Neurofibromatosis 1
Neurofibromatosis 2
Genes, Neurofibromatosis 1
Genes, Neurofibromatosis 2
Neurofibromatoses
Neurofibromin 1
Neurofibromin 2
Neurofibroma, Plexiform
Neurofibroma
Cafe-au-Lait Spots
Optic Nerve Glioma
Nerve Sheath Neoplasms
Neurilemmoma
Optic Nerve Neoplasms
Neuroma, Acoustic
Peripheral Nervous System Neoplasms
Neurofibrosarcoma
Cranial Nerve Neoplasms
Nervous System Neoplasms
Meningioma
Auditory Brain Stem Implants
Meningocele
Hamartoma
Pseudarthrosis
Chromosomes, Human, Pair 17
Schwann Cells
Learning Disorders
Noonan Syndrome
Facial Neoplasms
Mosaicism
Bone Diseases, Developmental
Chromosomes, Human, Pair 22
Neoplasms, Multiple Primary
Piebaldism
Pheochromocytoma
Magnetic Resonance Imaging
Meningeal Neoplasms
Somatostatinoma
Decalcification, Pathologic
Mutation
Pigmentation Disorders
Glomus Tumor
Optic Chiasm
Thoracic Neoplasms
Pedigree
Mixed Tumor, Malignant
Astrocytoma
Proteus Syndrome
Leukemia, Myelomonocytic, Juvenile
Tomography, X-Ray Computed
Iris Neoplasms
Angiomatosis
Arachnoid
The duty to recontact: attitudes of genetics service providers. (1/767)
The term "duty to recontact" refers to the possible ethical and/or legal obligation of genetics service providers (GSPs) to recontact former patients about advances in research that might be relevant to them. Although currently this practice is not part of standard care, some argue that such an obligation may be established in the future. Little information is available, however, on the implications of this requirement, from the point of view of GSPs. To explore the opinions of genetics professionals on this issue, we sent a self-administered questionnaire to 1,000 randomly selected U.S. and Canadian members of the American Society of Human Genetics. We received 252 completed questionnaires. The major categories of respondents were physician geneticist (41%), Ph.D. geneticist (30%), and genetic counselor (18%); 72% of the total stated that they see patients. Respondents indicated that responsibility for staying in contact should be shared between health professionals and patients. Respondents were divided about whether recontacting patients should be the standard of care: 46% answered yes, 43% answered no, and 11% did not know. Those answering yes included 44% of physician geneticists, 53% of Ph.D. geneticists, and 31% of genetic counselors; answers were statistically independent of position or country of practice but were dependent on whether the respondent sees patients (43% answered yes) or not (54% answered yes). There also was a lack of consensus about the possible benefits and burdens of recontacting patients and about various alternative methods of informing patients about research advances. Analysis of qualitative data suggested that most respondents consider recontacting patients an ethically desirable, but not feasible, goal. Points to consider in the future development of guidelines for practice are presented. (+info)Neurological complications of neurofibromatosis type 1 in adulthood. (2/767)
Neurofibromatosis type 1 (NF1) is a genetic disease with a wide range of neurological manifestations. To examine these, and to evaluate neurological morbidity in adulthood of patients with NF1, we studied a hospital-based series of 158 patients that included 138 adult patients aged >18 years and 20 children. NF1 evaluation included a multidisciplinary clinical and a clinically oriented radiological investigation. Neurological events occurring during childhood (in both children and adults of the series) and adulthood were recorded. One or several neurological manifestations have been observed in 55% of patients (adults and children) (n = 87). These included: headache (28 patients); hydrocephalus (7); epilepsy (5); lacunar stroke (1); white matter disease (1); intraspinal neurofibroma (3); facial palsy (1); radiculopathy (5); and polyneuropathy (2). Tumours included: optic pathway tumours (20); meningioma (2); cerebral glioma (3); and malignant peripheral nerve sheath tumours (6). Life-threatening complications were observed in five adults and included four malignant peripheral nerve sheath tumours and one meningioma. Pain was the leading symptom in 11 adults and was related to malignant peripheral nerve sheath tumours, complications of intraspinal neurofibromas, subcutaneous neurofibromas and peripheral nerve neurofibromas. NF1 in adults was not associated with other disabling or life-threatening neurological complications. Symptomatic optic pathway tumours, cerebral gliomas, symptomatic aqueductal stenosis and spinal compression due to intraspinal NF were observed exclusively during childhood. In this series, the predominant neurological features of adults with NF1 were chronic pain and malignant peripheral nerve sheath tumours. (+info)Von Hippel's disease in association with von Recklinghausen's neurofibromatosis. (3/767)
Ten members of a large family who showed manifestations of either von Hippel-Lindau disease or von Recklinghausen's neurofibromatosis were examined. Three of 10 members were found to have retinal angiomas which had not been present on fundus examination 3 years previously. These angiomas were associated with ocular and systemic signs of neurofibromatosis. These cases show overlapping manifestations of different phakomatoses and provide support for the concept of a common aetiology for these diseases. (+info)Spontaneous haemothorax: a cause of sudden death in von Recklinghausen's disease. (4/767)
Vasculopathy is a relatively frequent but poorly recognised manifestation of von Recklinghausen's neurofibromatosis. One of its more dramatic presentations is as spontaneous haemothorax. Clinicians and pathologists should be aware of this syndrome as a cause of sudden death in patients with neurofibromatosis. (+info)A clinical study of type 1 neurofibromatosis in north west England. (5/767)
A clinical study of patients on the North West Regional Genetic Register with neurofibromatosis type 1 (NF1) identified 523 affected cases from 304 families. In those for whom relevant information was available, 86.7% (383 of 442) had more than six cafe au lait patches, 83.8% (310 of 370) had axillary freckling, 42.3% (151 of 357) had inguinal freckling, and 63% (157 of 249) had Lisch nodules. Cutaneous neurofibromas were present in 59.4% (217 of 365) and 45.5% (150 of 330) were noted to have subcutaneous tumours. Plexiform neurofibromas were present in 15.3% (80 of 523). A positive family history of NF1 was found in 71.2% (327 of 459) and 28.8% (132 of 459) of affected patients were considered to be the result of a new mutation. Learning difficulties of varying severity occurred in 62% (186 of 300). CNS tumours associated with NF1 were reported in 9.4% (49) of patients, optic gliomas occurring in 25 of these, 4.8% of patients. Some degree of scoliosis was reported for 11.7% (61), 1.9% (10) had pseudoarthrosis, 4.3% (23) had epilepsy, and 2.1% (11) had spinal neurofibromas. Actuarial analyses were carried out for both optic glioma and malignant nerve sheath tumours and the data are presented. (+info)The Nf1 tumor suppressor regulates mouse skin wound healing, fibroblast proliferation, and collagen deposited by fibroblasts. (6/767)
Neurofibromatosis type 1 patients develop peripheral nerve tumors (neurofibromas) composed mainly of Schwann cells and fibroblasts, in an abundant collagen matrix produced by fibroblasts. Trauma has been proposed to trigger neurofibroma formation. To test if loss of the neurofibromatosis type 1 gene (Nf1) compromises fibroblast function in vivo following trauma, skin wounding was performed in Nf1 knockout mice. The pattern and amount of collagen-rich granulation bed tissue, manufactured by fibroblasts, was grossly abnormal in 60% of Nf1+/- wounds. Nf1 mutant fibroblasts showed cell autonomous abnormalities in collagen deposition in vitro that were not mimicked by Ras activation in fibroblasts, even though some Nf1 effects are mediated through Ras. Nf1+/- skin wound fibroblasts also proliferated past the normal wound maturation phase; this in vivo effect was potentiated by muscle injury. In vitro, Nf1+/- fibroblasts showed higher proliferation in 10% serum than Nf1+/+ fibroblasts. Macrophage-conditioned media or epidermal growth factor potentiated Nf1+/- fibroblast proliferation in vitro, demonstrating abnormal response of mutant fibroblasts to wound cytokines. Thus Nf1 is a key regulator of fibroblast responses to injury, and Nf1 mutation in mouse fibroblasts causes abnormalities characteristic of human neurofibromas. (+info)Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. (7/767)
Individuals affected with neurofibromatosis 1 (NF1) harbor increased numbers of GFAP-immunoreactive cerebral astrocytes and develop astrocytomas that can lead to blindness and death. Mice heterozygous for a targeted Nf1 mutation (Nf1+/-) were employed as a model for the human disease to evaluate the hypothesis that reduced NF1 protein (neurofibromin) expression may confer a growth advantage for astrocytes, such that inactivation of only one NF1 allele is sufficient for abnormal astrocyte proliferation. Here, we report that Nf17+/- mice have increased numbers of cerebral astrocytes and increased astrocyte proliferation compared to wild-type littermates. Intriguingly, primary Nf1+/- astrocyte cultures failed to demonstrate a cell-autonomous growth advantage unless they were cocultured with C17 neuronal cells. This C17 neuronal cell-induced Nf1+/- increase in proliferation was blocked by MEK inhibition (PD98059), suggesting a p21-ras-dependent effect. Furthermore, mice heterozygous for a targeted mutation in another GAP molecule, p120-GAP, demonstrated no increases in cerebral astrocyte number. These findings suggest that reduced NF1 expression results in a cell context-dependent increase in astrocyte proliferation that may be sufficient for the development of astrocytic growth abnormalities in patients with NF1. (+info)Aberrant cutaneous expression of the angiogenic factor midkine is associated with neurofibromatosis type-1. (8/767)
Neurofibromatosis type 1 is a common autosomal dominant disorder (incidence 1:3500) characterized by lesions that include neural crest derivatives such as Schwann cells and melanocytes. A critical event in the pathogenesis of neurofibromatosis type 1 is the heterozygous germ-line loss of the tumor suppressor gene NF1. Additional genetic and/or epigenetic events have been posited, including various alterations in growth factor expression. By in situ hybridization and immunohistochemistry, we demonstrate aberrant expression of the angiogenic and tumorigenic growth factor midkine in the skin of patients with neurofibromatosis type 1, but not normal individuals. We demonstrate that midkine expression is independent of the presence of neurofibromas, and thus appears to be associated with mutations in the NF1 gene. Furthermore, midkine-containing culture media is shown to stimulate the growth of human endothelial and neurofibroma-derived cells. In conclusion, we introduce the skin as a source of dysregulated growth factors in neurofibromatosis type 1, and suggest the further study of the angiogenic factor midkine in neurofibromatosis type 1 pathogenesis. (+info)Neurofibromatosis 1 (NF1) is a genetic disorder that affects the development and growth of nerve tissue. It's also known as von Recklinghausen disease. NF1 is characterized by the growth of non-cancerous tumors on the nerves, as well as skin and bone abnormalities.
The symptoms of Neurofibromatosis 1 can vary widely, even among members of the same family. Some common features include:
* Multiple café au lait spots (flat, light brown patches on the skin)
* Freckles in the underarms and groin area
* Benign growths on or under the skin called neurofibromas
* Larger, more complex tumors called plexiform neurofibromas
* Optic gliomas (tumors that form on the optic nerve)
* Distinctive bone abnormalities, such as a curved spine (scoliosis) or an enlarged head (macrocephaly)
* Learning disabilities and behavioral problems
Neurofibromatosis 1 is caused by mutations in the NF1 gene, which provides instructions for making a protein called neurofibromin. This protein helps regulate cell growth and division. When the NF1 gene is mutated, the production of neurofibromin is reduced or absent, leading to uncontrolled cell growth and the development of tumors.
NF1 is an autosomal dominant disorder, which means that a person has a 50% chance of inheriting the mutated gene from an affected parent. However, about half of all cases are the result of new mutations in the NF1 gene, and occur in people with no family history of the disorder.
There is currently no cure for Neurofibromatosis 1, but treatments are available to manage the symptoms and complications of the disease. These may include medications to control pain or reduce the size of tumors, surgery to remove tumors or correct bone abnormalities, and physical therapy to improve mobility and strength. Regular monitoring by a healthcare team experienced in treating Neurofibromatosis 1 is also important to detect any changes in the condition and provide appropriate care.
Neurofibromatosis 2 (NF2) is a genetic disorder characterized by the development of non-cancerous tumors in the nervous system, particularly on the nerves related to hearing and balance. It's also known as central neurofibromatosis or bilateral acoustic neuroma syndrome.
The primary feature of NF2 is the growth of schwannomas, which are tumors that develop from the cells surrounding nerve fibers. These typically grow on the vestibular nerve, leading to hearing loss, ringing in the ears (tinnitus), and balance problems. Bilateral acoustic neuromas (schwannomas affecting both vestibular nerves) are a hallmark of this condition.
Other common features include:
1. Meningiomas: These are tumors that grow in the meninges, the protective layers surrounding the brain and spinal cord.
2. Ependymomas: These are tumors that develop from the ependymal cells lining the ventricles (fluid-filled spaces) in the brain or the spinal cord canal.
3. Neurofibromas: Unlike in Neurofibromatosis type 1, these are less common and typically don't become cancerous.
4. Skin changes: While not as prevalent as in NF1, some people with NF2 may have skin freckles, café-au-lait spots, or skin tumors.
5. Eye problems: Some individuals may experience cataracts, retinal abnormalities, or optic nerve tumors (optic gliomas).
6. Other potential symptoms: Headaches, facial weakness or numbness, and difficulty swallowing or speaking.
NF2 is an autosomal dominant disorder, meaning that a person has a 50% chance of inheriting the condition if one of their parents has it. However, about half of all NF2 cases result from spontaneous genetic mutations with no family history of the disorder.
Neurofibromatosis 1 (NF1) is a genetic disorder caused by mutations in the NF1 gene, which is located on chromosome 17 and encodes the protein neurofibromin. Neurofibromin is a tumor suppressor protein that regulates cell growth and differentiation.
The NF1 gene mutation leads to the development of benign (non-cancerous) tumors on nerves and skin, called neurofibromas, as well as other clinical features such as café-au-lait spots (light brown patches on the skin), freckling in the axillary or inguinal regions, Lisch nodules (harmless growths on the iris of the eye), and skeletal abnormalities.
Neurofibromatosis 1 is an autosomal dominant disorder, which means that a person has a 50% chance of inheriting the mutated gene from an affected parent. However, up to 50% of cases result from new mutations in the NF1 gene and occur in people with no family history of the condition.
The clinical manifestations of Neurofibromatosis 1 can vary widely among individuals, even within the same family. The diagnosis is typically made based on clinical criteria established by the National Institutes of Health (NIH). Treatment is generally focused on managing symptoms and addressing complications as they arise, although surgery may be necessary to remove large or symptomatic tumors.
Neurofibromatosis 2 (NF2) is a genetic disorder characterized by the development of non-cancerous tumors in the nervous system. It is caused by mutations in the NF2 gene, which provides instructions for making a protein called merlin or schwannomin. This protein helps regulate cell growth and plays a role in suppressing tumor formation.
In NF2, the lack of functional merlin protein leads to an increased risk of developing tumors on the nerves related to hearing and balance (vestibular schwannomas or acoustic neuromas), on the spine (schwannomas), and on the brain (meningiomas). These tumors can cause various symptoms, such as hearing loss, ringing in the ears, balance problems, numbness or weakness in the limbs, and visual changes.
NF2 is an autosomal dominant disorder, meaning that a person has a 50% chance of inheriting the mutated gene from an affected parent and developing the condition. However, about half of all NF2 cases result from new mutations in the NF2 gene, with no family history of the disorder.
Neurofibromatoses are a group of genetic disorders that primarily affect the nervous system. The term "neurofibromatosis" is often used to refer to two specific conditions: neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). These conditions are characterized by the growth of tumors on the nerves, called neurofibromas.
Neurofibromatosis type 1 (NF1): This is the most common form of neurofibromatosis, affecting about 1 in every 3,000 people worldwide. NF1 is caused by mutations in the NF1 gene and is characterized by the development of benign tumors on the nerves called neurofibromas. These tumors can develop anywhere on the body, including the skin, spinal cord, and brain. Other common features of NF1 include:
* Freckles in the underarms and groin area
* Lisch nodules (small, noncancerous growths) on the iris of the eye
* Bone abnormalities, such as scoliosis or bowing of the legs
* Learning disabilities or cognitive impairment
Neurofibromatosis type 2 (NF2): This form of neurofibromatosis is much rarer than NF1, affecting about 1 in every 30,000 people worldwide. NF2 is caused by mutations in the NF2 gene and is characterized by the development of benign tumors on the nerves that transmit sound from the inner ear to the brain (acoustic neuromas). These tumors can cause hearing loss, ringing in the ears, and balance problems. Other common features of NF2 include:
* Multiple schwannomas (tumors that develop on the protective covering of the nerves)
* Meningiomas (tumors that develop in the membranes surrounding the brain and spinal cord)
* Skin tumors called neurofibromas, although these are less common than in NF1
It is important to note that while neurofibromatoses can cause a range of symptoms and complications, most people with these conditions have a normal lifespan. With proper medical care and monitoring, it is possible to manage the symptoms and reduce the risk of complications.
Neurofibromin 1 is a protein that is encoded by the NF1 gene in humans. Neurofibromin 1 acts as a tumor suppressor, helping to regulate cell growth and division. It plays an important role in the nervous system, where it helps to control the development and function of nerve cells. Mutations in the NF1 gene can lead to neurofibromatosis type 1 (NF1), a genetic disorder characterized by the growth of non-cancerous tumors on the nerves (neurofibromas) and other symptoms.
Neurofibromin 2 is not a medical term itself, but Neurofibromin 1 and Neurofibromin 2 are related to a genetic disorder called Neurofibromatosis. Neurofibromin 1 is the correct term, which is a protein encoded by the NF1 gene in humans.
Neurofibromin 1 is a tumor suppressor protein that plays a crucial role in regulating cell growth and differentiation. Mutations in the NF1 gene can lead to Neurofibromatosis type 1 (NF1), a genetic disorder characterized by the development of benign tumors on the nerves, skin, and other parts of the body.
Neurofibromin 2, on the other hand, is not a recognized term in medical literature. It is possible that there is some confusion with Neurofibromatosis type 2 (NF2), which is a separate genetic disorder caused by mutations in the NF2 gene. The NF2 gene encodes a protein called Merlin, which also functions as a tumor suppressor and helps regulate cell growth and division.
Therefore, it is essential to clarify whether you are asking about Neurofibromin 1 or Neurofibromatosis type 2 when using the term "Neurofibromin 2."
A plexiform neurofibroma is a type of neurofibroma, which is a benign tumor that develops from the nerve sheath. In a plexiform neurofibroma, the tumor grows along the nerves and can involve multiple fascicles, leading to a large, diffuse mass. These tumors can occur anywhere in the body but are most commonly found in the head, neck, and trunk.
Plexiform neurofibromas can be associated with neurofibromatosis type 1 (NF1), a genetic disorder that affects approximately 1 in every 3,000 people worldwide. In individuals with NF1, plexiform neurofibromas can cause significant morbidity, including disfigurement, pain, and functional impairment. Additionally, there is a small risk of malignant transformation into a type of cancer called malignant peripheral nerve sheath tumor (MPNST).
The diagnosis of plexiform neurofibromas is typically made based on clinical examination, medical history, and imaging studies such as MRI. A biopsy may be necessary to confirm the diagnosis. Treatment options for plexiform neurofibromas include surgery, radiation therapy, and medication. The choice of treatment depends on several factors, including the size and location of the tumor, the presence of symptoms, and the risk of malignant transformation.
A neurofibroma is a benign (non-cancerous) tumor that develops from the nerve sheath, which is the protective covering around nerves. These tumors can grow anywhere on the body and can be found under the skin or deep inside the body. Neurofibromas can vary in size, and they may cause symptoms such as pain, numbness, or tingling if they press on nearby nerves.
Neurofibromas are a common feature of neurofibromatosis type 1 (NF1), a genetic disorder that affects approximately 1 in every 3,000 people worldwide. NF1 is characterized by the development of multiple neurofibromas and other tumors, as well as skin changes such as café-au-lait spots and freckling.
It's important to note that while most neurofibromas are benign, they can rarely undergo malignant transformation and become cancerous. If you have a neurofibroma or are concerned about your risk of developing one, it's important to seek medical advice from a healthcare professional who is familiar with this condition.
Café-au-lait spots are light to dark brown, flat patches on the skin that are benign and usually harmless. The term "café-au-lait" means "coffee with milk," which describes the color of these spots. They can vary in size from a few millimeters to several centimeters in diameter and can appear anywhere on the body, although they are most commonly found on the trunk and buttocks.
While café-au-lait spots are common and can occur in up to 20% of the general population, having multiple (more than six) such spots, especially if they are large or present at birth, may be a sign of an underlying medical condition, such as neurofibromatosis type 1 (NF1), a genetic disorder that affects the growth and development of nerve tissue.
Therefore, it is essential to monitor café-au-lait spots and report any changes or concerns to a healthcare provider.
An Optic Nerve Glioma is a type of brain tumor that arises from the glial cells (supportive tissue) within the optic nerve. It is most commonly seen in children, particularly those with neurofibromatosis type 1 (NF1). These tumors are typically slow-growing and may not cause any symptoms, especially if they are small. However, as they grow larger, they can put pressure on the optic nerve, leading to vision loss or other visual disturbances. In some cases, these tumors can also affect nearby structures in the brain, causing additional neurological symptoms. Treatment options may include observation, chemotherapy, radiation therapy, or surgery, depending on the size and location of the tumor, as well as the patient's age and overall health.
Nerve sheath neoplasms are a group of tumors that arise from the cells surrounding and supporting the nerves. These tumors can be benign or malignant and include schwannomas, neurofibromas, and malignant peripheral nerve sheath tumors (MPNSTs). Schwannomas develop from the Schwann cells that produce the myelin sheath of the nerve, while neurofibromas arise from the nerve's supporting cells called fibroblasts. MPNSTs are cancerous tumors that can grow rapidly and invade surrounding tissues. Nerve sheath neoplasms can cause various symptoms depending on their location and size, including pain, numbness, weakness, or paralysis in the affected area.
A neurilemmoma, also known as schwannoma or peripheral nerve sheath tumor, is a benign, slow-growing tumor that arises from the Schwann cells, which produce the myelin sheath that surrounds and insulates peripheral nerves. These tumors can occur anywhere along the course of a peripheral nerve, but they most commonly affect the acoustic nerve (vestibulocochlear nerve), leading to a type of tumor called vestibular schwannoma or acoustic neuroma. Neurilemmomas are typically encapsulated and do not invade the surrounding tissue, although larger ones may cause pressure-related symptoms due to compression of nearby structures. Rarely, these tumors can undergo malignant transformation, leading to a condition called malignant peripheral nerve sheath tumor or neurofibrosarcoma.
Optic nerve neoplasms refer to abnormal growths or tumors that develop within or near the optic nerve. These tumors can be benign (non-cancerous) or malignant (cancerous).
Benign optic nerve neoplasms include optic nerve meningiomas and schwannomas, which originate from the sheaths surrounding the optic nerve. They usually grow slowly and may not cause significant vision loss, but they can lead to compression of the optic nerve, resulting in visual field defects or optic disc swelling (papilledema).
Malignant optic nerve neoplasms are rare but more aggressive. The most common type is optic nerve glioma, which arises from the glial cells within the optic nerve. These tumors can quickly damage the optic nerve and cause severe vision loss.
It's important to note that any optic nerve neoplasm requires prompt medical evaluation and treatment, as they can potentially lead to significant visual impairment or even blindness if left untreated.
An acoustic neuroma, also known as vestibular schwannoma, is not actually a neuroma but rather a benign (noncancerous) tumor that develops on the vestibular nerve. This nerve is one of the two nerves that transmit sound and balance information from the inner ear to the brain. The tumor arises from an overproduction of Schwann cells, which normally provide a protective covering for the nerve fibers. As the tumor grows, it can press against the hearing and balance nerves, causing symptoms such as hearing loss, ringing in the ear (tinnitus), unsteadiness, and disequilibrium. In some cases, acoustic neuromas can become quite large and cause additional symptoms by pressing on nearby cranial nerves. Treatment options include observation, radiation therapy, or surgical removal of the tumor.
Peripheral nervous system (PNS) neoplasms refer to tumors that originate in the peripheral nerves, which are the nerves outside the brain and spinal cord. These tumors can be benign or malignant (cancerous). Benign tumors, such as schwannomas and neurofibromas, grow slowly and do not spread to other parts of the body. Malignant tumors, such as malignant peripheral nerve sheath tumors (MPNSTs), can invade nearby tissues and may metastasize (spread) to other organs.
PNS neoplasms can cause various symptoms depending on their location and size. Common symptoms include pain, weakness, numbness, or tingling in the affected area. In some cases, PNS neoplasms may not cause any symptoms until they become quite large. Treatment options for PNS neoplasms depend on several factors, including the type, size, and location of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Neurofibrosarcoma is a rare type of soft tissue sarcoma, which is a cancer that develops in the soft tissues of the body such as fat, muscle, tendons, blood vessels, and nerves. Neurofibrosarcoma specifically arises from the nerve sheath cells, also known as the Schwann cells, that cover and protect the peripheral nerves.
This type of cancer typically forms a painless mass or tumor in the affected area, which can grow and invade nearby tissues and organs over time. Neurofibrosarcoma can occur anywhere in the body but is most commonly found in the arms, legs, trunk, or head and neck region.
Neurofibrosarcoma can be classified into two main types: conventional and malignant peripheral nerve sheath tumor (MPNST). Conventional neurofibrosarcoma is more common and tends to occur in older adults, while MPNST is a more aggressive form that is associated with genetic disorders such as neurofibromatosis type 1.
Treatment for neurofibrosarcoma typically involves surgical removal of the tumor, along with radiation therapy and/or chemotherapy to help prevent recurrence and spread of the cancer. The prognosis for neurofibrosarcoma varies depending on several factors, including the size and location of the tumor, the patient's age and overall health, and the stage of the disease at diagnosis.
Cranial nerve neoplasms refer to abnormal growths or tumors that develop within or near the cranial nerves. These nerves are responsible for transmitting sensory and motor information between the brain and various parts of the head, neck, and trunk. There are 12 pairs of cranial nerves, each with a specific function and location in the skull.
Cranial nerve neoplasms can be benign or malignant and may arise from the nerve itself (schwannoma, neurofibroma) or from surrounding tissues that invade the nerve (meningioma, epidermoid cyst). The growth of these tumors can cause various symptoms depending on their size, location, and rate of growth. Common symptoms include:
* Facial weakness or numbness
* Double vision or other visual disturbances
* Hearing loss or tinnitus (ringing in the ears)
* Difficulty swallowing or speaking
* Loss of smell or taste
* Uncontrollable eye movements or drooping eyelids
Treatment for cranial nerve neoplasms depends on several factors, including the type, size, location, and extent of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. Regular follow-up care is essential to monitor for recurrence or complications.
Nervous system neoplasms are abnormal growths or tumors that occur within the nervous system, which includes the brain, spinal cord, and peripheral nerves. These tumors can be benign (non-cancerous) or malignant (cancerous), and their growth can compress or infiltrate surrounding tissues, leading to various neurological symptoms. The causes of nervous system neoplasms are not fully understood but may involve genetic factors, exposure to certain chemicals or radiation, and certain viral infections. Treatment options depend on the type, location, and size of the tumor and can include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
A meningioma is a type of slow-growing tumor that forms on the membranes (meninges) surrounding the brain and spinal cord. It's usually benign, meaning it doesn't spread to other parts of the body, but it can still cause serious problems if it grows and presses on nearby tissues.
Meningiomas most commonly occur in adults, and are more common in women than men. They can cause various symptoms depending on their location and size, including headaches, seizures, vision or hearing problems, memory loss, and changes in personality or behavior. In some cases, they may not cause any symptoms at all and are discovered only during imaging tests for other conditions.
Treatment options for meningiomas include monitoring with regular imaging scans, surgery to remove the tumor, and radiation therapy to shrink or kill the tumor cells. The best treatment approach depends on factors such as the size and location of the tumor, the patient's age and overall health, and their personal preferences.
An auditory brainstem implant (ABI) is a surgically placed device that provides a sense of sound to individuals who have severe hearing loss and cannot benefit from cochlear implants. Unlike cochlear implants, which stimulate the auditory nerve directly, ABIs transmit electrical signals directly to the brainstem, bypassing the inner ear entirely.
The ABI consists of a microphone, processor, and a series of electrodes that are surgically placed on the surface of the brainstem. The microphone picks up sounds from the environment, and the processor converts them into electrical signals. These signals are then sent to the electrodes, which stimulate the nearby nerve cells in the brainstem.
The brain interprets these stimuli as sound, allowing the individual to perceive some level of hearing. While ABIs do not provide the same level of hearing as cochlear implants, they can help individuals with profound hearing loss to communicate more effectively and improve their quality of life.
It's important to note that ABIs are typically reserved for individuals who have severe hearing loss due to damage to the inner ear or auditory nerve, and who are not candidates for cochlear implants. The procedure is complex and carries risks, so it is only recommended in cases where the potential benefits outweigh the risks.
A meningocele is a type of neural tube defect that results in the herniation of the meninges (the protective membranes covering the brain and spinal cord) through a defect in the vertebral column. The meninges protrude as a sac-like structure, which may be covered by skin or a thin layer of tissue. Meningoceles usually do not contain neural tissue, but cerebrospinal fluid is present within the sac. They are typically asymptomatic unless there is compression of surrounding structures or infection. Treatment generally involves surgical repair to prevent potential complications such as meningitis or neurological damage.
A hamartoma is a benign tumor-like growth that is composed of an unusual mixture of cells and tissues that are normally found in the affected area. These growths can occur anywhere in the body, but they are most commonly found in the skin, lungs, and brain. Hamartomas are typically slow growing and do not spread to other parts of the body (metastasize). They are usually harmless, but in some cases, they may cause symptoms or complications depending on their size and location. In general, hamartomas do not require treatment unless they are causing problems.
Pseudarthrosis is a medical term that refers to a false joint or a nonunion of bones, meaning that the broken bone ends do not heal properly and continue to move at the fracture site. This condition can cause pain, instability, and deformity in the affected limb. It may require additional treatment such as surgery to promote bone healing and stabilization.
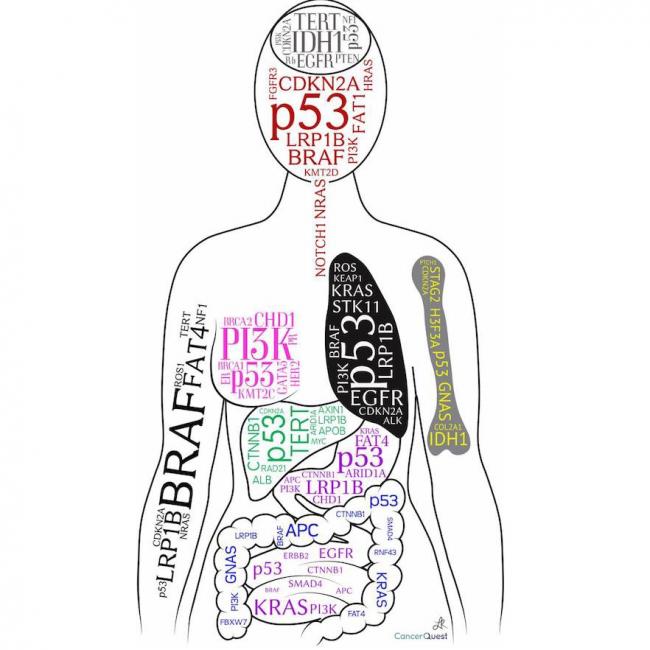
Human chromosome pair 17 consists of two rod-shaped structures present in the nucleus of each human cell. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex called chromatin. Chromosomes carry genetic information in the form of genes, which are segments of DNA that contain instructions for the development and function of an organism.
Human cells typically have 23 pairs of chromosomes, for a total of 46 chromosomes. Pair 17 is one of the autosomal pairs, meaning it is not a sex chromosome (X or Y). Chromosome 17 is a medium-sized chromosome and contains an estimated 800 million base pairs of DNA. It contains approximately 1,500 genes that provide instructions for making proteins and regulating various cellular processes.
Chromosome 17 is associated with several genetic disorders, including inherited cancer syndromes such as Li-Fraumeni syndrome and hereditary nonpolyposis colorectal cancer (HNPCC). Mutations in genes located on chromosome 17 can increase the risk of developing various types of cancer, including breast, ovarian, colon, and pancreatic cancer.
Schwann cells, also known as neurolemmocytes, are a type of glial cell that form the myelin sheath around peripheral nervous system (PNS) axons, allowing for the rapid and efficient transmission of nerve impulses. These cells play a crucial role in the maintenance and function of the PNS.
Schwann cells originate from the neural crest during embryonic development and migrate to the developing nerves. They wrap around the axons in a spiral fashion, forming multiple layers of myelin, which insulates the nerve fibers and increases the speed of electrical impulse transmission. Each Schwann cell is responsible for myelinating a single segment of an axon, with the gaps between these segments called nodes of Ranvier.
Schwann cells also provide structural support to the neurons and contribute to the regeneration of injured peripheral nerves by helping to guide the regrowth of axons to their targets. Additionally, Schwann cells can participate in immune responses within the PNS, such as releasing cytokines and chemokines to recruit immune cells during injury or infection.
A learning disorder is a neurodevelopmental disorder that affects an individual's ability to acquire, process, and use information in one or more academic areas despite normal intelligence and adequate instruction. It can manifest as difficulties with reading (dyslexia), writing (dysgraphia), mathematics (dyscalculia), or other academic skills. Learning disorders are not the result of low intelligence, lack of motivation, or environmental factors alone, but rather reflect a significant discrepancy between an individual's cognitive abilities and their academic achievement. They can significantly impact a person's ability to perform in school, at work, and in daily life, making it important to diagnose and manage these disorders effectively.
Noonan Syndrome is a genetic disorder that affects various parts of the body and is characterized by distinctive facial features, short stature, heart defects, and developmental delays. It is caused by mutations in genes responsible for regulating cell growth and division. The syndrome is often identified at birth or in early childhood due to its physical manifestations, which may include widely spaced eyes, low-set ears, a short neck, a broad or webbed neck, chest deformities, and pulmonary valve stenosis. Noonan Syndrome affects both sexes and all races equally, with an estimated prevalence of 1 in 1,000 to 1 in 2,500 live births.
Facial neoplasms refer to abnormal growths or tumors that develop in the tissues of the face. These growths can be benign (non-cancerous) or malignant (cancerous). Facial neoplasms can occur in any of the facial structures, including the skin, muscles, bones, nerves, and glands.
Benign facial neoplasms are typically slow-growing and do not spread to other parts of the body. Examples include papillomas, hemangiomas, and neurofibromas. While these tumors are usually harmless, they can cause cosmetic concerns or interfere with normal facial function.
Malignant facial neoplasms, on the other hand, can be aggressive and invasive. They can spread to other parts of the face, as well as to distant sites in the body. Common types of malignant facial neoplasms include basal cell carcinoma, squamous cell carcinoma, and melanoma.
Treatment for facial neoplasms depends on several factors, including the type, size, location, and stage of the tumor. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. It is important to seek medical attention promptly if you notice any unusual growths or changes in the skin or tissues of your face.
Mosaicism, in the context of genetics and medicine, refers to the presence of two or more cell lines with different genetic compositions in an individual who has developed from a single fertilized egg. This means that some cells have one genetic makeup, while others have a different genetic makeup. This condition can occur due to various reasons such as errors during cell division after fertilization.
Mosaicism can involve chromosomes (where whole or parts of chromosomes are present in some cells but not in others) or it can involve single genes (where a particular gene is present in one form in some cells and a different form in others). The symptoms and severity of mosaicism can vary widely, depending on the type and location of the genetic difference and the proportion of cells that are affected. Some individuals with mosaicism may not experience any noticeable effects, while others may have significant health problems.
Developmental bone diseases are a group of medical conditions that affect the growth and development of bones. These diseases are present at birth or develop during childhood and adolescence, when bones are growing rapidly. They can result from genetic mutations, hormonal imbalances, or environmental factors such as poor nutrition.
Some examples of developmental bone diseases include:
1. Osteogenesis imperfecta (OI): Also known as brittle bone disease, OI is a genetic disorder that affects the body's production of collagen, a protein necessary for healthy bones. People with OI have fragile bones that break easily and may also experience other symptoms such as blue sclerae (whites of the eyes), hearing loss, and joint laxity.
2. Achondroplasia: This is the most common form of dwarfism, caused by a genetic mutation that affects bone growth. People with achondroplasia have short limbs and a large head relative to their body size.
3. Rickets: A condition caused by vitamin D deficiency or an inability to absorb or use vitamin D properly. This leads to weak, soft bones that can bow or bend easily, particularly in children.
4. Fibrous dysplasia: A rare bone disorder where normal bone is replaced with fibrous tissue, leading to weakened bones and deformities.
5. Scoliosis: An abnormal curvature of the spine that can develop during childhood or adolescence. While not strictly a developmental bone disease, scoliosis can be caused by various underlying conditions such as cerebral palsy, muscular dystrophy, or spina bifida.
Treatment for developmental bone diseases varies depending on the specific condition and its severity. Treatment may include medication, physical therapy, bracing, or surgery to correct deformities and improve function. Regular follow-up with a healthcare provider is essential to monitor growth, manage symptoms, and prevent complications.
Human chromosome pair 22 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex structure called a chromatin.
Chromosome pair 22 is one of the 22 autosomal pairs of human chromosomes, meaning they are not sex chromosomes (X or Y). Chromosome 22 is the second smallest human chromosome, with each arm of the chromosome designated as p and q. The short arm is labeled "p," and the long arm is labeled "q."
Chromosome 22 contains several genes that are associated with various genetic disorders, including DiGeorge syndrome, velocardiofacial syndrome, and cat-eye syndrome, which result from deletions or duplications of specific regions on the chromosome. Additionally, chromosome 22 is the location of the NRXN1 gene, which has been associated with an increased risk for autism spectrum disorder (ASD) and schizophrenia when deleted or disrupted.
Understanding the genetic makeup of human chromosome pair 22 can provide valuable insights into human genetics, evolution, and disease susceptibility, as well as inform medical diagnoses, treatments, and research.
Multiple primary neoplasms refer to the occurrence of more than one primary malignant tumor in an individual, where each tumor is unrelated to the other and originates from separate cells or organs. This differs from metastatic cancer, where a single malignancy spreads to multiple sites in the body. Multiple primary neoplasms can be synchronous (occurring at the same time) or metachronous (occurring at different times). The risk of developing multiple primary neoplasms increases with age and is associated with certain genetic predispositions, environmental factors, and lifestyle choices such as smoking and alcohol consumption.
Piebaldism is a rare genetic disorder characterized by the presence of white patches of skin and hair due to a lack of melanin, the pigment that gives color to skin, hair, and eyes. These patches are present from birth and typically involve the forehead, chin, and midline of the body. The condition is caused by mutations in the KIT or SLC45A2 genes and is usually inherited in an autosomal dominant pattern, meaning only one copy of the altered gene is needed to cause the disorder. Piebaldism is not harmful to a person's overall health, but it can have significant psychological effects due to its impact on appearance.
Pheochromocytoma is a rare type of tumor that develops in the adrenal glands, which are triangular-shaped glands located on top of each kidney. These tumors produce excessive amounts of hormones called catecholamines, including adrenaline and noradrenaline. This can lead to a variety of symptoms such as high blood pressure, sweating, headaches, rapid heartbeat, and anxiety.
Pheochromocytomas are typically slow-growing and can be benign or malignant (cancerous). While the exact cause of these tumors is not always known, some genetic factors have been identified that may increase a person's risk. Treatment usually involves surgical removal of the tumor, along with medications to manage symptoms and control blood pressure before and after surgery.
Medical Definition:
Magnetic Resonance Imaging (MRI) is a non-invasive diagnostic imaging technique that uses a strong magnetic field and radio waves to create detailed cross-sectional or three-dimensional images of the internal structures of the body. The patient lies within a large, cylindrical magnet, and the scanner detects changes in the direction of the magnetic field caused by protons in the body. These changes are then converted into detailed images that help medical professionals to diagnose and monitor various medical conditions, such as tumors, injuries, or diseases affecting the brain, spinal cord, heart, blood vessels, joints, and other internal organs. MRI does not use radiation like computed tomography (CT) scans.
Meningeal neoplasms, also known as malignant meningitis or leptomeningeal carcinomatosis, refer to cancerous tumors that originate in the meninges, which are the membranes covering the brain and spinal cord. These tumors can arise primarily from the meningeal cells themselves, although they more commonly result from the spread (metastasis) of cancer cells from other parts of the body, such as breast, lung, or melanoma.
Meningeal neoplasms can cause a variety of symptoms, including headaches, nausea and vomiting, mental status changes, seizures, and focal neurological deficits. Diagnosis typically involves imaging studies (such as MRI) and analysis of cerebrospinal fluid obtained through a spinal tap. Treatment options may include radiation therapy, chemotherapy, or surgery, depending on the type and extent of the tumor. The prognosis for patients with meningeal neoplasms is generally poor, with a median survival time of several months to a year.
Somatostatinoma is a rare type of neuroendocrine tumor that originates from the delta cells (D cells) of the diffuse endocrine system, which are responsible for producing and secreting somatostatin, a hormone that inhibits the release of several other hormones. These tumors can occur in various organs, but they most commonly arise in the pancreas and the small intestine (duodenum).
Somatostatinomas are typically slow-growing and can be functional or nonfunctional. Functional somatostatinomas actively produce and secrete excessive amounts of somatostatin, which can lead to a variety of clinical symptoms due to the inhibition of other hormones' functions. Nonfunctional somatostatinomas do not secrete significant amounts of somatostatin and are often discovered incidentally during imaging studies or when they cause local mass effects.
Common symptoms associated with functional somatostatinomas include diarrhea, abdominal pain, weight loss, fat malabsorption, and steatorrhea (fatty stools). They can also lead to diabetes mellitus due to the inhibition of insulin secretion. Additionally, these tumors may cause symptoms related to hormone deficiencies or the compression of nearby structures, depending on their location.
Diagnosis typically involves imaging studies such as CT scans, MRI, and PET scans, along with biochemical tests to measure somatostatin levels in the blood. A definitive diagnosis usually requires a tissue biopsy or surgical removal of the tumor for histopathological examination. Treatment options include surgery, chemotherapy, radiation therapy, and targeted therapies, depending on the stage and location of the tumor.
Pathologic decalcification is a process that occurs when there is a loss of calcium salts from the bones or teeth. This can lead to weakening and structural damage in the affected area. It is often seen in conditions such as osteoporosis, Paget's disease, and tumors that involve bone. In dental contexts, decalcification can also refer to the loss of minerals from tooth enamel, which can lead to cavities and tooth decay. This is often caused by poor oral hygiene and a diet high in sugars.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Pigmentation disorders are conditions that affect the production or distribution of melanin, the pigment responsible for the color of skin, hair, and eyes. These disorders can cause changes in the color of the skin, resulting in areas that are darker (hyperpigmentation) or lighter (hypopigmentation) than normal. Examples of pigmentation disorders include melasma, age spots, albinism, and vitiligo. The causes, symptoms, and treatments for these conditions can vary widely, so it is important to consult a healthcare provider for an accurate diagnosis and treatment plan.
A Glomus tumor is a rare, benign (non-cancerous) neoplasm that arises from the glomus body, a specialized form of blood vessel found in the skin, particularly in the fingers and toes. These tumors are highly vascular and usually appear as small, blue or red nodules just beneath the nail bed or on the fingertips. They can also occur in other parts of the body such as the stomach, lung, and kidney, but these locations are much less common.
Glomus tumors typically present with symptoms like severe pain, especially when exposed to cold temperatures or pressure. The pain is often described as sharp, stabbing, or throbbing, and it can be debilitating for some individuals. Diagnosis of glomus tumors usually involves a physical examination, imaging studies such as MRI or CT scans, and sometimes biopsy. Treatment options include surgical excision, which is often curative, and in some cases, embolization or sclerotherapy may be used to reduce the blood flow to the tumor before surgery.
The optic chiasm is a structure in the brain where the optic nerves from each eye meet and cross. This allows for the integration of visual information from both eyes into the brain's visual cortex, creating a single, combined image of the visual world. The optic chiasm plays an important role in the processing of visual information and helps to facilitate depth perception and other complex visual tasks. Damage to the optic chiasm can result in various visual field deficits, such as bitemporal hemianopsia, where there is a loss of vision in the outer halves (temporal fields) of both eyes' visual fields.
Adrenal gland neoplasms refer to abnormal growths or tumors in the adrenal glands. These glands are located on top of each kidney and are responsible for producing hormones that regulate various bodily functions such as metabolism, blood pressure, and stress response. Adrenal gland neoplasms can be benign (non-cancerous) or malignant (cancerous).
Benign adrenal tumors are called adenomas and are usually small and asymptomatic. However, some adenomas may produce excessive amounts of hormones, leading to symptoms such as high blood pressure, weight gain, and mood changes.
Malignant adrenal tumors are called adrenocortical carcinomas and are rare but aggressive cancers that can spread to other parts of the body. Symptoms of adrenocortical carcinoma may include abdominal pain, weight loss, and hormonal imbalances.
It is important to diagnose and treat adrenal gland neoplasms early to prevent complications and improve outcomes. Diagnostic tests may include imaging studies such as CT scans or MRIs, as well as hormone level testing and biopsy. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Skin neoplasms refer to abnormal growths or tumors in the skin that can be benign (non-cancerous) or malignant (cancerous). They result from uncontrolled multiplication of skin cells, which can form various types of lesions. These growths may appear as lumps, bumps, sores, patches, or discolored areas on the skin.
Benign skin neoplasms include conditions such as moles, warts, and seborrheic keratoses, while malignant skin neoplasms are primarily classified into melanoma, squamous cell carcinoma, and basal cell carcinoma. These three types of cancerous skin growths are collectively known as non-melanoma skin cancers (NMSCs). Melanoma is the most aggressive and dangerous form of skin cancer, while NMSCs tend to be less invasive but more common.
It's essential to monitor any changes in existing skin lesions or the appearance of new growths and consult a healthcare professional for proper evaluation and treatment if needed.
Thoracic neoplasms refer to abnormal growths or tumors that develop in the thorax, which is the area of the body that includes the chest and lungs. These neoplasms can be benign (non-cancerous) or malignant (cancerous). Malignant thoracic neoplasms are often referred to as lung cancer, but they can also include other types of cancer such as mesothelioma, thymoma, and esophageal cancer.
Thoracic neoplasms can cause various symptoms depending on their location and size. Common symptoms include coughing, chest pain, shortness of breath, hoarseness, and difficulty swallowing. Treatment options for thoracic neoplasms depend on the type, stage, and location of the tumor, as well as the patient's overall health. Treatment may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
A "mixed tumor, malignant" is a rare and aggressive type of cancer that contains two or more different types of malignant tissue. It is also known as a "malignant mixed Mullerian tumor" (MMMT) or "carcinosarcoma." This type of tumor most commonly arises in the female reproductive organs, such as the uterus or ovaries, but can also occur in other parts of the body.
The malignant mixed Mullerian tumor is composed of both epithelial and mesenchymal components, which are two different types of tissue. The epithelial component is made up of cancerous glandular or squamous cells, while the mesenchymal component consists of cancerous connective tissue, such as muscle, fat, or bone.
Mixed tumors, malignant can be aggressive and have a high risk of recurrence and metastasis. Treatment typically involves surgical removal of the tumor, followed by radiation therapy and/or chemotherapy to kill any remaining cancer cells. The prognosis for mixed tumors, malignant varies depending on several factors, including the size and location of the tumor, the stage of the disease at diagnosis, and the patient's overall health.
Astrocytoma is a type of brain tumor that arises from astrocytes, which are star-shaped glial cells in the brain. These tumors can occur in various parts of the brain and can have different grades of malignancy, ranging from low-grade (I or II) to high-grade (III or IV). Low-grade astrocytomas tend to grow slowly and may not cause any symptoms for a long time, while high-grade astrocytomas are more aggressive and can grow quickly, causing neurological problems.
Symptoms of astrocytoma depend on the location and size of the tumor but may include headaches, seizures, weakness or numbness in the limbs, difficulty speaking or swallowing, changes in vision or behavior, and memory loss. Treatment options for astrocytomas include surgery, radiation therapy, chemotherapy, or a combination of these approaches. The prognosis for astrocytoma varies widely depending on the grade and location of the tumor, as well as the age and overall health of the patient.
Proteus Syndrome is a rare genetic disorder characterized by progressive overgrowth of skin, bones, muscles, and other tissues. It is caused by a mutation in the AKT1 gene, which regulates cell growth and division. The disorder is named after the Greek sea-god Proteus, who could change his shape at will, as people with this condition often have highly variable and asymmetric features.
The symptoms of Proteus Syndrome can vary widely from person to person, but may include:
1. Overgrowth of skin, which can lead to the formation of thickened, rough, or irregular areas of skin (known as "cerebriform" skin) and deep creases or folds.
2. Asymmetric overgrowth of bones, muscles, and other tissues, leading to differences in size and shape between the two sides of the body.
3. The formation of benign tumors (such as lipomas and lymphangiomas) and abnormal blood vessels.
4. Abnormalities of the brain, eyes, and other organs.
5. Increased risk of developing certain types of cancer.
Proteus Syndrome is typically diagnosed based on a combination of clinical features, medical imaging, and genetic testing. There is no cure for the disorder, but treatment is focused on managing symptoms and preventing complications. This may involve surgery to remove tumors or correct bone deformities, physical therapy to improve mobility and strength, and medications to control pain and other symptoms.
Juvenile Myelomonocytic Leukemia (JMML) is a rare and aggressive type of childhood leukemia, characterized by the overproduction of myeloid and monocytic white blood cells in the bone marrow. These cells accumulate in the bloodstream, leading to an increased risk of infection, anemia, and bleeding. JMML is different from other types of leukemia because it involves both the myeloid and monocytic cell lines, and it often affects younger children, typically those under 4 years old. The exact cause of JMML is not fully understood, but it has been linked to genetic mutations in certain genes, such as PTPN11, NRAS, KRAS, CBL, and NF1. Treatment for JMML usually involves a combination of chemotherapy, stem cell transplantation, and supportive care.
X-ray computed tomography (CT or CAT scan) is a medical imaging method that uses computer-processed combinations of many X-ray images taken from different angles to produce cross-sectional (tomographic) images (virtual "slices") of the body. These cross-sectional images can then be used to display detailed internal views of organs, bones, and soft tissues in the body.
The term "computed tomography" is used instead of "CT scan" or "CAT scan" because the machines take a series of X-ray measurements from different angles around the body and then use a computer to process these data to create detailed images of internal structures within the body.
CT scanning is a noninvasive, painless medical test that helps physicians diagnose and treat medical conditions. CT imaging provides detailed information about many types of tissue including lung, bone, soft tissue and blood vessels. CT examinations can be performed on every part of the body for a variety of reasons including diagnosis, surgical planning, and monitoring of therapeutic responses.
In computed tomography (CT), an X-ray source and detector rotate around the patient, measuring the X-ray attenuation at many different angles. A computer uses this data to construct a cross-sectional image by the process of reconstruction. This technique is called "tomography". The term "computed" refers to the use of a computer to reconstruct the images.
CT has become an important tool in medical imaging and diagnosis, allowing radiologists and other physicians to view detailed internal images of the body. It can help identify many different medical conditions including cancer, heart disease, lung nodules, liver tumors, and internal injuries from trauma. CT is also commonly used for guiding biopsies and other minimally invasive procedures.
In summary, X-ray computed tomography (CT or CAT scan) is a medical imaging technique that uses computer-processed combinations of many X-ray images taken from different angles to produce cross-sectional images of the body. It provides detailed internal views of organs, bones, and soft tissues in the body, allowing physicians to diagnose and treat medical conditions.
Iris neoplasms refer to abnormal growths or tumors that develop in the iris, which is the colored part of the eye. These neoplasms can be benign (non-cancerous) or malignant (cancerous). Benign iris neoplasms are typically slow-growing and do not spread to other parts of the body. Malignant iris neoplasms, on the other hand, can grow quickly and may spread to other parts of the eye or nearby structures, such as the ciliary body or choroid.
Iris neoplasms can cause various symptoms, including changes in the appearance of the eye, such as a visible mass or discoloration, pain, redness, light sensitivity, blurred vision, or changes in the size or shape of the pupil. The diagnosis of iris neoplasms typically involves a comprehensive eye examination, including a visual acuity test, refraction, slit-lamp examination, and sometimes imaging tests such as ultrasound or optical coherence tomography (OCT).
Treatment options for iris neoplasms depend on the type, size, location, and severity of the tumor. Small, benign iris neoplasms may not require treatment and can be monitored over time. Larger or malignant iris neoplasms may require surgical removal, radiation therapy, or other treatments to prevent complications or spread to other parts of the eye or body. It is essential to seek medical attention promptly if you experience any symptoms of iris neoplasms or notice any changes in your vision or the appearance of your eyes.
Angiomatosis is a medical term that refers to a benign condition characterized by the proliferation of blood vessels in various tissues and organs. It is typically composed of small, tangled blood vessels called capillaries, which can form clusters or networks. The condition can affect skin, internal organs, bones, and other tissues.
Angiomatosis is often asymptomatic and may be discovered incidentally during medical imaging or surgical procedures. In some cases, it may cause symptoms such as pain, swelling, or bleeding, depending on the location and extent of the lesions.
While angiomatosis is generally a benign condition, in rare cases, it can be associated with malignant tumors or other medical conditions. Treatment options for angiomatosis depend on the size, location, and symptoms of the lesions and may include observation, medication, or surgical removal.
The arachnoid is one of the three membranes that cover the brain and the spinal cord, known as the meninges. It is located between the dura mater (the outermost layer) and the pia mater (the innermost layer). The arachnoid is a thin, delicate membrane that is filled with cerebrospinal fluid, which provides protection and nutrition to the central nervous system.
The arachnoid has a spider-web like appearance, hence its name, and it is composed of several layers of collagen fibers and elastic tissue. It is highly vascularized, meaning that it contains many blood vessels, and it plays an important role in regulating the flow of cerebrospinal fluid around the brain and spinal cord.
In some cases, the arachnoid can become inflamed or irritated, leading to a condition called arachnoiditis. This can cause a range of symptoms, including pain, muscle weakness, and sensory changes, and it may require medical treatment to manage.
Genetic linkage is the phenomenon where two or more genetic loci (locations on a chromosome) tend to be inherited together because they are close to each other on the same chromosome. This occurs during the process of sexual reproduction, where homologous chromosomes pair up and exchange genetic material through a process called crossing over.
The closer two loci are to each other on a chromosome, the lower the probability that they will be separated by a crossover event. As a result, they are more likely to be inherited together and are said to be linked. The degree of linkage between two loci can be measured by their recombination frequency, which is the percentage of meiotic events in which a crossover occurs between them.
Linkage analysis is an important tool in genetic research, as it allows researchers to identify and map genes that are associated with specific traits or diseases. By analyzing patterns of linkage between markers (identifiable DNA sequences) and phenotypes (observable traits), researchers can infer the location of genes that contribute to those traits or diseases on chromosomes.
 Neurofibromatosis type I
Neurofibromatosis type I
Neurofibromatosis
Macrodystrophia lipomatosa
Proteus syndrome
Café au lait spot
Pheochromocytoma
Imatinib
Conditions comorbid to autism spectrum disorders
Noonan syndrome with multiple lentigines
Alcino J. Silva
Epilepsy
Friedrich Daniel von Recklinghausen
Crowe sign
Spinal tumor
Neurofibroma
Notching of the ribs
SPRED1
Phakomatosis
Ulisse Aldrovandi
Cro-Magnon rock shelter
Malignant peripheral nerve sheath tumor
Chiasmal syndrome
Santosh G. Honavar
Noonan syndrome
Legius syndrome
Watson syndrome
Microdeletion syndrome
Expressivity (genetics)
Megalencephaly
Schwannomatosis
 Neurofibromatosis Type 1: Practice Essentials, Background, Pathophysiology
Neurofibromatosis Type 1: Practice Essentials, Background, Pathophysiology
 Neurofibromatosis Type 1 | Denver Health
Neurofibromatosis Type 1 | Denver Health
 Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis
Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis
 Selumetinib Granted Orphan Drug Designation by the U.S. FDA for Neurofibromatosis Type 1 - Merck.com
Selumetinib Granted Orphan Drug Designation by the U.S. FDA for Neurofibromatosis Type 1 - Merck.com
 Modulation of the neurofibromatosis type 1 gene product, neurofibromin, during Schwann cell differentiation
Modulation of the neurofibromatosis type 1 gene product, neurofibromin, during Schwann cell differentiation
 Intraoral presentation of multiple malignant peripheral nerve sheath tumors associated with neurofibromatosis-1 - Amrita Vishwa...
Intraoral presentation of multiple malignant peripheral nerve sheath tumors associated with neurofibromatosis-1 - Amrita Vishwa...
A clinical study of type 1 neurofibromatosis in north west England | Journal of Medical Genetics
Brain structure and function in neurofibromatosis type 1: current concepts and future directions | Journal of Neurology,...
2013 ICD-9-CM Diagnosis Code 237.71 : Neurofibromatosis, type 1 [von recklinghausen's disease]
 Neurofibromatosis Type 1 and Legius Syndrome Panel, Sequencing and Deletion/Duplication | Test Fact Sheet
Neurofibromatosis Type 1 and Legius Syndrome Panel, Sequencing and Deletion/Duplication | Test Fact Sheet
Spinal and Paraspinal Plexiform Neurofibromas in Patients with Neurofibromatosis Type 1: A Novel Scoring System for...
Neurofibromatosis-1: Overview, Causes - Health32
HL-085 in Adults With Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas - CheckOrphan
 Neurofibromatosis | NF | MedlinePlus
Neurofibromatosis | NF | MedlinePlus
 Neurofibromatosis News, Articles and Research
Neurofibromatosis News, Articles and Research
 Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome.
Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome.
 Neurofibromatosis 1 and intracranial neoplasms of childhood | MedLink Neurology
Neurofibromatosis 1 and intracranial neoplasms of childhood | MedLink Neurology
Neurofibromatosis type I - Wikipedia
 Neurofibromatosis 1 :: CSHL DNA Learning Center
Neurofibromatosis 1 :: CSHL DNA Learning Center
 Understanding neurofibromatosis type 1 (NF1) - M4RD
Understanding neurofibromatosis type 1 (NF1) - M4RD
 New Research Tools Available in the Search for Therapies for Neurofibromatosis Type 1 Plexiform Neurofibromas - NTAP
New Research Tools Available in the Search for Therapies for Neurofibromatosis Type 1 Plexiform Neurofibromas - NTAP
 Mila's Story - pRVH PCOR Collaborative - Neurofibromatosis Type 1
Mila's Story - pRVH PCOR Collaborative - Neurofibromatosis Type 1
Idaho Medical Home Portal - Neurofibromatosis Type 1 (FAQ)
 Genetic Disorder Reference Sheet: Neurofibromatosis Type 1 | ONS Voice
Genetic Disorder Reference Sheet: Neurofibromatosis Type 1 | ONS Voice
 Koselugo (selumetinib) for the Treatment of Neurofibromatosis Type 1
Koselugo (selumetinib) for the Treatment of Neurofibromatosis Type 1
 GINGIVAL NEUROFIBROMA IN NON SYNDROMIC NEUROFIBROMATOSIS TYPE 1 - Authorea
GINGIVAL NEUROFIBROMA IN NON SYNDROMIC NEUROFIBROMATOSIS TYPE 1 - Authorea
 Refractory Neurofibromatosis Type 1 (Concept Id: C4744489)
- MedGen - NCBI
Refractory Neurofibromatosis Type 1 (Concept Id: C4744489)
- MedGen - NCBI
 The Difference Between Neurofibromatosis Type 1 and Type 2
The Difference Between Neurofibromatosis Type 1 and Type 2
 Neurofibromatosis Type 1 with Cutaneous Neurofibromas: Facts, Symptoms, Treatments | Carenity
Neurofibromatosis Type 1 with Cutaneous Neurofibromas: Facts, Symptoms, Treatments | Carenity
 Neurocutaneous Syndromes in Children | University Hospitals
Neurocutaneous Syndromes in Children | University Hospitals
Tumors18
- Neurofibromatosis type 1 (NF1) is a multisystem genetic disorder that is characterized by cutaneous findings, most notably café-au-lait spots and axillary freckling (see the images below), by skeletal dysplasias, and by the growth of both benign and malignant nervous system tumors, most notably benign neurofibromas. (medscape.com)
- Neurofibromatosis (NF) is a neurocutaneous syndrome characterized by the development of tumors of the central or peripheral nervous system including the brain, spinal cord, organs, skin, and bones. (nih.gov)
- Three independent factors were found to be associated with increased risk for neurologic deficit: 1) bilateral tumors at the same level in the cervical region that approximated each other, 2) paraspinal tumors at the lumbar region, and 3) intradural lesions. (ajnr.org)
- Neurofibromatosis-1 is an inherited disorder in which nerve tissue tumors (neurofibromas) form in the skin, bottom layer of skin (subcutaneous tissue), and nerves from the brain (cranial nerves) and spinal cord (spinal root nerves). (health32.com)
- Neurofibromatosis is a genetic condition in which tumors form on the nerves of the inner ear and cause loss of hearing and balance. (news-medical.net)
- Neurofibromatosis 1 is associated with a higher incidence of primary central nervous system tumors. (medlink.com)
- NF-1 causes tumors along the nervous system which can grow anywhere on the body. (wikipedia.org)
- Common symptoms of NF-1 include brownish-red spots in the colored part of the eye called Lisch nodules, benign skin tumors called neurofibromas, and larger benign tumors of nerves called plexiform neurofibromas, scoliosis (curvature of the spine), learning disabilities, vision disorders, mental disabilities, multiple café au lait spots and epilepsy. (wikipedia.org)
- The results of this work set the stage for future experiments to: (1) test effective, already-approved drugs for plexiform neurofibromas and other "benign" tumors, (2) discover new pathway interactions that drive tumor growth, and (3) show how cells with different genetic signatures react to various drug combinations. (n-tap.org)
- Neurofibromatosis type 1 (NF1) is a genetic disorder of the nervous system which can cause tumors to form on the nerves anywhere in the body at any time resulting in a variety of medical problems, primarily affecting the skin and nervous systems. (medicalhomeportal.org)
- For children with certain inherited syndromes that put them at higher risk for brain tumors, such as neurofibromatosis or tuberous sclerosis, doctors often recommend frequent physical exams and other tests. (cancer.org)
- Schwannomas and meningiomas are nervous system tumors that can occur sporadically or in patients with neurofibromatosis type 2 (NF2). (medworm.com)
- Mutations of the Neurofibromatosis 2 (NF2) gene are frequently observed in these tumors. (medworm.com)
- Treatment with antiangiogenesis drugs may improve the effectiveness of radiation treatment of nervous system tumors that interfere with the hearing of patients with the genetic disorder neurofibromatosis 2, investigators report. (medworm.com)
- Neurofibromatosis is a set of complex genetic conditions that can affect nearly every part of the body: It causes tumors to grow on nerves throughout the body and in the brain. (stlouischildrens.org)
- JUPITER, FL- Neurofibromatosis type 1, a disease that features nerve tumors, an elevated risk of autism, and many other symptoms, has long been tied to mutations in a gene called NF1 . (scripps.edu)
- Benign tumors that grow from nerves anywhere in the body are one of the classic features of neurofibromatosis type 1, but the disorder can include epilepsy, learning disabilities, vision problems, and skin lesions, along with a higher risk of autism spectrum disorder, cancers, and heart disease. (scripps.edu)
- She helped develop the first medical therapy approved by the U.S. Food and Drug Administration (FDA) to treat tumors in children with neurofibromatosis type 1 (NF1). (medlineplus.gov)
Neurofibromas12
- Intraoral neurofibromas associated with NF-1 are quite common, but the occurrence of malignant peripheral nerve sheath tumor (MPNST) in the oral cavity is very rare. (amrita.edu)
- 1 , 2 The syndrome is characterized by a combination of clinical traits: café au lait macules, Lisch nodules (iris hamartomas), neurofibromas (cutaneous, subcutaneous, plexiform), optic pathway gliomas, and bone dysplasia. (ajnr.org)
- 07 ). Neurofibromatosis 1, also known as von Recklinghausen disease, is the most common and is characterized by multiple peripheral neurofibromas and the classical hyperpigmented macules, historically described as café-au-lait spots. (medlink.com)
- Plexiform neurofibromas affect up to 50 percent of people with neurofibromatosis type I (NF1), a rare disease of the nervous system for which there are no approved drug therapies. (n-tap.org)
- Neurofibromatosis type 1 is a rare and untreatable genetic disease-causing irregular skin colour (pigmentation) and development of benign tumours on nerves, skin (neurofibromas), brain and other body parts. (clinicaltrialsarena.com)
- NF1 is also known as Von Recklinghausen's disease, peripheral plexiform neurofibromas (NF) and Von Recklinghausen neurofibromatosis. (clinicaltrialsarena.com)
- Less than 1 in 100 people with NF1 will have cancer (malignant) in the neurofibromas. (uhhospitals.org)
- Selumetinib for symptomatic, inoperable plexiform neurofibromas in children with neurofibromatosis type 1: A national real-world case series. (bvsalud.org)
- Neurofibromatosis type 1 -associated plexiform neurofibromas can cause debilitating symptoms and be life threatening. (bvsalud.org)
- The majority of isolated or solitary neurofibromas are sporadic, and a small minority may be associated with the NF-1 syndrome. (medscape.com)
- Plexiform neurofibromas are considered to be pathognomonic of for NF-1. (medscape.com)
- Complicações como neurofibromas viscerais estão presentes em apenas 1% dos casos de NF1. (bvsalud.org)
Recklinghausen5
- Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system human disorder caused by the mutation of Neurofibromin 1, a gene on chromosome 17 that is responsible for production of a protein (neurofibromin) which is needed for normal function in many human cell types. (wikipedia.org)
- NF-1 was formerly known as von Recklinghausen disease, after the researcher (Friedrich Daniel von Recklinghausen) who first documented the disorder. (wikipedia.org)
- Neurofibromatosis Type 1 , also called von Recklinghausen disease, is one of the most frequent genetic conditions: it affects about 1 in 4,000 people. (carenity.co.uk)
- Neurofibromatosis type 1 (NF1, or von Recklinghausen disease) is most prevalent, occurring in 1 of 2500 to 3000 people. (msdmanuals.com)
- Neurofibroma is seen either as a solitary lesion or as part of the generalized syndrome of neurofibromatosis (usually neurofibromatosis type 1 [NF-1], also called von Recklinghausen disease of the skin). (medscape.com)
Schwannomatosis2
- Schwannomatosis 1. (uhhospitals.org)
- Schwannomatosis, a rare disorder, is classified as a 3rd type of neurofibromatosis. (msdmanuals.com)
Mutations10
- CONTEXT: Autosomal dominant inactivating sprouty-related EVH1 domain-containing protein 1 (SPRED1) mutations have recently been described in individuals presenting mainly with café au lait macules (CALMs), axillary freckling, and macrocephaly. (duke.edu)
- NF-1 is a developmental syndrome caused by germline mutations in neurofibromin, a gene that is involved in the RAS pathway (RASopathy). (wikipedia.org)
- NF1 has an incidence of 1/2600 - 1/3000 and arises from mutations in the NF1 gene whose protein, neurofibromin, is normally involved in suppressing cell division. (n-tap.org)
- Neurofibromatosis 1 occurs due to mutations in the NF1 gene found on chromosome 17. (clinicaltrialsarena.com)
- Neurofibromatosis 1 is caused by mutations in the NF1 gene found on chromosome 17. (clinicaltrialsarena.com)
- The autosomal dominant monogenetic disease neurofibromatosis type 1 (NF1) affects approximately one in 3,000 individuals and is caused by mutations in the NF1 tumour suppressor gene, leading to dysfunction in the protein neurofibromin (Nf1) 1 , 2 . (nature.com)
- Somatic Nf1 mutations are also present in 5-10% of cancers, demonstrating the role of Nf1 as a tumour suppressor 1 . (nature.com)
- Neurofibromatosis 1 is a disorder caused by mutations in a gene that regulates the production of a tumor suppressor. (moremedtech.com)
- Following recognition of linkage of the gene for juvenile glaucoma on chromosome 1 (band 1q21-q31), the gene itself was identified and related to mutations found in the trabecular meshwork inducible glucocorticoid response (TIGR) gene in patients with juvenile glaucoma. (medscape.com)
- La neurofibromatosis tipo 1 es una enfermedad hereditaria disease with complete penetrance and is related to mutations in the NF1 de carácter autosómico dominante, con penetración completa y relacionada gene (17q11.2). (bvsalud.org)
Patients21
- NF type 1 (NF1) is differentiated from central NF or NF type 2 in which patients demonstrate a relative paucity of cutaneous findings but have a high incidence of meningiomas and acoustic neuromas (which are frequently bilateral). (medscape.com)
- Dr. Roy Baynes, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories, said, "This is an important collaborative effort with our colleagues at AstraZeneca addressing an area of significant unmet medical need to potentially benefit patients with neurofibromatosis type 1. (merck.com)
- The potential benefit of selumetinib in NF1 is being explored in the U.S. National Cancer Institute-sponsored phase 1/2 SPRINT trial in pediatric patients with symptomatic NF1-related PNs. (merck.com)
- The aim of this study was to characterize the radiologic presentation of patients with neurofibromatosis type 1 with widespread spinal disease and to correlate it to clinical presentation and outcome. (ajnr.org)
- We conducted a historical cohort study of adult patients with neurofibromatosis type 1 with spinal involvement. (ajnr.org)
- Two hundred fifty-seven adult patients with neurofibromatosis type 1 are followed in our center. (ajnr.org)
- The Children's Tumor Foundation (CTF) has announced a groundbreaking 3-year study, which it will fund for nearly $2 million, to determine if a DNA-based blood test can offer better understanding and ultimately earlier diagnosis of cancer predisposition in neurofibromatosis type 1 (NF1) patients. (news-medical.net)
- OBJECTIVE: To determine the frequency, mutational spectrum, and phenotype of neurofibromatosis type 1-like syndrome (NFLS) in a large cohort of patients. (duke.edu)
- In a second cross-sectional study, 1318 unrelated anonymous samples collected in 2003-2007 from patients with a broad range of signs typically found in neurofibromatosis type 1 (NF1) but no detectable NF1 germline mutation underwent SPRED1 mutation analysis. (duke.edu)
- Due to the potential increased susceptibility of patients with neurofibromatosis 1 to the deleterious side effects of radiation therapy, such as mutagenesis and vasculopathy, alternative treatments are required for patients with progressive disease. (medlink.com)
- 20% of NF-1 patients have moderate cases, with several symptoms that have little more than cosmetic effects. (wikipedia.org)
- Focal scoliosis and/or kyphosis are the most common skeletal manifestation of NF-1, occurring in 20% of affected patients. (wikipedia.org)
- The primary endpoint measure in SPRINT Stratum 1 trial was overall response rate (ORR), defined as the percentage of patients with confirmed complete response (vanishing of the target PN) or partial response of at least 20% reduction in PN volume on MRI that was confirmed on a subsequent MRI within 3-6 months. (clinicaltrialsarena.com)
- Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. (nih.gov)
- Consecutive NF1 patients referred to the National French Referral Center for Neurofibromatoses were included. (biomedcentral.com)
- In FLAIR images of neurofibromatosis 1 patients, there often are abnormally bright spots that likely indicate the presence of fluid between layers of myelin sheaths-the insulation surrounding certain nerve cells-which may contribute to the cognitive deficits observed in patients. (moremedtech.com)
- Additionally, applying tools like LST to examine the relationships between UBO volumes and cognitive abilities will help create and refine targeted pharmacological, academic, and behavioral interventions that may improve neurofibromatosis 1 patients' quality of life. (moremedtech.com)
- Patients with neurofibromatosis 1 have been reported to be at an increased risk for several malignancies. (medworm.com)
- The findings dovetail with prior hints of metabolic abnormalities in neurofibromatosis type 1 patients, and suggest the possibility that these abnormalities are key features and drivers of the neurofibromatosis disease process. (scripps.edu)
- NF-1) in patients with JXG [ 3,4,6 ]. (who.int)
- Oral cavity involvement by a solitary and peripheral plexiform neurofibroma in patients with no other signs of neurofibromatosis is uncommon. (medscape.com)
20211
- Through collaboration with the Children's Hospital on or after June 1, 2021, irrespective of etiology. (cdc.gov)
Genetic disorder7
- Neurofibromatosis type 1 (NF1) is a multisystem genetic disorder that commonly is associated with cutaneous, neurologic, and orthopedic manifestations. (medscape.com)
- Neurofibromatosis is a genetic disorder of the nervous system. (medlineplus.gov)
- Neurofibromatosis 1 is an autosomally dominant inherited genetic disorder that has variable clinical manifestations. (medlink.com)
- At around two months old, Mila was diagnosed with Neurofibromatosis Type 1, a genetic disorder that occurs in one in every 3,000 births. (prvh-pcor.org)
- Neurofibromatosis, also known as NF, is a genetic disorder that affects the growth of nerve tissue. (trendyvouge.com)
- Neurofibromatosis type 1 is an autosomal dominant, common genetic disorder that affects many systems, including the skeleton and neurocutaneous system. (ksbu.edu.tr)
- Tia Leigh, 14, from Cwmbran, Wales, was diagnosed with the genetic disorder neurofibromatosis type 1 as a baby. (medworm.com)
Syndrome9
- Neurofibromatosis type 1 is a common tumor predisposition syndrome. (ajnr.org)
- Scholars@Duke publication: Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. (duke.edu)
- Although neurofibromatosis was brought into the public consciousness by Treves' depiction of Merrick in a widely performed play, and later a movie on the Englishman's life, it is now generally accepted that Merrick probably had Proteus Syndrome rather than neurofibromatosis 1. (medlink.com)
- Due to its rarity, and to the fact that genetic diagnosis has been used only in recent years, in the past NF-1 was in some cases confused with Legius syndrome, another syndrome with vaguely similar symptoms, including cafe-au-lait spots. (wikipedia.org)
- The transition between closed, self-inhibited states of Nf1 and open states provides guidance for targeted studies deciphering the complex molecular mechanism behind the widespread neurofibromatosis syndrome and Nf1 dysfunction in carcinogenesis. (nature.com)
- Neurofibromatosis is a neurocutaneous syndrome (a syndrome with neurologic and cutaneous manifestations). (msdmanuals.com)
- Here we present a case of Cotard syndrome in a patient with neurofibromatosis. (archivespp.pl)
- Neurofibromatosis 2 (NF2) is an inherited autosomal dominant syndrome characterized by multiple schwannomas, meningiomas, and ependymomas. (medscape.com)
- Many familial cancer syndromes increase glioma risk including neurofibromatosis 1 (NF1), NF2, tuberous sclerosis 1 (TSC1), TSC2, Lynch Syndrome, and Li-Fraumeni syndrome. (cdc.gov)
Patient with neurofibromatosis4
- We report a case of synchronous multiple colon adenocarcinomas in a patient with neurofibromatosis type 1 (NF1). (elsevierpure.com)
- Kim, IY , Cho, MY & Kim, YW 2014, ' Synchronous multiple colonic adenocarcinomas arising in patient with neurofibromatosis type 1 ', Annals of Surgical Treatment and Research , vol. 87, no. 3, pp. 156-160. (elsevierpure.com)
- A case of non-Hodgkin's lymphoma in a patient with neurofibromatosis type 1. (korea.ac.kr)
- Dive into the research topics of 'A case of non-Hodgkin's lymphoma in a patient with neurofibromatosis type 1. (korea.ac.kr)
20221
- Neuro Oncol 2022 Aug 1;24(8):1377-1386. (nih.gov)
Mutation5
- It is associated with the mutation of NF-1 gene, a tumor suppressor gene located on chromosome 17q11.2. (amrita.edu)
- You can get neurofibromatosis from your parents, or it can happen because of a mutation (change) in your genes. (medlineplus.gov)
- NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy (or allele) of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1. (wikipedia.org)
- A parent with NF has a 1 in 2 chance of passing on the genetic mutation and disease to each child. (uhhospitals.org)
- From 3 in 10 to 1 in 2 cases of NF are caused by a new mutation and not inherited. (uhhospitals.org)
Disease9
- Neurofibromatosis-1 (NF-1) is a relatively common autosomal dominant disease characterized by multiple cutaneous fibromatoses and café au lait spots. (amrita.edu)
- The author reviews the role of chemotherapy for gliomas associated with neurofibromatosis 1 and its efficacy on disease control and visual outcome. (medlink.com)
- Neurofibromatosis 2, also called central neurofibromatosis, is another disease entity with features that overlap with neurofibromatosis 1. (medlink.com)
- One of these types, NF Type 1, is the most common type of disease. (trendyvouge.com)
- However, if a person has NF Type 1, it is also important to keep the underlying disease in check by observing any changes in their nervous system. (trendyvouge.com)
- Neurofibromatosis is a congenital disease and its symptoms appear in childhood. (carenity.co.uk)
- However, in most cases neurofibromatosis is a disease with cutaneous symptoms and some benign tumours. (carenity.co.uk)
- Neurofibromatosis is a genetic disease. (carenity.co.uk)
- In the new study, Tomchik and his colleagues, including first author Valentina Botero, a graduate student in the Tomchik laboratory, investigated the mechanisms of neurofibromatosis type 1's metabolic impact with detailed experiments on Drosophila fruit flies-which, despite their evolutionary distance from humans, model many aspects of the human disease. (scripps.edu)
Diagnosis5
- A clinical diagnosis can be made in 50% of affected children by 1 year of age and in nearly all by 8 years of age. (arupconsult.com)
- The following is a list of conditions and complications associated with NF-1, and, where available, age range of onset and progressive development, occurrence percentage of NF-1 population, method of earliest diagnosis, and treatments and related medical specialties. (wikipedia.org)
- Concurrent COVID-19 diagnosis was defined as past year and the possible contributing role of SARS-CoV-2 having International Classification of Diseases, Tenth Revision infection ( 1 ). (cdc.gov)
- In 1988, the National Institutes of Health released a conference statement regarding neurofibromatosis that addressed the diagnosis of NF2. (medscape.com)
- Subsequently, a neurofibromatosis type 1 diagnosis was performed. (bvsalud.org)
Cutaneous manifestations1
- Finally, even if they are not harmful, cutaneous manifestations of neurofibromatosis are difficult to accept because of their unattractiveness. (carenity.co.uk)
Complications3
- Sean Bohen, executive vice president, global medicines development and chief medical officer, AstraZeneca, said, "Neurofibromatosis type 1 is a devastating condition that can lead to life-threatening complications. (merck.com)
- To avoid possible complications of neurofibromatosis, a medical follow-up is established. (carenity.co.uk)
- Specialised healthcare professionals take care of the neurofibromatosis complications . (carenity.co.uk)
Treatment of neurofibromatosis4
- AstraZeneca and Merck (NYSE:MRK), known as MSD outside the U.S. and Canada, today announced that the U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation (ODD) for selumetinib, a MEK 1/2 inhibitor, for the treatment of neurofibromatosis type 1 (NF1). (merck.com)
- He also discusses the potential role of molecularly targeted therapy in the treatment of neurofibromatosis 1-associated gliomas. (medlink.com)
- Koselugo® (selumetinib) is the first FDA-approved drug indicated for the treatment of neurofibromatosis type 1 (NFI), a rare and incurable genetic condition, developed and commercialised globally by AstraZeneca and Merck (MSD) under a licensing agreement. (clinicaltrialsarena.com)
- Koselugo® (selumetinib) is the first FDA approved drug indicated for the treatment of neurofibromatosis type 1 (NFI). (clinicaltrialsarena.com)
Child with neurofibromatosis2
- Assessment and management information for the primary care clinician caring for the child with neurofibromatosis type 1 (NF1). (medicalhomeportal.org)
- The second patient was a child with neurofibromatosis type 1 who developed a pigmented peripapillary lesion following excision of an optic nerve glioma. (nih.gov)
Neurofibromin3
- Neurofibromatosis type 1 (NF1) is an autosomal dominant condition that stems from a pathogenic variant in the NF1 gene , which regulates the production of the tumor-suppressing neurofibromin protein. (ons.org)
- The neurofibromatosis 1 (NF1) gene product, neurofibromin, is a tumor suppressor gene product capable of inhibiting the growth of cells in culture. (wustl.edu)
- If neurofibromin suppresses cell growth by arresting cells in G 0 or G 1 , its expression might be regulated in a cell cycle-dependent fashion. (wustl.edu)
Abnormalities1
- In recent years, studies of neurofibromatosis type 1 in people and in animal models have begun turning up evidence that the disorder also involves abnormalities in metabolism. (scripps.edu)
Symptoms of neurofibromatosis4
- The symptoms of neurofibromatosis vary widely between individuals, with some people only experiencing mild health problems and others finding they are severely affected on a day-to-day basis. (news-medical.net)
- What are the Symptoms of Neurofibromatosis Type 1? (trendyvouge.com)
- Some of the most serious symptoms of neurofibromatosis, apart from cutaneous disorders, are neurological disorders and the development of tumours . (carenity.co.uk)
- Skeletal modifications (bone tumours like pseudoarthrosis, scoliosis), as well as endocrine adenomas (benign tumours that develop on the endocrine glands) and vascular disorders (blood vessels, renal artery) which can lead to arterial hypertension, are also possible symptoms of neurofibromatosis. (carenity.co.uk)
Individuals with neurofibromatosis2
- The open access version of 'ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1' is available online! (ern-cranio.eu)
- St. Louis Children's Hospital is a national referral center for individuals with neurofibromatosis. (stlouischildrens.org)
Selumetinib3
- Selumetinib, is an investigational MEK 1/2 inhibitor licensed by AstraZeneca from Array BioPharma Inc. in 2003. (merck.com)
- Selumetinib is a mitogen-activated protein kinases 1 and 2 (MEK1/2) inhibitor. (clinicaltrialsarena.com)
- Selumetinib is an oral selective inhibitor of RAS - mitogen-activated protein kinase (MAPK) 1 and 2, which has shown efficacy for tumour shrinkage/stabilisation and symptom improvement. (bvsalud.org)
Type of neurofibromatosis1
- This is the more common type of neurofibromatosis. (uhhospitals.org)
Cure for neurofibromatosis2
- There is no cure for neurofibromatosis. (medscape.com)
- There is no known cure for neurofibromatosis and there are limited treatment options to manage symptoms. (merck.com)
Lesions2
- The best treatment a person with NF Type 1 can get is total excision of the lesions. (trendyvouge.com)
- Lesions may thigh (Figure 1). (who.int)
30001
- Neurofibromatosis type 1 (NF1) is a common genetic condition, affecting 1 in 3000 individuals, and people with NF1 are at greater risk of developing a rare, aggressive form of cancer. (news-medical.net)
Glioma3
- In distinction, an individual is diagnosed to have neurofibromatosis 2 if the person has bilateral eighth nerve masses seen with appropriate imaging techniques or a first degree relative with neurofibromatosis 2 and either: (1) a unilateral eighth nerve mass, (2) Two or more of the following: neurofibroma, meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity. (medlink.com)
- Risk factors for treatment-refractory and relapsed optic pathway glioma in children with neurofibromatosis type 1. (nih.gov)
- Trametinib therapy for children with neurofibromatosis type 1 and life-threatening plexiform neurofibroma or treatment-refractory low-grade glioma. (nih.gov)
Manifestations2
- Neurofibromatosis 1 has protean manifestations, of which intracranial gliomas are one of the most common. (medlink.com)
- Neurofibromatosis refers to several related disorders that have overlapping clinical manifestations but that are now understood to have distinct genetic causes. (msdmanuals.com)
3,0003
- NF1 is one of the most common genetic disorders in the United States (approximately 1 in every 2,500 to 3,000 births). (medicalhomeportal.org)
- Signs and symptoms vary widely, but NF1 disorders occur in 1 in about 3,000-4,000 people . (ons.org)
- Neurofibromatosis Type 1 (NF1) occurs in about 1 in 3,000 to 4,000 babies in the U.S. NF1 is an autosomal dominant disorder. (uhhospitals.org)
Bilateral1
- Axial T1-weighted postcontrast image demonstrates bilateral internal auditory canal-enhancing masses that are diagnostic for neurofibromatosis type 2 (NF2). (medscape.com)
Chromosome3
- The NF-1 gene is located on chromosome 17. (medicalhomeportal.org)
- Physicians may also recommend that a person with NF Type 1 take a chromosome and genetic test to determine the cause of their symptoms. (trendyvouge.com)
- It affects about 1 in 25,000 babies in the U.S. The gene change that causes NF2 is on chromosome 22. (uhhospitals.org)
Vestibular Schwannoma1
- In some cases of vestibular schwannoma, a sometimes-lethal tumor often associated with neurofibromatosis 2 (NF2), secretions from the tumor contain toxic molecules that damage the inner ear. (medworm.com)
Affects1
- NF Type 2 is much rarer than NF Type 1, and it affects only men. (trendyvouge.com)
Cognitive2
- Neurofibromatosis type 1 (NF1) is a common neurogenetic condition associated with cognitive dysfunction and learning disability. (bmj.com)
- More information: Emily M. Harriott et al, Using a semi-automated approach to quantify unidentified bright objects in Neurofibromatosis type 1 and linkages to cognitive and academic outcomes, Magnetic Resonance Imaging (2023). (moremedtech.com)
Neurogenetic condition1
- Neurofibromatosis type 1 (NF1) is a neurogenetic condition that approximately 1 in every 2,700 people are born with. (m4rd.org)
Multisystem1
- Multisystem burden of neurofibromatosis 1 in Denmark: registry- and population-based rates of hospitalizations over the life span. (cdc.gov)
Predisposition1
- It presents extremely variable expression and predisposition con mutaciones en el gen NF1 (17q11.2). (bvsalud.org)
Chemotherapy1
- Gliomas in children with neurofibromatosis 1, if requiring treatment, may be chemotherapy-sensitive, and radiotherapy should be used as a last resort, given potential long-term sequelae. (medlink.com)
Morbidity1
- The complex course of the neurofibromatosis predisposes the individual towards higher physical and psychiatric morbidity. (archivespp.pl)
Children7
- Clinical findings in people with NF-1 include: Reduced skeletal muscle size Reduced exercise capacity Muscle weakness (The most recent study reports between 30-50% reduced upper and lower limb muscle strength in NF-1 children compare with matched controls). (wikipedia.org)
- How is NF Type 1 Diagnosed in Children? (trendyvouge.com)
- According to Healthline, there are several ways physicians can diagnose NF Type 1 in children. (trendyvouge.com)
- They are most common in small children suffering with neurofibromatosis type 1. (carenity.co.uk)
- Reading difficulties are one of the most significant challenges for children with neurofibromatosis type 1 (NF1). (edu.au)
- The current work, led by the first author and neuroscience Ph.D. student Emily Harriott and co-author and recent neuroscience Ph.D. graduate Tin Nguyen, tested and validated a semi-automated approach for detecting and segmenting UBOs in children and adolescents with neurofibromatosis 1. (moremedtech.com)
- The patient was ing in diameter from 3 mm to 15 mm the head and neck of infants and young the product of full-term pregnancy and with regular borders, and a giant CALM children and resolves spontaneously measuring 15 × 10 cm on the right caesarean section delivery due to failure without treatment [ 1 ]. (who.int)
Occurs1
- Las occurs in only 1% of NF1 cases. (bvsalud.org)
Nervous system1
- Inclusion of exon 23a is found in most human tissues but predominately skipped in neurons of the central nervous system 3 , and variation of the isoform 1/2 splicing ratio leads to disturbed neuronal differentiation 11 . (nature.com)
20181
- 2018). Worries and needs of adults and parents of adults with neurofibromatosis type 1. (bvsalud.org)
Treatments1
- It is also being explored as a monotherapy and in combination with other treatments in phase 1 trials. (merck.com)
People4
- However, 60% of people with NF-1 have mild cases, with few symptoms that have very little effect in their day-to-day lives. (wikipedia.org)
- Some people with NF Type 1 have other symptoms. (trendyvouge.com)
- Neurofibromatosis type 2 (NF2) accounts for 10% of cases, occurring in about 1 of 35,000 people. (msdmanuals.com)
- This study certainly has given us an idea of the populations of neurons in people that we should look at in future studies for their potential role in mediating metabolic effects of neurofibromatosis type 1," he says. (scripps.edu)
Schwannomas1
- Vestibular schwannomas related to neurofibromatosis type 2 (NF2) are difficult to manage and are sometimes treated with a noninvasive option, stereotactic radiosurgery. (news-medical.net)
Neoplasms1
- Neurofibromatosis 1 is an autosomally dominated inherited genetic condition that predisposes those involved to the development of intracranial neoplasms. (medlink.com)
20171
- Mila Gray Roomberg was born at 1:11 am on September 16, 2017 to her very adoring parents, Jessica and Dan. (prvh-pcor.org)
Pharmacological1
- These advances are described in the manuscript "Pharmacological and genomic profiling of neurofibromatosis type 1 plexiform neurofibroma-derived Schwann cells," published in Scientific Data on June 12. (n-tap.org)