Peroxisomal Disorders
Zellweger Syndrome
Refsum Disease
Adrenoleukodystrophy
Microbodies
Chondrodysplasia Punctata, Rhizomelic
Phytanic Acid
Plasmalogens
Peroxisomes
Chondrodysplasia Punctata
Peroxisomal Multifunctional Protein-2
Fatty Acids
Acetyl-CoA C-Acetyltransferase
Lipid Metabolism, Inborn Errors
Gas Chromatography-Mass Spectrometry
Oxidation-Reduction
Acyl-CoA Oxidase
Fibroblasts
Enoyl-CoA Hydratase
Peroxisomal Bifunctional Enzyme
Acetyl-CoA C-Acyltransferase
Bipolar Disorder
3-Hydroxyacyl CoA Dehydrogenases
Mental Disorders
Clofibrate
Pichia
Metabolic control of peroxisome abundance. (1/105)
Zellweger syndrome and related disorders represent a group of lethal, genetically heterogeneous diseases. These peroxisome biogenesis disorders (PBDs) are characterized by defective peroxisomal matrix protein import and comprise at least 10 complementation groups. The genes defective in seven of these groups and more than 90% of PBD patients are now known. Here we examine the distribution of peroxisomal membrane proteins in fibroblasts from PBD patients representing the seven complementation groups for which the mutant gene is known. Peroxisomes were detected in all PBD cells, indicating that the ability to form a minimal peroxisomal structure is not blocked in these mutants. We also observed that peroxisome abundance was reduced fivefold in PBD cells that are defective in the PEX1, PEX5, PEX12, PEX6, PEX10, and PEX2 genes. These cell lines all display a defect in the import of proteins with the type-1 peroxisomal targeting signal (PTS1). In contrast, peroxisome abundance was unaffected in cells that are mutated in PEX7 and are defective only in the import of proteins with the type-2 peroxisomal targeting signal. Interestingly, a fivefold reduction in peroxisome abundance was also observed for cells lacking either of two PTS1-targeted peroxisomal beta-oxidation enzymes, acyl-CoA oxidase and 2-enoyl-CoA hydratase/D-3-hydroxyacyl-CoA dehydrogenase. These results indicate that reduced peroxisome abundance in PBD cells may be caused by their inability to import these PTS1-containing enzymes. Furthermore, the fact that peroxisome abundance is influenced by peroxisomal 105-oxidation activities suggests that there may be metabolic control of peroxisome abundance. (+info)Accumulation of glycolipids in mutant Chinese hamster ovary cells (Z65) with defective peroxisomal assembly and comparison of the metabolic rate of glycosphingolipids between Z65 cells and wild-type CHO-K1 cells. (2/105)
The influence of peroxisomal dysfunction on glycosphingolipid metabolism was investigated using mutant Chinese hamster ovary (CHO) cells (Z65) with defective assembly of the peroxisomal membranes. In accordance with previous observations, the concentration of very long chain fatty acid (C24:0) was shown to be higher in Z65 cells than in control cells. We then compared the composition of glycolipids in Z65 cells with that in CHO-K1 cells, which are wild-type Chinese hamster ovary cells with intact peroxisomes, and found significantly increased concentrations of ceramide monohexoside (CMH) and ganglioside GM3 in Z65 cells. However, there were no differences in the concentrations of glycerophospholipids, triglycerides, free fatty acids and cholesterol between Z65 and CHO-K1 cells. Further, to investigate the metabolic rate of the major lipids, Z65 and CHO-K1 cells were pulse-labeled with [3-14C]serine. [3-14C]Serine was incorporated into phosphatidylserine, phosphatidylethanolamine and sphingomyelin more quickly in CHO-K1 than in Z65 cells. However, after 48 h, the radioactivity incorporated into those lipids, including CMH, was greater in Z65 cells than in CHO-K1 cells. Thus, the altered metabolism of glycosphingolipids, probably due to peroxisomal dysfunction, was thought to be responsible for the change in glycosphingolipid composition in Z65 cells. (+info)Nonsense and temperature-sensitive mutations in PEX13 are the cause of complementation group H of peroxisome biogenesis disorders. (3/105)
Peroxisome biogenesis disorders, including Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD) and infantile Refsum disease, are lethal hereditary diseases caused by abnormalities in peroxisomal assembly. To date, 12 genotypes have been identified. We now have evidence that the complete human cDNA encoding Pex13p, an SH3 protein of a docking factor for the peroxisome targeting signal 1 receptor (Pex5p), rescues peroxisomal matrix protein import and its assembly in fibroblasts from PBD patients of complementation group H. In addition, we detected mutations on the human PEX13 cDNA in two patients of group H. A severe phenotype of a ZS patient (H-02) was homozygous for a nonsense mutation, W234ter, which results in the loss of not only the SH3 domain but also the putative transmembrane domain of Pex13p. A more mildly affected NALD patient (H-01), whose fibroblasts showed the temperature-sensitive (TS) phenotype, was homozygous for a missense mutation in the SH3 domain of Pex13p, I326T. This mutant PEX13 cDNA expression in a PEX13-defective CHO mutant showed I326T to be a TS mutation and thus suggested that Pex13p with the I326T mutation in the SH3 domain is stable at 30 degrees C but is somewhat unstable at 37 degrees C. (+info)Enoyl-CoA hydratase deficiency: identification of a new type of D-bifunctional protein deficiency. (4/105)
D-bifunctional protein is involved in the peroxisomal beta-oxidation of very long chain fatty acids, branched chain fatty acids and bile acid intermediates. In line with the central role of D-bifunctional protein in the beta-oxidation of these three types of fatty acids, all patients with D-bifunctional protein deficiency so far reported in the literature show elevated levels of very long chain fatty acids, branched chain fatty acids and bile acid inter-mediates. In contrast, we now report two novel patients with D-bifunctional protein deficiency who both have normal levels of bile acid intermediates. Complementation analysis and D-bifunctional protein activity measurements revealed that both patients had an isolated defect in the enoyl-CoA hydratase domain of D-bifunctional protein. Subsequent mutation analysis showed that both patients are homozygous for a missense mutation (N457Y), which is located in the enoyl-CoA hydratase coding part of the D-bifunctional protein gene. Expression of the mutant protein in the yeast Saccharomyces cerevisiae confirmed that the N457Y mutation is the disease-causing mutation. Immunoblot analysis of patient fibroblast homogenates showed that the protein levels of full-length D-bifunctional protein were strongly reduced while the enoyl-CoA hydratase component produced after processing within the peroxisome was undetectable, which indicates that the mutation leads to an unstable protein. (+info)PEX13 is mutated in complementation group 13 of the peroxisome-biogenesis disorders. (5/105)
The peroxisome-biogenesis disorders (PBDs) are a genetically and phenotypically diverse group of diseases caused by defects in peroxisome assembly. One of the milder clinical variants within the PBDs is neonatal adrenoleukodystrophy (NALD), a disease that is usually associated with partial defects in the import of peroxisomal matrix proteins that carry the type 1 or type 2 peroxisomal targeting signals. Here, we characterize the sole representative of complementation group 13 of the PBDs, a patient with NALD (patient PBD222). Skin fibroblasts from patient PBD222 display defects in the import of multiple peroxisomal matrix proteins. However, residual matrix-protein import can be detected in cells from patient PBD222, consistent with the relatively mild phenotypes of the patient. PEX13 encodes a peroxisomal membrane protein with a cytoplasmically exposed SH3 domain, and we find that expression of human PEX13 restores peroxisomal matrix-protein import in cells from patient PBD222. Furthermore, these cells are homozygous for a missense mutation at a conserved position in the PEX13 SH3 domain. This mutation attenuated the activity of human PEX13, and an analogous mutation in yeast PEX13 also reduced its activity. The mutation was absent in >100 control alleles, indicating that it is not a common polymorphism. Previous studies have demonstrated extragenic suppression in the PBDs, but the phenotypes of patient PBD222 cells could not be rescued by expression of any other human PEX genes. Taken together, these results provide strong evidence that mutations in PEX13 are responsible for disease in patient PBD222 and, by extension, in complementation group 13 of the PBDs. (+info)Chronic peroxisome proliferation and hepatomegaly associated with the hepatocellular tumorigenesis of di(2-ethylhexyl)phthalate and the effects of recovery. (6/105)
This study compared the levels of cell proliferation and peroxisome proliferation in rodent liver with tumor incidence, to provide more information on the relationship between these events following chronic exposure. Fischer 344 rats were treated with 0, 100, 500, 2500, or 12,500 ppm DEHP, and B6C3F1 mice were treated with 0, 100, 500, 1500, or 6000 ppm DEHP in the diet for up to 104 weeks. Additional groups of rats and mice received the highest concentration for 78 weeks and then the control diet for an additional 26 weeks (recovery groups). Animals were terminated at weeks 79 and 105 for histopathologic examination. Elevated palmitoyl CoA oxidation activity and higher liver-to-body weight ratios were observed for the 2500- and 12,500-ppm groups of rats, and for the 500-, 1500-, and 6000-ppm groups of mice at Week 105. No increase in palmitoyl CoA oxidation activity was evident in the recovery group, and relative liver weights were near control levels following recovery. No hepatic cell proliferation was detected at Weeks 79 or 105 in either species although preliminary data indicated that cell proliferation did occur within the first 13 weeks of exposure. A significantly higher incidence of hepatocellular tumors was only observed for the 2500- and 12,500-ppm group and its recovery group of rats, and for the 500-, 1500-, and 6000-ppm groups and the recovery group of mice. The tumor incidences were reduced for the recovery groups compared with the groups fed DEHP continuously for 104 weeks. The data indicate that high levels of peroxisome proliferation and hepatomegaly are associated with DEHP hepatocarcinogenesis in rodent liver, and that the tumorigenic process may be arrested by cessation of DEHP treatment, suggesting that extended treatment with DEHP acts to promote tumor growth. (+info)Defective PEX gene products correlate with the protein import, biochemical abnormalities, and phenotypic heterogeneity in peroxisome biogenesis disorders. (7/105)
Peroxisome biogenesis disorders (PBD) comprise three phenotypes including Zellweger syndrome (ZS) (the most severe), neonatal adrenoleucodystrophy, and infantile Refsum disease (IRD) (the most mild), and can be classified into at least 12 genetic complementation groups, which are not predictive of the phenotypes. Several pathogenic genes for PBD groups have been identified, but the relationship between the defective gene products and phenotypic heterogeneity has remained unclear. We identified a mutation in the PEX2 gene in an IRD patient with compound heterozygosity for a missense mutation and the known nonsense mutation detected in ZS patients. In transfection experiments using the peroxisome deficient CHO mutant, Z65 with a nonsense mutation in the PEX2 gene, we noted the E55K mutation had mosaic activities of peroxisomal protein import machinery and residual activities of peroxisomal functions, including dihydroxyacetone phosphate acyltransferase and beta oxidation of very long chain fatty acids. The nonsense mutation severely affects these peroxisomal functions as well as the protein import. These data suggest that allelic heterogeneity of the PEX gene affects the peroxisomal protein import and functions and regulates the clinical severity in PBD. (+info)PEX12 interacts with PEX5 and PEX10 and acts downstream of receptor docking in peroxisomal matrix protein import. (8/105)
Peroxisomal matrix protein import requires PEX12, an integral peroxisomal membrane protein with a zinc ring domain at its carboxy terminus. Mutations in human PEX12 result in Zellweger syndrome, a lethal neurological disorder, and implicate the zinc ring domain in PEX12 function. Using two-hybrid studies, blot overlay assays, and coimmunoprecipitation experiments, we observed that the zinc-binding domain of PEX12 binds both PEX5, the PTS1 receptor, and PEX10, another integral peroxisomal membrane protein required for peroxisomal matrix protein import. Furthermore, we identified a patient with a missense mutation in the PEX12 zinc-binding domain, S320F, and observed that this mutation reduces the binding of PEX12 to PEX5 and PEX10. Overexpression of either PEX5 or PEX10 can suppress this PEX12 mutation, providing genetic evidence that these interactions are biologically relevant. PEX5 is a predominantly cytoplasmic protein and previous PEX5-binding proteins have been implicated in docking PEX5 to the peroxisome surface. However, we find that loss of PEX12 or PEX10 does not reduce the association of PEX5 with peroxisomes, demonstrating that these peroxins are not required for receptor docking. These and other results lead us to propose that PEX12 and PEX10 play direct roles in peroxisomal matrix protein import downstream of the receptor docking event. (+info)Peroxisomal disorders are a group of inherited metabolic diseases caused by defects in the function or structure of peroxisomes, which are specialized subcellular organelles found in the cells of animals, plants, and humans. These disorders can affect various aspects of metabolism, including fatty acid oxidation, bile acid synthesis, and plasma cholesterol levels.
Peroxisomal disorders can be classified into two main categories: single peroxisomal enzyme deficiencies and peroxisome biogenesis disorders (PBDs). Single peroxisomal enzyme deficiencies are characterized by a defect in a specific enzyme found within the peroxisome, while PBDs are caused by problems with the formation or assembly of the peroxisome itself.
Examples of single peroxisomal enzyme deficiencies include X-linked adrenoleukodystrophy (X-ALD), Refsum disease, and acyl-CoA oxidase deficiency. PBDs include Zellweger spectrum disorders, such as Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease.
Symptoms of peroxisomal disorders can vary widely depending on the specific disorder and the severity of the enzyme or biogenesis defect. They may include neurological problems, vision and hearing loss, developmental delays, liver dysfunction, and skeletal abnormalities. Treatment typically focuses on managing symptoms and addressing any underlying metabolic imbalances.
Zellweger Syndrome is a rare genetic disorder that affects the development and function of multiple organ systems in the body. It is part of a group of conditions known as peroxisome biogenesis disorders (PBDs), which are characterized by abnormalities in the structure and function of peroxisomes, which are cellular structures that break down fatty acids and other substances in the body.
Zellweger Syndrome is caused by mutations in one or more genes involved in the formation and maintenance of peroxisomes. As a result, people with this condition have reduced levels of certain enzymes that are necessary for normal brain development, as well as for the breakdown of fats and other substances in the body.
Symptoms of Zellweger Syndrome typically appear within the first few months of life and may include:
* Severe developmental delays and intellectual disability
* Hypotonia (low muscle tone) and poor motor skills
* Vision and hearing problems
* Facial abnormalities, such as a high forehead, wide-set eyes, and a prominent nasal bridge
* Liver dysfunction and jaundice
* Seizures
* Feeding difficulties and failure to thrive
There is no cure for Zellweger Syndrome, and treatment is focused on managing the symptoms of the condition. The prognosis for people with this disorder is generally poor, with most individuals not surviving beyond the first year of life. However, some individuals with milder forms of the condition may live into early childhood or adolescence.
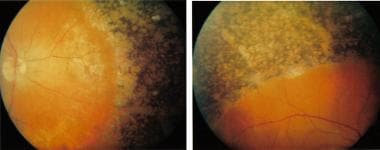
Refsum Disease is a rare inherited neurological disorder characterized by the accumulation of phytanic acid in various tissues of the body due to impaired breakdown of this fatty acid. This is caused by a deficiency in the enzyme phytanoyl-CoA hydroxylase or the transporter protein peroxisomal biogenesis factor 7 (PEX7).
The symptoms of Refsum Disease can vary but often include progressive neurological dysfunction, retinitis pigmentosa leading to decreased vision and night blindness, hearing loss, ichthyosis (dry, scaly skin), and cardiac abnormalities. The onset of symptoms is usually in childhood or adolescence, but milder cases may not become apparent until later in life.
The treatment for Refsum Disease involves a strict diet that limits the intake of phytanic acid, which is found in dairy products, beef, and certain fish. Plasmapheresis, a procedure to remove harmful substances from the blood, may also be used to reduce the levels of phytanic acid in the body. Early diagnosis and treatment can help slow down or prevent the progression of the disease.
Adrenoleukodystrophy (ADL) is a rare genetic disorder that affects the nervous system and adrenal glands. It is characterized by the accumulation of very long-chain fatty acids (VLCFAs) in the brain, leading to progressive neurological symptoms such as behavioral changes, visual loss, hearing loss, seizures, and difficulties with coordination and movement.
ADL is caused by mutations in the ABCD1 gene, which provides instructions for making a protein involved in the breakdown of VLCFA. Without this protein, VLCFAs accumulate in the brain and adrenal glands, leading to damage and dysfunction.
There are several forms of ADL, including:
* Childhood cerebral ADL: This is the most severe form of the disorder, typically affecting boys between the ages of 4 and 8. It progresses rapidly and can lead to significant neurological impairment within a few years.
* Adrenomyeloneuropathy (AMN): This form of ADL affects both men and women and is characterized by progressive stiffness, weakness, and spasticity in the legs. It typically develops in adulthood and progresses slowly over many years.
* Addison's disease: This is a condition that affects the adrenal glands, leading to hormonal imbalances and symptoms such as fatigue, weight loss, and low blood pressure.
There is no cure for ADL, but treatments can help manage the symptoms and slow down the progression of the disorder. These may include dietary changes, medications to control seizures or hormone levels, and physical therapy. In some cases, stem cell transplantation may be recommended as a treatment option.
Microbodies are small, membrane-bound organelles found in the cells of eukaryotic organisms. They typically measure between 0.2 to 0.5 micrometers in diameter and play a crucial role in various metabolic processes, particularly in the detoxification of harmful substances and the synthesis of lipids.
There are several types of microbodies, including:
1. Peroxisomes: These are the most common type of microbody. They contain enzymes that help break down fatty acids and amino acids, producing hydrogen peroxide as a byproduct. Another set of enzymes within peroxisomes then converts the harmful hydrogen peroxide into water and oxygen, thus detoxifying the cell.
2. Glyoxysomes: These microbodies are primarily found in plants and some fungi. They contain enzymes involved in the glyoxylate cycle, a metabolic pathway that helps convert stored fats into carbohydrates during germination.
3. Microbody-like particles (MLPs): These are smaller organelles found in certain protists and algae. Their functions are not well understood but are believed to be involved in lipid metabolism.
It is important to note that microbodies do not have a uniform structure or function across all eukaryotic cells, and their specific roles can vary depending on the organism and cell type.
Chondrodysplasia punctata, rhizomelic is a rare genetic disorder that affects the development of bones and cartilage. The condition is characterized by shortened limbs (rhizomelia), particularly the upper arms and thighs, and multiple small punctate calcifications in the cartilage of the body, including the ears, nose, and other areas.
The disorder is caused by mutations in the gene PEX7, which is involved in the transport of enzymes to peroxisomes, cellular organelles that break down fatty acids and other substances. Without functional PEX7, these enzymes cannot reach the peroxisomes, leading to abnormal accumulation of lipids and other substances in various tissues, including bone and cartilage.
Chondrodysplasia punctata, rhizomelic is typically diagnosed in infancy or early childhood based on clinical features and imaging studies. The condition can be associated with a range of complications, including developmental delays, respiratory problems, hearing loss, and visual impairment. There is no cure for the disorder, and treatment is focused on managing symptoms and addressing specific complications as they arise.
Phytanic acid is a branched-chain fatty acid that is primarily found in animal products, such as dairy foods and meat, but can also be present in some plants. It is a secondary plant metabolite that originates from the breakdown of phytol, a component of chlorophyll.
Phytanic acid is unique because it contains a methyl group branching off from the middle of the carbon chain, making it difficult for the body to break down and metabolize. Instead, it must be degraded through a process called α-oxidation, which takes place in peroxisomes.
In some cases, impaired phytanic acid metabolism can lead to a rare genetic disorder known as Refsum disease, which is characterized by the accumulation of phytanic acid in various tissues and organs, leading to neurological symptoms, retinal degeneration, and cardiac dysfunction.
Plasmalogens are a type of complex lipid called glycerophospholipids, which are essential components of cell membranes. They are characterized by having a unique chemical structure that includes a vinyl ether bond at the sn-1 position of the glycerol backbone and an ester bond at the sn-2 position, with the majority of them containing polyunsaturated fatty acids. The headgroup attached to the sn-3 position is typically choline or ethanolamine.
Plasmalogens are abundant in certain tissues, such as the brain, heart, and skeletal muscle. They have been suggested to play important roles in cellular functions, including membrane fluidity, signal transduction, and protection against oxidative stress. Reduced levels of plasmalogens have been associated with various diseases, including neurological disorders, cardiovascular diseases, and aging-related conditions.
Peroxisomes are membrane-bound subcellular organelles found in the cytoplasm of eukaryotic cells. They play a crucial role in various cellular processes, including the breakdown of fatty acids and the detoxification of harmful substances such as hydrogen peroxide (H2O2). Peroxisomes contain numerous enzymes, including catalase, which converts H2O2 into water and oxygen, thus preventing oxidative damage to cellular components. They also participate in the biosynthesis of ether phospholipids, a type of lipid essential for the structure and function of cell membranes. Additionally, peroxisomes are involved in the metabolism of reactive oxygen species (ROS) and contribute to the regulation of intracellular redox homeostasis. Dysfunction or impairment of peroxisome function has been linked to several diseases, including neurological disorders, developmental abnormalities, and metabolic conditions.
Chondrodysplasia punctata is a group of genetic disorders that affect the development of bones and cartilage. The condition is characterized by stippled calcifications, or spots of calcium deposits, in the cartilage that can be seen on X-rays. These spots are typically found at the ends of long bones, in the sternum, and in the pelvis.
The symptoms of chondrodysplasia punctata can vary widely depending on the specific type of the disorder. Some people with the condition may have short stature, bowed legs, and other skeletal abnormalities, while others may have only mild symptoms or no symptoms at all. The condition can also be associated with developmental delays, intellectual disability, and other health problems.
There are several different types of chondrodysplasia punctata, each caused by a different genetic mutation. Some forms of the disorder are inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) in order to develop the condition. Other forms of chondrodysplasia punctata are inherited in an X-linked dominant manner, meaning that a single copy of the mutated gene (on the X chromosome) is enough to cause the disorder in females. Males, who have only one X chromosome, will typically be more severely affected by X-linked dominant disorders.
There is no cure for chondrodysplasia punctata, and treatment is focused on managing the symptoms of the condition. This may include physical therapy, bracing or surgery to correct skeletal abnormalities, and medications to manage pain or other health problems.
Peroxisomal multifunctional protein-2 (MFP2) is a key enzyme found within peroxisomes, which are membrane-bound organelles present in eukaryotic cells. MFP2 plays a crucial role in the breakdown of fatty acids and the detoxification of harmful substances within peroxisomes. It is involved in multiple steps of these processes, hence the term "multifunctional."
MFP2 catalyzes several reactions during the beta-oxidation of fatty acids, a process that breaks down long-chain fatty acids into shorter ones to generate energy for the cell. Specifically, MFP2 helps convert the breakdown products from earlier steps into forms that can enter subsequent steps of the beta-oxidation pathway.
Additionally, MFP2 is involved in the detoxification of molecules such as methanol and formaldehyde by facilitating their conversion to less harmful substances. This enzyme helps convert methanol into formic acid and then further metabolizes it, while formaldehyde is converted to formate.
Deficiencies in MFP2 or other peroxisomal proteins can lead to severe inherited metabolic disorders known as peroxisome biogenesis disorders (PBDs). These conditions can affect multiple organ systems and may cause neurological symptoms, developmental delays, vision loss, and hearing impairment.
Fatty acids are carboxylic acids with a long aliphatic chain, which are important components of lipids and are widely distributed in living organisms. They can be classified based on the length of their carbon chain, saturation level (presence or absence of double bonds), and other structural features.
The two main types of fatty acids are:
1. Saturated fatty acids: These have no double bonds in their carbon chain and are typically solid at room temperature. Examples include palmitic acid (C16:0) and stearic acid (C18:0).
2. Unsaturated fatty acids: These contain one or more double bonds in their carbon chain and can be further classified into monounsaturated (one double bond) and polyunsaturated (two or more double bonds) fatty acids. Examples of unsaturated fatty acids include oleic acid (C18:1, monounsaturated), linoleic acid (C18:2, polyunsaturated), and alpha-linolenic acid (C18:3, polyunsaturated).
Fatty acids play crucial roles in various biological processes, such as energy storage, membrane structure, and cell signaling. Some essential fatty acids cannot be synthesized by the human body and must be obtained through dietary sources.
Acetyl-CoA C-acetyltransferase (also known as acetoacetyl-CoA thiolase or just thiolase) is an enzyme involved in the metabolism of fatty acids and ketone bodies. Specifically, it catalyzes the reaction that converts two molecules of acetyl-CoA into acetoacetyl-CoA, which is a key step in the breakdown of fatty acids through beta-oxidation.
The enzyme works by bringing together two acetyl-CoA molecules and removing a coenzyme A (CoA) group from one of them, forming a carbon-carbon bond between the two molecules to create acetoacetyl-CoA. This reaction is reversible, meaning that the enzyme can also catalyze the breakdown of acetoacetyl-CoA into two molecules of acetyl-CoA.
There are several different isoforms of Acetyl-CoA C-acetyltransferase found in various tissues throughout the body, with differing roles and regulation. For example, one isoform is highly expressed in the liver and plays a key role in ketone body metabolism, while another isoform is found in mitochondria and is involved in fatty acid synthesis.
Inborn errors of lipid metabolism refer to genetic disorders that affect the body's ability to break down and process lipids (fats) properly. These disorders are caused by defects in genes that code for enzymes or proteins involved in lipid metabolism. As a result, toxic levels of lipids or their intermediates may accumulate in the body, leading to various health issues, which can include neurological problems, liver dysfunction, muscle weakness, and cardiovascular disease.
There are several types of inborn errors of lipid metabolism, including:
1. Disorders of fatty acid oxidation: These disorders affect the body's ability to convert long-chain fatty acids into energy, leading to muscle weakness, hypoglycemia, and cardiomyopathy. Examples include medium-chain acyl-CoA dehydrogenase deficiency (MCAD) and very long-chain acyl-CoA dehydrogenase deficiency (VLCAD).
2. Disorders of cholesterol metabolism: These disorders affect the body's ability to process cholesterol, leading to an accumulation of cholesterol or its intermediates in various tissues. Examples include Smith-Lemli-Opitz syndrome and lathosterolosis.
3. Disorders of sphingolipid metabolism: These disorders affect the body's ability to break down sphingolipids, leading to an accumulation of these lipids in various tissues. Examples include Gaucher disease, Niemann-Pick disease, and Fabry disease.
4. Disorders of glycerophospholipid metabolism: These disorders affect the body's ability to break down glycerophospholipids, leading to an accumulation of these lipids in various tissues. Examples include rhizomelic chondrodysplasia punctata and abetalipoproteinemia.
Inborn errors of lipid metabolism are typically diagnosed through genetic testing and biochemical tests that measure the activity of specific enzymes or the levels of specific lipids in the body. Treatment may include dietary modifications, supplements, enzyme replacement therapy, or gene therapy, depending on the specific disorder and its severity.
Gas Chromatography-Mass Spectrometry (GC-MS) is a powerful analytical technique that combines the separating power of gas chromatography with the identification capabilities of mass spectrometry. This method is used to separate, identify, and quantify different components in complex mixtures.
In GC-MS, the mixture is first vaporized and carried through a long, narrow column by an inert gas (carrier gas). The various components in the mixture interact differently with the stationary phase inside the column, leading to their separation based on their partition coefficients between the mobile and stationary phases. As each component elutes from the column, it is then introduced into the mass spectrometer for analysis.
The mass spectrometer ionizes the sample, breaks it down into smaller fragments, and measures the mass-to-charge ratio of these fragments. This information is used to generate a mass spectrum, which serves as a unique "fingerprint" for each compound. By comparing the generated mass spectra with reference libraries or known standards, analysts can identify and quantify the components present in the original mixture.
GC-MS has wide applications in various fields such as forensics, environmental analysis, drug testing, and research laboratories due to its high sensitivity, specificity, and ability to analyze volatile and semi-volatile compounds.
Oxidation-Reduction (redox) reactions are a type of chemical reaction involving a transfer of electrons between two species. The substance that loses electrons in the reaction is oxidized, and the substance that gains electrons is reduced. Oxidation and reduction always occur together in a redox reaction, hence the term "oxidation-reduction."
In biological systems, redox reactions play a crucial role in many cellular processes, including energy production, metabolism, and signaling. The transfer of electrons in these reactions is often facilitated by specialized molecules called electron carriers, such as nicotinamide adenine dinucleotide (NAD+/NADH) and flavin adenine dinucleotide (FAD/FADH2).
The oxidation state of an element in a compound is a measure of the number of electrons that have been gained or lost relative to its neutral state. In redox reactions, the oxidation state of one or more elements changes as they gain or lose electrons. The substance that is oxidized has a higher oxidation state, while the substance that is reduced has a lower oxidation state.
Overall, oxidation-reduction reactions are fundamental to the functioning of living organisms and are involved in many important biological processes.
Acyl-CoA oxidase is an enzyme that plays a crucial role in the breakdown of fatty acids within the body. It is located in the peroxisomes, which are small organelles found in the cells of living organisms. The primary function of acyl-CoA oxidase is to catalyze the initial step in the beta-oxidation of fatty acids, a process that involves the sequential removal of two-carbon units from fatty acid molecules in the form of acetyl-CoA.
The reaction catalyzed by acyl-CoA oxidase is as follows:
acyl-CoA + FAD → trans-2,3-dehydroacyl-CoA + FADH2 + H+
In this reaction, the enzyme removes a hydrogen atom from the fatty acyl-CoA molecule and transfers it to its cofactor, flavin adenine dinucleotide (FAD). This results in the formation of trans-2,3-dehydroacyl-CoA, FADH2, and a proton. The FADH2 produced during this reaction can then be used to generate ATP through the electron transport chain, while the trans-2,3-dehydroacyl-CoA undergoes further reactions in the beta-oxidation pathway.
There are two main isoforms of acyl-CoA oxidase found in humans: ACOX1 and ACOX2. ACOX1 is primarily responsible for oxidizing straight-chain fatty acids, while ACOX2 specializes in the breakdown of branched-chain fatty acids. Mutations in the genes encoding these enzymes can lead to various metabolic disorders, such as peroxisomal biogenesis disorders and Refsum disease.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
Enoyl-CoA hydratase is an enzyme that catalyzes the second step in the fatty acid oxidation process, also known as the beta-oxidation pathway. The systematic name for this reaction is (3R)-3-hydroxyacyl-CoA dehydratase.
The function of Enoyl-CoA hydratase is to convert trans-2-enoyl-CoA into 3-hydroxyacyl-CoA by adding a molecule of water (hydration) across the double bond in the substrate. This reaction forms a chiral center, resulting in the production of an (R)-stereoisomer of 3-hydroxyacyl-CoA.
The gene that encodes for Enoyl-CoA hydratase is called ECHS1, and mutations in this gene can lead to a rare genetic disorder known as Enoyl-CoA Hydratase Deficiency or ECHS1 Deficiency. This condition affects the breakdown of fatty acids in the body and can cause neurological symptoms such as developmental delay, seizures, and movement disorders.
A peroxisomal bifunctional enzyme is not a specific medical term, but it refers to a type of enzyme that has two distinct functional domains and is located within peroxisomes. Peroxisomes are small membrane-bound organelles found in the cells of many organisms, including humans, where they play a crucial role in various metabolic processes, such as fatty acid oxidation and detoxification of harmful substances.
The term "bifunctional" indicates that this enzyme possesses two distinct catalytic activities or functions within the same polypeptide chain. In the context of peroxisomal enzymes, bifunctional enzymes often participate in the breakdown of specific fatty acids, particularly very-long-chain fatty acids (VLCFAs) and branched-chain fatty acids (BCFAs).
An example of a peroxisomal bifunctional enzyme is the D-bifunctional protein (DBP), which has two catalytic domains: one for the oxidation of long-chain 2-enoyl-CoA intermediates and another for the hydratase/dehydrogenase activity. DBP plays a critical role in peroxisomal fatty acid beta-oxidation, particularly for VLCFAs and BCFAs, which cannot be efficiently metabolized by mitochondria.
Defects in peroxisomal bifunctional enzymes can lead to various genetic disorders, such as peroxisome biogenesis disorders (PBDs) or peroxisomal fatty acid oxidation disorders, which may result in severe neurological symptoms and developmental delays.
Acetyl-CoA C-acyltransferase is also known as acyl-CoA synthetase or thiokinase. It is an enzyme that plays a crucial role in the metabolism of fatty acids. Specifically, it catalyzes the formation of an acyl-CoA molecule from a free fatty acid and coenzyme A (CoA).
The reaction catalyzed by Acetyl-CoA C-acyltransferase is as follows:
R-COOH + CoA-SH + ATP → R-CO-SCoA + AMP + PPi
where R-COOH represents a free fatty acid, and R-CO-SCoA is an acyl-CoA molecule.
This enzyme exists in several forms, each specific to different types of fatty acids. Acetyl-CoA C-acyltransferase is essential for the metabolism of fatty acids because it activates them for further breakdown in the cell through a process called beta-oxidation. This enzyme is found in various tissues, including the liver, muscle, and adipose tissue.
Bipolar disorder, also known as manic-depressive illness, is a mental health condition that causes extreme mood swings that include emotional highs (mania or hypomania) and lows (depression). When you become depressed, you may feel sad or hopeless and lose interest or pleasure in most activities. When your mood shifts to mania or hypomania (a less severe form of mania), you may feel euphoric, full of energy, or unusually irritable. These mood swings can significantly affect your job, school, relationships, and overall quality of life.
Bipolar disorder is typically characterized by the presence of one or more manic or hypomanic episodes, often accompanied by depressive episodes. The episodes may be separated by periods of normal mood, but in some cases, a person may experience rapid cycling between mania and depression.
There are several types of bipolar disorder, including:
* Bipolar I Disorder: This type is characterized by the occurrence of at least one manic episode, which may be preceded or followed by hypomanic or major depressive episodes.
* Bipolar II Disorder: This type involves the presence of at least one major depressive episode and at least one hypomanic episode, but no manic episodes.
* Cyclothymic Disorder: This type is characterized by numerous periods of hypomania and depression that are not severe enough to meet the criteria for a full manic or depressive episode.
* Other Specified and Unspecified Bipolar and Related Disorders: These categories include bipolar disorders that do not fit the criteria for any of the other types.
The exact cause of bipolar disorder is unknown, but it appears to be related to a combination of genetic, environmental, and neurochemical factors. Treatment typically involves a combination of medication, psychotherapy, and lifestyle changes to help manage symptoms and prevent relapses.
3-Hydroxyacyl CoA Dehydrogenases (3-HADs) are a group of enzymes that play a crucial role in the beta-oxidation of fatty acids. These enzymes catalyze the third step of the beta-oxidation process, which involves the oxidation of 3-hydroxyacyl CoA to 3-ketoacyl CoA. This reaction is an essential part of the energy-generating process that occurs in the mitochondria of cells and allows for the breakdown of fatty acids into smaller molecules, which can then be used to produce ATP, the primary source of cellular energy.
There are several different isoforms of 3-HADs, each with specific substrate preferences and tissue distributions. The most well-known isoform is the mitochondrial 3-hydroxyacyl CoA dehydrogenase (M3HD), which is involved in the oxidation of medium and long-chain fatty acids. Other isoforms include the short-chain 3-hydroxyacyl CoA dehydrogenase (SCHAD) and the long-chain 3-hydroxyacyl CoA dehydrogenase (LCHAD), which are involved in the oxidation of shorter and longer chain fatty acids, respectively.
Deficiencies in 3-HADs can lead to serious metabolic disorders, such as 3-hydroxyacyl-CoA dehydrogenase deficiency (3-HAD deficiency), which is characterized by the accumulation of toxic levels of 3-hydroxyacyl CoAs in the body. Symptoms of this disorder can include hypoglycemia, muscle weakness, cardiomyopathy, and developmental delays. Early diagnosis and treatment of 3-HAD deficiency are essential to prevent serious complications and improve outcomes for affected individuals.
A mental disorder is a syndrome characterized by clinically significant disturbance in an individual's cognition, emotion regulation, or behavior. It's associated with distress and/or impaired functioning in social, occupational, or other important areas of life, often leading to a decrease in quality of life. These disorders are typically persistent and can be severe and disabling. They may be related to factors such as genetics, early childhood experiences, or trauma. Examples include depression, anxiety disorders, bipolar disorder, schizophrenia, and personality disorders. It's important to note that a diagnosis should be made by a qualified mental health professional.
Clofibrate is a medication that belongs to the class of drugs known as fibrates. It is primarily used to lower elevated levels of cholesterol and other fats (lipids) in the blood, specifically low-density lipoprotein (LDL), or "bad" cholesterol, and triglycerides, while increasing high-density lipoprotein (HDL), or "good" cholesterol. Clofibrate works by reducing the production of very-low-density lipoproteins (VLDL) in the liver, which in turn lowers triglyceride levels and indirectly reduces LDL cholesterol levels.
Clofibrate is available in oral tablet form and is typically prescribed for patients with high cholesterol or triglycerides who are at risk of cardiovascular disease, such as those with a history of heart attacks, strokes, or peripheral artery disease. It is important to note that clofibrate should be used in conjunction with lifestyle modifications, including a healthy diet, regular exercise, and smoking cessation.
Like all medications, clofibrate can have side effects, some of which may be serious. Common side effects include stomach upset, diarrhea, gas, and changes in taste. Less commonly, clofibrate can cause more severe side effects such as liver or muscle damage, gallstones, and an increased risk of developing certain types of cancer. Patients taking clofibrate should be monitored regularly by their healthcare provider to ensure that the medication is working effectively and to monitor for any potential side effects.
Anxiety disorders are a category of mental health disorders characterized by feelings of excessive and persistent worry, fear, or anxiety that interfere with daily activities. They include several different types of disorders, such as:
1. Generalized Anxiety Disorder (GAD): This is characterized by chronic and exaggerated worry and tension, even when there is little or nothing to provoke it.
2. Panic Disorder: This is characterized by recurring unexpected panic attacks and fear of experiencing more panic attacks.
3. Social Anxiety Disorder (SAD): Also known as social phobia, this is characterized by excessive fear, anxiety, or avoidance of social situations due to feelings of embarrassment, self-consciousness, and concern about being judged or viewed negatively by others.
4. Phobias: These are intense, irrational fears of certain objects, places, or situations. When a person with a phobia encounters the object or situation they fear, they may experience panic attacks or other severe anxiety responses.
5. Agoraphobia: This is a fear of being in places where it may be difficult to escape or get help if one has a panic attack or other embarrassing or incapacitating symptoms.
6. Separation Anxiety Disorder (SAD): This is characterized by excessive anxiety about separation from home or from people to whom the individual has a strong emotional attachment (such as a parent, sibling, or partner).
7. Selective Mutism: This is a disorder where a child becomes mute in certain situations, such as at school, but can speak normally at home or with close family members.
These disorders are treatable with a combination of medication and psychotherapy (cognitive-behavioral therapy, exposure therapy). It's important to seek professional help if you suspect that you or someone you know may have an anxiety disorder.
Mood disorders are a category of mental health disorders characterized by significant and persistent changes in mood, affect, and emotional state. These disorders can cause disturbances in normal functioning and significantly impair an individual's ability to carry out their daily activities. The two primary types of mood disorders are depressive disorders (such as major depressive disorder or persistent depressive disorder) and bipolar disorders (which include bipolar I disorder, bipolar II disorder, and cyclothymic disorder).
Depressive disorders involve prolonged periods of low mood, sadness, hopelessness, and a lack of interest in activities. Individuals with these disorders may also experience changes in sleep patterns, appetite, energy levels, concentration, and self-esteem. In severe cases, they might have thoughts of death or suicide.
Bipolar disorders involve alternating episodes of mania (or hypomania) and depression. During a manic episode, individuals may feel extremely elated, energetic, or irritable, with racing thoughts, rapid speech, and impulsive behavior. They might engage in risky activities, have decreased sleep needs, and display poor judgment. In contrast, depressive episodes involve the same symptoms as depressive disorders.
Mood disorders can be caused by a combination of genetic, biological, environmental, and psychological factors. Proper diagnosis and treatment, which may include psychotherapy, medication, or a combination of both, are essential for managing these conditions and improving quality of life.
"Pichia" is a genus of single-celled yeast organisms that are commonly found in various environments, including on plant and animal surfaces, in soil, and in food. Some species of Pichia are capable of causing human infection, particularly in individuals with weakened immune systems. These infections can include fungemia (bloodstream infections), pneumonia, and urinary tract infections.
Pichia species are important in a variety of industrial processes, including the production of alcoholic beverages, biofuels, and enzymes. They are also used as model organisms for research in genetics and cell biology.
It's worth noting that Pichia was previously classified under the genus "Candida," but it has since been reclassified due to genetic differences between the two groups.
Catalase is a type of enzyme that is found in many living organisms, including humans. Its primary function is to catalyze the decomposition of hydrogen peroxide (H2O2) into water (H2O) and oxygen (O2). This reaction helps protect cells from the harmful effects of hydrogen peroxide, which can be toxic at high concentrations.
The chemical reaction catalyzed by catalase can be represented as follows:
H2O2 + Catalase → H2O + O2 + Catalase
Catalase is a powerful antioxidant enzyme that plays an important role in protecting cells from oxidative damage. It is found in high concentrations in tissues that produce or are exposed to hydrogen peroxide, such as the liver, kidneys, and erythrocytes (red blood cells).
Deficiency in catalase activity has been linked to several diseases, including cancer, neurodegenerative disorders, and aging. On the other hand, overexpression of catalase has been shown to have potential therapeutic benefits in various disease models, such as reducing inflammation and oxidative stress.
 Peroxisomal disorder
Peroxisomal disorder
Acyl-CoA oxidase deficiency
Infantile Refsum disease
Zellweger syndrome
Peroxisome
Primary hyperoxaluria
Alpha-methylacyl-CoA racemase
PEX19
Phytol
PEX10
Bisulfite sequencing
PEX3
Peroxin
Plasmalogen
Jean Bennett
Rhizomelic chondrodysplasia punctata
Hugo Moser (scientist)
Protein targeting
PEX1
PEX13
Neonatal adrenoleukodystrophy
Epicanthic fold
Myelin
D-bifunctional protein deficiency
PEX6
EHHADH
Pipecolic acidemia
Very long chain fatty acid
Phytanoyl-CoA dioxygenase
Peroxisomal biogenesis factor 2
Peroxisomal disorder - Wikipedia
 Peroxisomal Disorders: Background, Pathophysiology, Epidemiology
Peroxisomal Disorders: Background, Pathophysiology, Epidemiology
Complementation analysis in patients with the clinical phenotype of a generalised peroxisomal disorder. | Journal of Medical...
 Peroxisomal Disorders - Children's Health Issues - MSD Manual Consumer Version
Peroxisomal Disorders - Children's Health Issues - MSD Manual Consumer Version
 Peroxisomal acyl-CoA oxidase deficiency: MedlinePlus Genetics
Peroxisomal acyl-CoA oxidase deficiency: MedlinePlus Genetics
Plasma and red blood cell fatty acids in peroxisomal disorders<...
Peroxisomal Disorders: Background, Pathophysiology, Epidemiology
 October, 2022 - The Global Foundation for Peroxisomal Disorders
October, 2022 - The Global Foundation for Peroxisomal Disorders
March, 2018 - The Global Foundation for Peroxisomal Disorders
 Disorders with deficiency of a single peroxisomal enzyme | Rare Diseases | RareGuru
Disorders with deficiency of a single peroxisomal enzyme | Rare Diseases | RareGuru
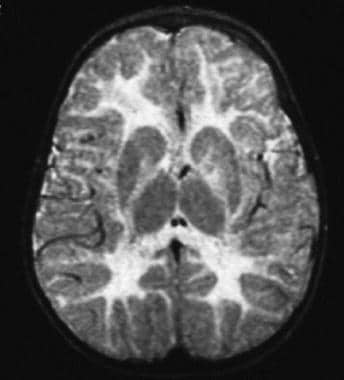
 Peroxisomal leukoencephalopathy
Peroxisomal leukoencephalopathy
 Adrenoleukodystrophy News, Research
Adrenoleukodystrophy News, Research
Very long-chain fatty acids in diagnosis, pathogenesis, and therapy of peroxisomal disorders<...
GNPAT gene: MedlinePlus Genetics
AMACR gene: MedlinePlus Genetics
 Support for Firefighters, Inc. helped by your shopping
Support for Firefighters, Inc. helped by your shopping
CHILD Syndrome Differential Diagnoses
Plus it
 Kristina Cusmano-Ozog's Profile | Stanford Profiles
Kristina Cusmano-Ozog's Profile | Stanford Profiles
 BIT: Forschung
BIT: Forschung
BIT: Publikationen
Refsum Disease: Practice Essentials, Background, Pathophysiology
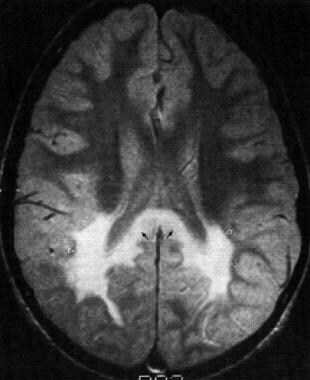
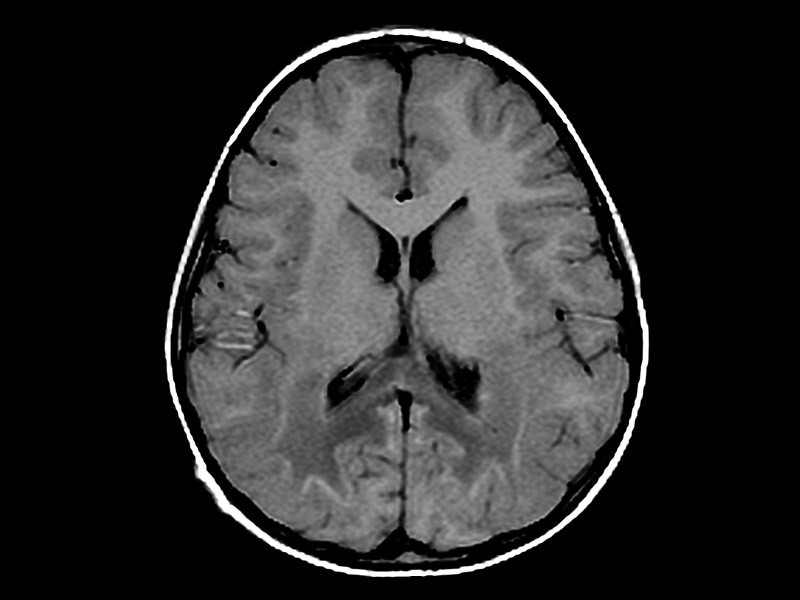
 Septo-optic dysplasia | Radiology Reference Article | Radiopaedia.org
Septo-optic dysplasia | Radiology Reference Article | Radiopaedia.org
 Drug Trials Snapshot: CHOLBAM (bile acid synthesis disorders) | FDA
Drug Trials Snapshot: CHOLBAM (bile acid synthesis disorders) | FDA
KEGG PATHWAY: hsa04146
 Ghent University Academic Bibliography
Ghent University Academic Bibliography
 Recent departmental publications - Ophthalmology & Visual Sciences
Recent departmental publications - Ophthalmology & Visual Sciences
 Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
 Structural and functional insights of the human peroxisomal ABC transporter ALDP | eLife
Structural and functional insights of the human peroxisomal ABC transporter ALDP | eLife
 Adult onset seizures in learning disability | Royal College of Physicians of Edinburgh
Adult onset seizures in learning disability | Royal College of Physicians of Edinburgh
Enzyme13
- Peroxisomal acyl-CoA oxidase deficiency is caused by mutations in the ACOX1 gene, which provides instructions for making an enzyme called peroxisomal straight-chain acyl-CoA oxidase. (medlineplus.gov)
- The peroxisomal straight-chain acyl-CoA oxidase enzyme plays a role in the breakdown of certain fat molecules called very long-chain fatty acids (VLCFAs). (medlineplus.gov)
- ACOX1 gene mutations prevent the peroxisomal straight-chain acyl-CoA oxidase enzyme from breaking down VLCFAs efficiently. (medlineplus.gov)
- Connect with other caregivers and patients with Disorders with deficiency of a single peroxisomal enzyme and get the support you need. (rareguru.com)
- Peroxisomal leukoencephalopathies include diseases belonging to the Zellweger spectrum and the rhizomelic chondrodysplasia punctata spectrum, as well as some single enzyme defects of peroxisomal β-oxidation. (nih.gov)
- AMACR gene mutations that result in a lack of functional AMACR enzyme have also been identified in infants with a life-threatening disorder called congenital bile acid synthesis defect type 4. (medlineplus.gov)
- Patients with RD are unable to degrade phytanic acid because of a deficient activity of phytanoyl-CoA hydroxylase (PhyH), a peroxisomal enzyme catalyzing the first step of phytanic acid alpha-oxidation. (medscape.com)
- The first group is disorders of peroxisome biogenesis which include Zellweger syndrome, and the second group is single peroxisomal enzyme deficiencies. (genome.jp)
- CHOLBAM was studied for the treatment of bile acid synthesis disorders due to what are called single enzyme defects (SEDs). (fda.gov)
- A peroxisomal enzyme. (leedsth.nhs.uk)
- PubChem] Cholic acid, formulated as Cholbam capsules, is approved by the United States Food and Drug Administration as a treatment for children and adults with bile acid synthesis disorders due to single enzyme defects, and for peroxisomal disorders (such as Zellweger syndrome). (rcsb.org)
- Cholbam is FDA-approved for the treatment of bile acid synthesis disorders due to single enzyme deficiencies and adjunctive treatment of peroxisomal disorders in patients who show signs or symptoms or liver disease. (businesswire.com)
- Patients with Refsum disease are unable to degrade phytanic acid because of a deficient activity of phytanoyl-CoA hydroxylase (PhyH), a peroxisomal enzyme catalyzing the first step of phytanic acid alpha-oxidation. (medscape.com)
Peroxisomes8
- Peroxisomal disorders are a group of genetically heterogeneous metabolic diseases that share dysfunction of peroxisomes. (medscape.com)
- The generalised peroxisomal disorders (GPDs) Zellweger syndrome (ZS), neonatal adrenoleucodystrophy (NALD), and infantile Refsum's disease (IRD) are autosomal recessive disorders associated with a failure to assemble mature peroxisomes. (bmj.com)
- Peroxisomes' deficiencies lead to severe and often fatal inherited peroxisomal disorders (PD). (genome.jp)
- Generalized peroxisome-deficient disorders including cerebro-hepato-renal Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease are autosomal recessive diseases, where catalase-containing particles (peroxisomes) are morphologically absent. (jci.org)
- Zellweger syndrome is also known as cerebrohepatorenal syndrome is a rare, congenital disorder, characterized by the reduction or absence of peroxisomes in the cells of the liver , kidneys, and brain. (healthcaremagic.com)
- PEX1, also known as Peroxisome biogenesis factor 1, is a 1,283 amino acid protein that is 143 kDa, cytoplasmic but is often anchored to a peroxisomal membrane where it forms a heteromeric complex and plays a role in the import of proteins into peroxisomes and peroxisome biogenesis. (novusbio.com)
- Peroxisomes can arise de novo through the endoplasmic reticulum (ER) derived pre-peroxisomal vesicles ( Agrawal and Subramani, 2016 ). (biorxiv.org)
- As peroxisomes grow and mature, cargo import into the peroxisomal matrix takes place. (biorxiv.org)
Adjunctive treatment of peroxisomal disorders1
- and as adjunctive treatment of peroxisomal disorders including Zellweger spectrum disorders in patients who exhibit manifestations of liver disease, steatorrhea or complications from decreased fat soluble vitamin absorption. (rcsb.org)
Adrenoleukodystrophy16
- PBD-ZSD represents a continuum of disorders including infantile Refsum disease, neonatal adrenoleukodystrophy, and Zellweger syndrome. (wikipedia.org)
- Treatment of the peroxisomal leukoencephalopathies is largely symptomatic, except for boys affected by the cerebral form of X-linked adrenoleukodystrophy in whom a bone marrow/hematopoietic stem cell transplant can be lifesaving, at least in the early stages of the disease. (nih.gov)
- Adrenoleukodystrophy (ALD) is one of a group of genetic disorders called the leukodystrophies that cause damage to the myelin sheath, an insulating membrane that surrounds nerve cells in the brain. (news-medical.net)
- Lorenzo's Oil" was to help a seriously ill boy suffering from a peroxisomal disorder (adrenoleukodystrophy/ALD). (news-medical.net)
- In a recent clinical trial, a gene therapy to treat cerebral adrenoleukodystrophy (CALD) -- a neurodegenerative disease that typically claims young boys' lives within 10 years of diagnosis -- effectively stabilized the disease's progression in 88 percent of patients, researchers from the Dana-Farber/Boston Children's Cancer and Blood Disorders Center and Massachusetts General Hospital report today. (news-medical.net)
- X-linked adrenoleukodystrophy (ALD) is a rare peroxisomal disorder that affects the white matter of the CNS, adrenal cortex, and testes (1-3). (ajnr.org)
- Adrenoleukodystrophy protein (ALDP) is responsible for the transport of very-long-chain fatty acids (VLCFAs) and corresponding CoA-esters across the peroxisomal membrane. (elifesciences.org)
- Dysfunction of ALDP leads to peroxisomal metabolic disorder exemplified by X-linked adrenoleukodystrophy (ALD). (elifesciences.org)
- The adrenoleukodystrophy protein (ALDP) or ABCD1 is an ABC transporter that participates in the transport of free very long-chain fatty acids and their CoA esters across the peroxisomal membrane. (elifesciences.org)
- By determining the cryo EM structure of human ABCD1 the study represents a valuable insight into its transport mechanism and the mechanistic basis for mutations causing the severe neurodegenerative disorder, X-linked adrenoleukodystrophy. (elifesciences.org)
- Early in the course of the disease, one may consider bone marrow transplantation in some lysosomal disorders and in the cerebral form of X-linked adrenoleukodystrophy. (vumc.com)
- Other peroxisome biogenesis disorders are Infantile Refsum disease, neonatal adrenoleukodystrophy and rhizomelic chondrodysplasia. (healthcaremagic.com)
- The zellweger syndrome spectrum is one of the peroxisomal biogenesis disorders and x-adrenoleukodystrophy belongs to a group that is characterized by individual peroxisomal protein dysfunctions. (uni-goettingen.de)
- Due to the complex diagnostics and the lack of therapy, there is still a lot to learn about peroxisomal diseases such as Zellweger syndrome and x-adrenoleukodystrophy. (uni-goettingen.de)
- Biologic, morphologic, and biochemical investigations performed in 2 patients demonstrate multiple peroxisomal deficiencies in the cerebrohepatorenal syndrome of Zellweger (CHRS) and neonatal adrenoleukodystrophy (NALD). (lookformedical.com)
- X-linked adrenoleukodystrophy is a peroxisomal disorder caused by a mutation in ABCD1 gene. (elsevierpure.com)
Diseases5
- Scientists from the University of Bonn, the German Center for Neurodegenerative Diseases and the German Cancer Research Center investigated such peroxisomal diseases on fruit flies. (news-medical.net)
- Although decreased citrulline is used as a newborn screening (NBS) marker to identify proximal urea cycle disorders (UCDs), it is also a feature of some mitochondrial diseases, including MT-ATP6 mitochondrial disease. (stanford.edu)
- This group includes lysosomal storage disorders, various mitochondrial diseases, other neurometabolic disorders, and several other miscellaneous disorders. (medscape.com)
- 1 In diseases involving peroxisomal impairment the branched chain fatty acids phytanic acid and pristanic acid accumulate in high amounts due to deficiencies in alpha- and beta-oxidation. (matreya.com)
- Lara Bloom is the President and CEO of The Ehlers-Danlos Society and responsible for globally raising awareness of rare, chronic, and invisible diseases, specializing in the Ehlers-Danlos syndromes, hypermobility spectrum disorders (HSD), and related conditions. (globalgenes.org)
Deficiency13
- PAF deficiency impairs glutaminergic signaling and has been implicated in human lissencephaly and neuronal migration disorders. (medscape.com)
- One further patient (BOX-1) had the clinical phenotype of ZS, but biochemical investigations indicated an isolated deficiency of peroxisomal beta oxidation. (bmj.com)
- Peroxisomal acyl-CoA oxidase deficiency is a disorder that causes deterioration of nervous system functions (neurodegeneration) beginning in infancy. (medlineplus.gov)
- Newborns with peroxisomal acyl-CoA oxidase deficiency have weak muscle tone (hypotonia) and seizures. (medlineplus.gov)
- Most babies with peroxisomal acyl-CoA oxidase deficiency learn to walk and begin speaking, but they experience a gradual loss of these skills (developmental regression), usually beginning between the ages of 1 and 3. (medlineplus.gov)
- Most children with peroxisomal acyl-CoA oxidase deficiency do not survive past early childhood. (medlineplus.gov)
- Peroxisomal acyl-CoA oxidase deficiency is a rare disorder. (medlineplus.gov)
- It is unclear exactly how VLCFA accumulation leads to the specific features of peroxisomal acyl-CoA oxidase deficiency. (medlineplus.gov)
- Leukodystrophy is likely involved in the development of the neurological abnormalities that occur in peroxisomal acyl-CoA oxidase deficiency. (medlineplus.gov)
- Some researchers consider congenital bile acid synthesis defect type 4 and AMACR deficiency (see above) to be variations of the same disorder. (medlineplus.gov)
- Human dihydroxyacetonephosphate acyltransferase deficiency: a new peroxisomal disorder. (medlineplus.gov)
- Deficiency of plasmalogens causes profound abnormalities in the myelination of nerve cells, which is one reason why many peroxisomal disorders lead to neurological disease. (nih.gov)
- Multiple peroxisomal enzymatic deficiency disorders. (lookformedical.com)
Beta-oxidation2
- Another major function of the peroxisomal beta-oxidation system is related to the biosynthesis of polyunsaturated fatty acid (C22:6w3). (medscape.com)
- Specifically, it is involved in the first step of a process called the peroxisomal fatty acid beta-oxidation pathway. (medlineplus.gov)
Defects4
- Peroxisomal disorders represent a class of medical conditions caused by defects in peroxisome functions. (wikipedia.org)
- One of the most challenging aspects in pathogenesis of these disorders is the mechanism responsible for neuronal migration defects. (medscape.com)
- Molecular basis of peroxisomal biogenesis disorders caused by defects in peroxisomal matrix protein import. (ruhr-uni-bochum.de)
- Enzymatic deficiencies reflected the peroxisomal defects. (lookformedical.com)
Peroxisome biogenesis disorder5
- RD can be classified as a peroxisome biogenesis disorder. (medscape.com)
- Infantile Refsum's disease is a peroxisome biogenesis disorder that falls within the Zellweger disorder spectrum, sharing similar clinical and biochemical features but the clinical picture is less severe such that some patients survive to adulthood. (rcpe.ac.uk)
- Late onset white matter disease in peroxisome biogenesis disorder. (rcpe.ac.uk)
- Refsum disease can be classified as a peroxisome biogenesis disorder. (medscape.com)
- [ 1 ] Infantile Refsum disease is a peroxisome biogenesis disorder. (medscape.com)
Phytanic acid alpha-oxidation1
- Peroxisomal disorders affecting phytanic acid alpha-oxidation: a review. (medlineplus.gov)
Alpha-oxidation1
- Refsum disease is a recessive disorder characterized by defective peroxisomal alpha-oxidation of phytanic acid. (medscape.com)
Gene9
- Inheritance of Single-Gene Disorders Genes are segments of deoxyribonucleic acid (DNA) that contain the code for a specific protein that functions in one or more types of cells in the body or code for functional RNA molecules. (msdmanuals.com)
- In most peroxisomal disorders, both parents of the affected child carry a copy of the abnormal gene. (msdmanuals.com)
- Because usually two copies of the abnormal gene are necessary for the disorder to occur, usually neither parent has the disorder. (msdmanuals.com)
- which means only one copy of the abnormal gene can cause the disorder in boys. (msdmanuals.com)
- Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. (medlineplus.gov)
- This gene normally encodes for a peroxisomal membrane protein called ALD-P. (ajnr.org)
- Several single gene disorders share clinical and radiologic characteristics with multiple sclerosis and have the potential to be overlooked in the differential diagnostic evaluation of both adult and paediatric patients with multiple sclerosis. (medscape.com)
- Recognition of a single-gene disorder as causal for a patient's 'multiple sclerosis-like' phenotype is critically important for accurate direction of patient management, and evokes broader genetic counselling implications for affected families. (medscape.com)
- Here we review single gene disorders that have the potential to mimic multiple sclerosis, provide an overview of clinical and investigational characteristics of each disorder, and present guidelines for when clinicians should suspect an underlying heritable disorder that requires diagnostic confirmation in a patient with a definite or probable diagnosis of multiple sclerosis. (medscape.com)
Induces peroxisomal2
- Administration of phenobarbital and clofibrate, an agent that induces peroxisomal proliferation and enzymatic activities, to the NALD patient did not bring about any changes in plasma metabolites, liver peroxisome population, or oxidizing activities. (lookformedical.com)
- Depletion of HNRNPA1 induces peroxisomal autophagy by regulating PEX1 expression. (bvsalud.org)
Infantile Refsum's disease1
- The cause of his learning disability was uncertain but a peroxisomal disorder, possibly infantile Refsum's disease, had been suspected because of early onset blindness although metabolic investigations in childhood had been nondiagnostic. (rcpe.ac.uk)
Rhizomelic1
- Peroxisome biogenesis disorders (PBDs) include the Zellweger syndrome spectrum (PBD-ZSD) and rhizomelic chondrodysplasia punctata type 1 (RCDP1). (wikipedia.org)
Biochemical4
- The diagnosis of a peroxisomal disorder can be determined by a battery of biochemical assays in blood and/or urine, and should be confirmed in cultured fibroblasts and DNA analysis. (nih.gov)
- Biochemical genetic testing and newborn screening are essential laboratory services for the screening, detection, diagnosis, and monitoring of inborn errors of metabolism or inherited metabolic disorders. (cdc.gov)
- These recommendations are intended for laboratories that perform biochemical genetic testing to improve the quality of laboratory services and for newborn screening laboratories to ensure the quality of laboratory practices for inherited metabolic disorders. (cdc.gov)
- An infantile form of Refsum disease also exists and is an autosomal recessive disorder of peroxisomal biogenesis, leading to many biochemical abnormalities, including elevated plasma concentration of phytanic acid, pristanic acid, very long chain fatty acids, and C27 bile acids. (medscape.com)
Mutations4
- Peroxisome biogenesis disorders are usually caused by biallelic mutations in the 13 PEX genes. (rcpe.ac.uk)
- Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. (rcpe.ac.uk)
- This is an autosomal recessive peroxisomal biogenesis disorder (PBD) resulting from mutations in a number of PEX genes ( PEX1 , PEX2 , PEX3 , PEX12 , PEX26 ). (arizona.edu)
- Various mutations in the PEX genes are genetic causes for the development of inheritable peroxisomal-biogenesis disorders, such as Zellweger syndrome . (bvsalud.org)
Metabolism4
- Overview of Hereditary Metabolic Disorders Hereditary metabolic disorders are inherited genetic conditions that cause metabolism problems. (msdmanuals.com)
- Human disorders of peroxisome metabolism and biogenesis. (rcpe.ac.uk)
- To detect disorders of hormone production that may affect baby's metabolism, response to infection, ability to regulate salt levels and sex characteristics. (kkh.com.sg)
- These results establish Hif-2α as a negative regulator of peroxisome abundance and metabolism and suggest a mechanism by which cells attune peroxisomal function with oxygen availability. (uzh.ch)
Diagnosis1
- The demonstration of abnormal levels of fatty acids or plasmalogens in plasma or red blood cells is key to the diagnosis of peroxisomal disorders. (psu.edu)
Autophagy1
- Hif-2α activation augments peroxisome turnover by selective autophagy (pexophagy) and thereby changes lipid composition reminiscent of peroxisomal disorders. (uzh.ch)
PEX12
- The peroxisomal PTS1-Import defect of PEX1-deficient cells is independent of pexophagy in Saccharomyces cerevisiae. (ruhr-uni-bochum.de)
- Taken together, our results suggest that depletion of HNRNPA1 increases peroxisomal ROS and pexophagy by downregulating PEX1 expression. (bvsalud.org)
Receptor5
- VK5211, the company's lead program for muscle and bone disorders, is an orally available, non-steroidal selective androgen receptor modulator (SARM) being developed for the treatment of patients recovering from non-elective hip fracture surgery. (news-medical.net)
- The deubiquitination of the PTS1-import receptor Pex5p is required for peroxisomal matrix protein import. (ruhr-uni-bochum.de)
- Cysteine-specific ubiquitination protects the peroxisomal import receptor Pex5p against proteasomal degradation. (ruhr-uni-bochum.de)
- The cycling peroxisomal targeting signal type 1 - receptor Pex5p: reaching the circle`s end with ubiquitin. (ruhr-uni-bochum.de)
- The peroxisomal receptor dislocation pathway: to the exportomer and beyond. (ruhr-uni-bochum.de)
Zellweger Syndrome1
- Zellweger syndrome is the most severe form of peroxisome biogenesis disorders. (healthcaremagic.com)
Fatty acids1
- Abnormally high levels of very long-chain fatty acids (VLCFA) are a feature in nine of the fifteen peroxisomal disorders that have been identified so far. (johnshopkins.edu)
Newborns1
- Although most newborns with these disorders look healthy at birth, they may be at risk of having serious health problems later in life such as learning difficulties, recurrent sicknesses and even death if their disorder is not detected and treated early. (kkh.com.sg)
Matrix protein import2
- ATP-driven processes of peroxisomal matrix protein import. (ruhr-uni-bochum.de)
- Regulation of peroxisomal matrix protein import by ubiquitination. (ruhr-uni-bochum.de)
Proteins2
- Matrix proteins in the cytosol are recognized by peroxisomal targeting signals (PTS) and transported to the docking complex at the peroxisomal membrane. (genome.jp)
- Several peroxisomal proteins , known as peroxins (PEXs), control peroxisome biogenesis and degradation. (bvsalud.org)
Neurological1
- This disorder leads to a variety of neurological problems that begin in adulthood, including gradual loss in intellectual functioning (cognitive decline), seizures, and weakness and loss of sensation in the limbs due to nerve damage (sensorimotor neuropathy). (medlineplus.gov)
Autosomal recessive disorders1
- Zellweger spectrum disorder (ZSD) is a group of autosomal recessive disorders caused by biallelic pathogenic variants in any one of the 13 PEX genes essential for peroxisomal biogenesis. (stanford.edu)
Cytosol1
- Therefore, we are interested in two central transport processes: (1) the biogenetic transport of newly synthesised peroxisomal enzymes from the cytosol over the peroxisomal membrane into the matrix as well as (2) the transport of the entire organelle to the vacuolar lumen in the context of selective autophagic degradation. (ruhr-uni-bochum.de)
Fission3
- The novel peroxin Pex37: the Pxmp2-family joins the peroxisomal fission machinery. (ruhr-uni-bochum.de)
- Here we probe these fundamental questions using peroxisomal compartmentalization of the last steps of lysine and histidine biosynthesis in the fission yeast Schizosaccharomyces japonicus . (biorxiv.org)
- The fission yeast clade has lost the peroxisomal FA β-oxidation system. (biorxiv.org)
PEX61
- A homozygous Gly470Ala variant in PEX6 causes severe Zellweger spectrum disorder. (stanford.edu)
Pexophagy1
- Inhibition of the generation of peroxisomal ROS by treatment with NAC significantly suppressed pexophagy in HNRNPA1-deficient cells . (bvsalud.org)
Refsum1
- Of all the above mentioned peroxisomal disorders infantile Refsum disease is the least severe form of the disease. (healthcaremagic.com)
Abnormalities1
- Migrational abnormalities are the most likely causes of the severe seizures and psychomotor retardation associated with many types of peroxisomal disorders. (medscape.com)
Liver3
- Collectively, PBDs are autosomal recessive developmental brain disorders that also result in skeletal and craniofacial dysmorphism, liver dysfunction, progressive sensorineural hearing loss, and retinopathy. (wikipedia.org)
- Viking Therapeutics, Inc., a clinical-stage biopharmaceutical company focused on the development of novel, first-in-class or best-in-class therapies for metabolic and endocrine disorders, today announced that it has submitted an investigational new drug (IND) application to the U.S. Food and Drug Administration to conduct a Phase 2 study of VK2809 in patients with hypercholesterolemia and fatty liver disease. (news-medical.net)
- In the trials that supported the FDA approval of CHOLBAM, 64% of patients with rare bile acid synthesis disorders had improvement in body weight as a measure of growth as well as improvements in liver tests that are expected to be related to less liver damage. (fda.gov)
Spectrum disorders2
- CHOLBAM was also studied for the treatment of what is called peroxisomal disorders (PDs) including Zellweger's spectrum disorders. (fda.gov)
- Zellweger spectrum disorders: clinical overview and management approach. (rcpe.ac.uk)
Neuromuscular disorders2
- neurodevelopmental, metabolic and neuromuscular disorders. (nih.gov)
- Neuromuscular Disorders , 29 (2), 146-149. (elsevierpure.com)
Fibroblasts1
- Complementation analysis by cell fusion of the CHO mutants with cultured fibroblasts from patients with generalized peroxisomal disorders revealed that two CHO mutants (Z24 and ZP92) represent the human complementation groups, E (the same as group 1 in the U.S.) and C (the same as group 4), respectively. (jci.org)
Clinical4
- Complementation analysis in patients with the clinical phenotype of a generalised peroxisomal disorder. (bmj.com)
- Viking Therapeutics, Inc., a clinical-stage biopharmaceutical company focused on the development of novel, first-in-class or best-in-class therapies for metabolic and endocrine disorders, today announced the initiation of dosing in the company's Phase 2 clinical trial of VK5211 in patients who recently suffered a hip fracture. (news-medical.net)
- Viking Therapeutics, Inc., a clinical-stage biopharmaceutical company focused on the development of novel, first-in-class or best-in-class therapies for metabolic and endocrine disorders, today announced the successful completion of a short-term safety, tolerability, and pharmacokinetic study of VK5211 in healthy elderly subjects. (news-medical.net)
- Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. (medscape.com)
Infants2
- CHOLBAM is a treatment for infants, children and adults who have a rare condition called bile acid synthesis disorders. (fda.gov)
- Fifty years ago, these infants' disorders would have gone undetected until symptoms appeared, leading to possible death or lifelong disability. (myadlm.org)