCR to quantify the mRNA expression of 489 selected genes in MPNSTs, in comparison with plexiform neurofibromas. The expression of 28 (5.7%) of the 489 genes was significantly different between MPNSTs and plexiform neurofibromas; 16 genes were upregulated and 12 were downregulated in MPNSTs. The altered genes were mainly involved in cell proliferation (MKI67, TOP2A, CCNE2), senescence (TERT, TERC), apoptosis (BIRC5/Survivin, TP73) and extracellular matrix remodeling (MMP13, MMP9, TIMP4, ITGB4). More interestingly, other genes were involved in the Ras signaling pathway (RASSF2, HMMR/RHAMM) and the Hedgehog-Gli signaling pathway (DHH, PTCH2). Several of the down-regulated genes were Schwann cell-specific (L1CAM, MPZ, S100B, SOX10, ERBB3) or mast cell-specific (CMA1, TPSB), pointing to a depletion and/or dedifferentiation of Schwann cells and mast cells during malignant transformation of plexiform neurofibromas. CONCLUSION: These data suggest that a limited number of signaling pathways, and particularly the Hedgehog-Gli signaling pathway, may be involved in malignant transformation of plexiform neurofibromas. Some of the relevant genes or their products warrant further investigation as potential therapeutic targets in NF1. (+info)A neurofibroma is a benign (non-cancerous) tumor that develops from the nerve sheath, which is the protective covering around nerves. These tumors can grow anywhere on the body and can be found under the skin or deep inside the body. Neurofibromas can vary in size, and they may cause symptoms such as pain, numbness, or tingling if they press on nearby nerves.
Neurofibromas are a common feature of neurofibromatosis type 1 (NF1), a genetic disorder that affects approximately 1 in every 3,000 people worldwide. NF1 is characterized by the development of multiple neurofibromas and other tumors, as well as skin changes such as café-au-lait spots and freckling.
It's important to note that while most neurofibromas are benign, they can rarely undergo malignant transformation and become cancerous. If you have a neurofibroma or are concerned about your risk of developing one, it's important to seek medical advice from a healthcare professional who is familiar with this condition.
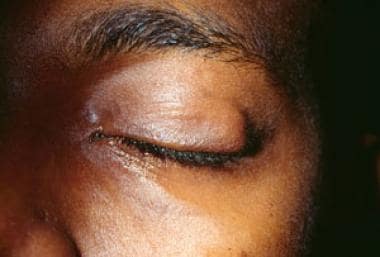
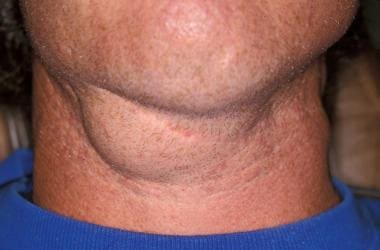
A plexiform neurofibroma is a type of neurofibroma, which is a benign tumor that develops from the nerve sheath. In a plexiform neurofibroma, the tumor grows along the nerves and can involve multiple fascicles, leading to a large, diffuse mass. These tumors can occur anywhere in the body but are most commonly found in the head, neck, and trunk.
Plexiform neurofibromas can be associated with neurofibromatosis type 1 (NF1), a genetic disorder that affects approximately 1 in every 3,000 people worldwide. In individuals with NF1, plexiform neurofibromas can cause significant morbidity, including disfigurement, pain, and functional impairment. Additionally, there is a small risk of malignant transformation into a type of cancer called malignant peripheral nerve sheath tumor (MPNST).
The diagnosis of plexiform neurofibromas is typically made based on clinical examination, medical history, and imaging studies such as MRI. A biopsy may be necessary to confirm the diagnosis. Treatment options for plexiform neurofibromas include surgery, radiation therapy, and medication. The choice of treatment depends on several factors, including the size and location of the tumor, the presence of symptoms, and the risk of malignant transformation.
Neurofibromatosis 1 (NF1) is a genetic disorder that affects the development and growth of nerve tissue. It's also known as von Recklinghausen disease. NF1 is characterized by the growth of non-cancerous tumors on the nerves, as well as skin and bone abnormalities.
The symptoms of Neurofibromatosis 1 can vary widely, even among members of the same family. Some common features include:
* Multiple café au lait spots (flat, light brown patches on the skin)
* Freckles in the underarms and groin area
* Benign growths on or under the skin called neurofibromas
* Larger, more complex tumors called plexiform neurofibromas
* Optic gliomas (tumors that form on the optic nerve)
* Distinctive bone abnormalities, such as a curved spine (scoliosis) or an enlarged head (macrocephaly)
* Learning disabilities and behavioral problems
Neurofibromatosis 1 is caused by mutations in the NF1 gene, which provides instructions for making a protein called neurofibromin. This protein helps regulate cell growth and division. When the NF1 gene is mutated, the production of neurofibromin is reduced or absent, leading to uncontrolled cell growth and the development of tumors.
NF1 is an autosomal dominant disorder, which means that a person has a 50% chance of inheriting the mutated gene from an affected parent. However, about half of all cases are the result of new mutations in the NF1 gene, and occur in people with no family history of the disorder.
There is currently no cure for Neurofibromatosis 1, but treatments are available to manage the symptoms and complications of the disease. These may include medications to control pain or reduce the size of tumors, surgery to remove tumors or correct bone abnormalities, and physical therapy to improve mobility and strength. Regular monitoring by a healthcare team experienced in treating Neurofibromatosis 1 is also important to detect any changes in the condition and provide appropriate care.
Neurofibromin 1 is a protein that is encoded by the NF1 gene in humans. Neurofibromin 1 acts as a tumor suppressor, helping to regulate cell growth and division. It plays an important role in the nervous system, where it helps to control the development and function of nerve cells. Mutations in the NF1 gene can lead to neurofibromatosis type 1 (NF1), a genetic disorder characterized by the growth of non-cancerous tumors on the nerves (neurofibromas) and other symptoms.
Neurofibrosarcoma is a rare type of soft tissue sarcoma, which is a cancer that develops in the soft tissues of the body such as fat, muscle, tendons, blood vessels, and nerves. Neurofibrosarcoma specifically arises from the nerve sheath cells, also known as the Schwann cells, that cover and protect the peripheral nerves.
This type of cancer typically forms a painless mass or tumor in the affected area, which can grow and invade nearby tissues and organs over time. Neurofibrosarcoma can occur anywhere in the body but is most commonly found in the arms, legs, trunk, or head and neck region.
Neurofibrosarcoma can be classified into two main types: conventional and malignant peripheral nerve sheath tumor (MPNST). Conventional neurofibrosarcoma is more common and tends to occur in older adults, while MPNST is a more aggressive form that is associated with genetic disorders such as neurofibromatosis type 1.
Treatment for neurofibrosarcoma typically involves surgical removal of the tumor, along with radiation therapy and/or chemotherapy to help prevent recurrence and spread of the cancer. The prognosis for neurofibrosarcoma varies depending on several factors, including the size and location of the tumor, the patient's age and overall health, and the stage of the disease at diagnosis.
Nerve sheath neoplasms are a group of tumors that arise from the cells surrounding and supporting the nerves. These tumors can be benign or malignant and include schwannomas, neurofibromas, and malignant peripheral nerve sheath tumors (MPNSTs). Schwannomas develop from the Schwann cells that produce the myelin sheath of the nerve, while neurofibromas arise from the nerve's supporting cells called fibroblasts. MPNSTs are cancerous tumors that can grow rapidly and invade surrounding tissues. Nerve sheath neoplasms can cause various symptoms depending on their location and size, including pain, numbness, weakness, or paralysis in the affected area.
Neurofibromatoses are a group of genetic disorders that primarily affect the nervous system. The term "neurofibromatosis" is often used to refer to two specific conditions: neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). These conditions are characterized by the growth of tumors on the nerves, called neurofibromas.
Neurofibromatosis type 1 (NF1): This is the most common form of neurofibromatosis, affecting about 1 in every 3,000 people worldwide. NF1 is caused by mutations in the NF1 gene and is characterized by the development of benign tumors on the nerves called neurofibromas. These tumors can develop anywhere on the body, including the skin, spinal cord, and brain. Other common features of NF1 include:
* Freckles in the underarms and groin area
* Lisch nodules (small, noncancerous growths) on the iris of the eye
* Bone abnormalities, such as scoliosis or bowing of the legs
* Learning disabilities or cognitive impairment
Neurofibromatosis type 2 (NF2): This form of neurofibromatosis is much rarer than NF1, affecting about 1 in every 30,000 people worldwide. NF2 is caused by mutations in the NF2 gene and is characterized by the development of benign tumors on the nerves that transmit sound from the inner ear to the brain (acoustic neuromas). These tumors can cause hearing loss, ringing in the ears, and balance problems. Other common features of NF2 include:
* Multiple schwannomas (tumors that develop on the protective covering of the nerves)
* Meningiomas (tumors that develop in the membranes surrounding the brain and spinal cord)
* Skin tumors called neurofibromas, although these are less common than in NF1
It is important to note that while neurofibromatoses can cause a range of symptoms and complications, most people with these conditions have a normal lifespan. With proper medical care and monitoring, it is possible to manage the symptoms and reduce the risk of complications.
Peripheral nervous system (PNS) neoplasms refer to tumors that originate in the peripheral nerves, which are the nerves outside the brain and spinal cord. These tumors can be benign or malignant (cancerous). Benign tumors, such as schwannomas and neurofibromas, grow slowly and do not spread to other parts of the body. Malignant tumors, such as malignant peripheral nerve sheath tumors (MPNSTs), can invade nearby tissues and may metastasize (spread) to other organs.
PNS neoplasms can cause various symptoms depending on their location and size. Common symptoms include pain, weakness, numbness, or tingling in the affected area. In some cases, PNS neoplasms may not cause any symptoms until they become quite large. Treatment options for PNS neoplasms depend on several factors, including the type, size, and location of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Neurofibromatosis 1 (NF1) is a genetic disorder caused by mutations in the NF1 gene, which is located on chromosome 17 and encodes the protein neurofibromin. Neurofibromin is a tumor suppressor protein that regulates cell growth and differentiation.
The NF1 gene mutation leads to the development of benign (non-cancerous) tumors on nerves and skin, called neurofibromas, as well as other clinical features such as café-au-lait spots (light brown patches on the skin), freckling in the axillary or inguinal regions, Lisch nodules (harmless growths on the iris of the eye), and skeletal abnormalities.
Neurofibromatosis 1 is an autosomal dominant disorder, which means that a person has a 50% chance of inheriting the mutated gene from an affected parent. However, up to 50% of cases result from new mutations in the NF1 gene and occur in people with no family history of the condition.
The clinical manifestations of Neurofibromatosis 1 can vary widely among individuals, even within the same family. The diagnosis is typically made based on clinical criteria established by the National Institutes of Health (NIH). Treatment is generally focused on managing symptoms and addressing complications as they arise, although surgery may be necessary to remove large or symptomatic tumors.
Schwann cells, also known as neurolemmocytes, are a type of glial cell that form the myelin sheath around peripheral nervous system (PNS) axons, allowing for the rapid and efficient transmission of nerve impulses. These cells play a crucial role in the maintenance and function of the PNS.
Schwann cells originate from the neural crest during embryonic development and migrate to the developing nerves. They wrap around the axons in a spiral fashion, forming multiple layers of myelin, which insulates the nerve fibers and increases the speed of electrical impulse transmission. Each Schwann cell is responsible for myelinating a single segment of an axon, with the gaps between these segments called nodes of Ranvier.
Schwann cells also provide structural support to the neurons and contribute to the regeneration of injured peripheral nerves by helping to guide the regrowth of axons to their targets. Additionally, Schwann cells can participate in immune responses within the PNS, such as releasing cytokines and chemokines to recruit immune cells during injury or infection.
Café-au-lait spots are light to dark brown, flat patches on the skin that are benign and usually harmless. The term "café-au-lait" means "coffee with milk," which describes the color of these spots. They can vary in size from a few millimeters to several centimeters in diameter and can appear anywhere on the body, although they are most commonly found on the trunk and buttocks.
While café-au-lait spots are common and can occur in up to 20% of the general population, having multiple (more than six) such spots, especially if they are large or present at birth, may be a sign of an underlying medical condition, such as neurofibromatosis type 1 (NF1), a genetic disorder that affects the growth and development of nerve tissue.
Therefore, it is essential to monitor café-au-lait spots and report any changes or concerns to a healthcare provider.
A neurilemmoma, also known as schwannoma or peripheral nerve sheath tumor, is a benign, slow-growing tumor that arises from the Schwann cells, which produce the myelin sheath that surrounds and insulates peripheral nerves. These tumors can occur anywhere along the course of a peripheral nerve, but they most commonly affect the acoustic nerve (vestibulocochlear nerve), leading to a type of tumor called vestibular schwannoma or acoustic neuroma. Neurilemmomas are typically encapsulated and do not invade the surrounding tissue, although larger ones may cause pressure-related symptoms due to compression of nearby structures. Rarely, these tumors can undergo malignant transformation, leading to a condition called malignant peripheral nerve sheath tumor or neurofibrosarcoma.
A neuroma is not a specific type of tumor, but rather refers to a benign (non-cancerous) growth or swelling of nerve tissue. The most common type of neuroma is called a Morton's neuroma, which typically occurs between the third and fourth toes in the foot. It develops as a result of chronic irritation, compression, or trauma to the nerves leading to the toes, causing them to thicken and enlarge.
Morton's neuroma can cause symptoms such as pain, numbness, tingling, or burning sensations in the affected area. Treatment options for Morton's neuroma may include rest, ice, orthotics, physical therapy, medication, or in some cases, surgery. It is essential to consult a healthcare professional if you suspect you have a neuroma or are experiencing related symptoms.
Myelography is a medical imaging technique used to examine the spinal cord and surrounding structures, such as the spinal nerves, intervertebral discs, and the spinal column. This procedure involves the injection of a contrast dye into the subarachnoid space, which is the area surrounding the spinal cord filled with cerebrospinal fluid (CSF). The dye outlines the spinal structures, making them visible on X-ray or CT scan images.
The primary purpose of myelography is to diagnose various spinal conditions, including herniated discs, spinal stenosis, tumors, infection, and traumatic injuries. It can help identify any compression or irritation of the spinal cord or nerves that may be causing pain, numbness, weakness, or other neurological symptoms.
The procedure typically requires the patient to lie flat on their stomach or side while the radiologist inserts a thin needle into the subarachnoid space, usually at the lower lumbar level. Once the contrast dye is injected, the patient will be repositioned for various X-ray views or undergo a CT scan to capture detailed images of the spine. After the procedure, patients may experience headaches, nausea, or discomfort at the injection site, but these symptoms usually resolve within a few days.
Vagus nerve diseases, also known as vagus nerve disorders, refer to conditions that affect the functioning of the vagus nerve. The vagus nerve is the tenth cranial nerve and extends from the brainstem to the abdomen, playing a crucial role in regulating various automatic functions of the body such as heart rate, digestion, respiratory rate, and sweating.
Diseases of the vagus nerve can result from various causes, including inflammation, infection, trauma, compression, or degeneration. Some common vagus nerve disorders include:
1. Vagus nerve dysfunction: This is a general term used to describe any abnormality in the functioning of the vagus nerve. Symptoms may vary depending on the specific functions affected but can include difficulty swallowing, hoarseness, voice changes, and abnormal heart rate or blood pressure.
2. Vagus nerve neuropathy: This is a condition that results from damage to the vagus nerve fibers. It can cause symptoms such as difficulty swallowing, voice changes, and abnormal digestive function.
3. Gastroparesis: This is a condition in which the stomach muscles fail to contract properly, leading to delayed gastric emptying. Vagus nerve dysfunction is a common cause of gastroparesis.
4. Orthostatic hypotension: This is a condition characterized by a drop in blood pressure when standing up from a sitting or lying down position. Vagus nerve dysfunction can contribute to this condition by causing an abnormal response in the heart rate and blood vessels.
5. Inflammatory disorders: Certain inflammatory conditions such as rheumatoid arthritis, lupus, and sarcoidosis can affect the vagus nerve and cause various symptoms.
Treatment for vagus nerve diseases depends on the underlying cause and may include medications, surgery, or lifestyle changes.
Mandibular neoplasms refer to abnormal growths or tumors that develop in the mandible, which is the lower jawbone. These growths can be benign (non-cancerous) or malignant (cancerous). Benign neoplasms are typically slow-growing and rarely spread to other parts of the body, while malignant neoplasms can invade surrounding tissues and may metastasize (spread) to distant sites.
Mandibular neoplasms can have various causes, including genetic mutations, exposure to certain chemicals or radiation, and infection with certain viruses. The symptoms of mandibular neoplasms may include swelling or pain in the jaw, difficulty chewing or speaking, numbness in the lower lip or chin, loose teeth, and/or a lump or mass in the mouth or neck.
The diagnosis of mandibular neoplasms typically involves a thorough clinical examination, imaging studies such as X-rays, CT scans, or MRI scans, and sometimes a biopsy to confirm the type and extent of the tumor. Treatment options depend on the type, stage, and location of the neoplasm, and may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. Regular follow-up care is essential to monitor for recurrence or metastasis.
Gingival neoplasms refer to abnormal growths or tumors that occur in the gingiva, which are the part of the gums that surround the teeth. These growths can be benign (non-cancerous) or malignant (cancerous). Benign neoplasms include conditions such as fibromas, papillomas, and hemangiomas, while malignant neoplasms are typically squamous cell carcinomas.
Gingival neoplasms can present with a variety of symptoms, including swelling, bleeding, pain, and loose teeth. They may also cause difficulty with chewing, speaking, or swallowing. The exact cause of these neoplasms is not always known, but risk factors include tobacco use, alcohol consumption, poor oral hygiene, and certain viral infections.
Diagnosis of gingival neoplasms typically involves a thorough clinical examination, including a dental exam and biopsy. Treatment options depend on the type and stage of the neoplasm, but may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. Regular dental check-ups and good oral hygiene practices can help to detect gingival neoplasms at an early stage and improve treatment outcomes.
The scalp is the anatomical region located at the upper part of the human head, covering the skull except for the face and the ears. It is made up of several layers: the skin, the connective tissue, the galea aponeurotica (a strong, flat, tendinous sheet), loose areolar tissue, and the periosteum (the highly vascularized innermost layer that attaches directly to the skull bones). The scalp has a rich blood supply and is home to numerous sensory receptors, including those for touch, pain, and temperature. It also contains hair follicles, sebaceous glands, and sweat glands.
Spinal cord neoplasms refer to abnormal growths or tumors within the spinal cord. These can be benign (non-cancerous) or malignant (cancerous). They originate from the cells within the spinal cord itself (primary tumors), or they may spread to the spinal cord from other parts of the body (metastatic tumors). Spinal cord neoplasms can cause various symptoms depending on their location and size, including back pain, neurological deficits, and even paralysis. Treatment options include surgery, radiation therapy, and chemotherapy.
A lipoma is a common, benign (non-cancerous) soft tissue growth. It is composed of adipose or fatty tissue and typically found just beneath the skin, but they can also occur deeper within the body. Lipomas are usually round, moveable, and painless, although they may cause discomfort if they grow large enough to put pressure on nearby nerves or if they're located in a sensitive area. They generally grow slowly over time. Surgical removal is an option if the lipoma becomes bothersome or grows significantly in size. It's important to note that while lipomas are typically harmless, any new lumps or bumps should be evaluated by a healthcare professional to confirm the diagnosis and rule out other more serious conditions.
S100 proteins are a family of calcium-binding proteins that are involved in the regulation of various cellular processes, including cell growth and differentiation, intracellular signaling, and inflammation. They are found in high concentrations in certain types of cells, such as nerve cells (neurons), glial cells (supporting cells in the nervous system), and skin cells (keratinocytes).
The S100 protein family consists of more than 20 members, which are divided into several subfamilies based on their structural similarities. Some of the well-known members of this family include S100A1, S100B, S100 calcium-binding protein A8 (S100A8), and S100 calcium-binding protein A9 (S100A9).
Abnormal expression or regulation of S100 proteins has been implicated in various pathological conditions, such as neurodegenerative diseases, cancer, and inflammatory disorders. For example, increased levels of S100B have been found in the brains of patients with Alzheimer's disease, while overexpression of S100A8 and S100A9 has been associated with the development and progression of certain types of cancer.
Therefore, understanding the functions and regulation of S100 proteins is important for developing new diagnostic and therapeutic strategies for various diseases.
Neurofibromatosis 2 (NF2) is a genetic disorder characterized by the development of non-cancerous tumors in the nervous system, particularly on the nerves related to hearing and balance. It's also known as central neurofibromatosis or bilateral acoustic neuroma syndrome.
The primary feature of NF2 is the growth of schwannomas, which are tumors that develop from the cells surrounding nerve fibers. These typically grow on the vestibular nerve, leading to hearing loss, ringing in the ears (tinnitus), and balance problems. Bilateral acoustic neuromas (schwannomas affecting both vestibular nerves) are a hallmark of this condition.
Other common features include:
1. Meningiomas: These are tumors that grow in the meninges, the protective layers surrounding the brain and spinal cord.
2. Ependymomas: These are tumors that develop from the ependymal cells lining the ventricles (fluid-filled spaces) in the brain or the spinal cord canal.
3. Neurofibromas: Unlike in Neurofibromatosis type 1, these are less common and typically don't become cancerous.
4. Skin changes: While not as prevalent as in NF1, some people with NF2 may have skin freckles, café-au-lait spots, or skin tumors.
5. Eye problems: Some individuals may experience cataracts, retinal abnormalities, or optic nerve tumors (optic gliomas).
6. Other potential symptoms: Headaches, facial weakness or numbness, and difficulty swallowing or speaking.
NF2 is an autosomal dominant disorder, meaning that a person has a 50% chance of inheriting the condition if one of their parents has it. However, about half of all NF2 cases result from spontaneous genetic mutations with no family history of the disorder.
Soft tissue neoplasms refer to abnormal growths or tumors that develop in the soft tissues of the body. Soft tissues include muscles, tendons, ligaments, fascia, nerves, blood vessels, fat, and synovial membranes (the thin layer of cells that line joints and tendons). Neoplasms can be benign (non-cancerous) or malignant (cancerous), and their behavior and potential for spread depend on the specific type of neoplasm.
Benign soft tissue neoplasms are typically slow-growing, well-circumscribed, and rarely spread to other parts of the body. They can often be removed surgically with a low risk of recurrence. Examples of benign soft tissue neoplasms include lipomas (fat tumors), schwannomas (nerve sheath tumors), and hemangiomas (blood vessel tumors).
Malignant soft tissue neoplasms, on the other hand, can grow rapidly, invade surrounding tissues, and may metastasize (spread) to distant parts of the body. They are often more difficult to treat than benign neoplasms and require a multidisciplinary approach, including surgery, radiation therapy, and chemotherapy. Examples of malignant soft tissue neoplasms include sarcomas, such as rhabdomyosarcoma (arising from skeletal muscle), leiomyosarcoma (arising from smooth muscle), and angiosarcoma (arising from blood vessels).
It is important to note that soft tissue neoplasms can occur in any part of the body, and their diagnosis and treatment require a thorough evaluation by a healthcare professional with expertise in this area.