Porphyrias
Porphyrias, Hepatic
Porphyria, Acute Intermittent
Porphyria, Erythropoietic
Porphyria, Variegate
Porphyria Cutanea Tarda
Hydroxymethylbilane Synthase
Porphobilinogen
Uroporphyrinogen Decarboxylase
Protoporphyrinogen Oxidase
Porphyrins
Uroporphyrinogen III Synthetase
Uroporphyrins
Porphyria, Hepatoerythropoietic
Coproporphyrins
5-Aminolevulinate Synthetase
Coproporphyria, Hereditary
Uroporphyrinogens
Aminolevulinic Acid
Porphyrinogens
Porphobilinogen Synthase
Ammonia-Lyases
Flavoproteins
Ferrochelatase
Heme
Coproporphyrinogen Oxidase
Coproporphyrinogens
Dicarbethoxydihydrocollidine
Skin Diseases
Oxidoreductases Acting on CH-CH Group Donors
Protoporphyria, Erythropoietic
Photosensitivity Disorders
Bloodletting
Protoporphyrins
Allylisopropylacetamide
Precipitating Factors
Hemochromatosis
Erythrocytes
Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. (1/11)
Protoporphyrinogen IX oxidase (PPO), the last common enzyme of haem and chlorophyll biosynthesis, catalyses the oxidation of protoporphyrinogen IX to protoporphyrin IX. The membrane-embedded flavoprotein is the target of a large class of herbicides. In humans, a defect in PPO is responsible for the dominantly inherited disease variegate porphyria. Here we present the crystal structure of mitochondrial PPO from tobacco complexed with a phenyl-pyrazol inhibitor. PPO forms a loosely associated dimer and folds into an FAD-binding domain of the p-hydroxybenzoate-hydrolase fold and a substrate-binding domain that enclose a narrow active site cavity beneath the FAD and an alpha-helical membrane-binding domain. The active site architecture suggests a specific substrate-binding mode compatible with the unusual six-electron oxidation. The membrane-binding domains can be docked onto the dimeric structure of human ferrochelatase, the next enzyme in haem biosynthesis, embedded in the opposite side of the membrane. This modelled transmembrane complex provides a structural explanation for the uncoupling of haem biosynthesis observed in variegate porphyria patients and in plants after inhibiting PPO. (+info)Recovery from a variegate porphyria by a liver transplantation. (2/11)
The porphyrias are a group of inherited or acquired enzymatic defects of heme biosynthesis. Each type of porphyria has a characteristic pattern of overproduction and accumulation of heme precursors based on the location of dysfunctional enzyme in the heme synthetic pathway. Variegate porphyria, one of the acute hepatic porphyrias, is characterized by a partial reduction in protoporphyrinogen oxidase, the seventh enzyme of the heme biosynthetic pathway. A case of liver transplantation is described with a recovery from a variegate porphyria. Acute porphyria is commonly worsened by a wide variety of medications. We describe a step-by-step perioperative management protocol. (+info)Swiss patients with variegate porphyria have unique mutations. (3/11)
BACKGROUND: Variegate porphyria (VP), also known as South African porphyria, is a low-penetrance, autosomal dominant disorder as the result of a partial deficiency of protoporphyrinogen oxidase (PPOX). Clinically, VP is characterised by photosensitivity and neurovisceral attacks whereby the two symptoms can appear separately or together in patients. VP is little known in Switzerland. In this study, we report a clinical, biochemical and mutational study of eight Swiss VP patients and their families. RESULTS: Six of the eight index patients presented with only skin symptoms, and one with only neurological symptoms. Another patient had both skin and neurological symptoms. Faecal porphyrin excretion was elevated in all patients thus enabling diagnosis. Four different mutations including three novel mutations (G11D, 1041-1042 ins T and 1262-1263 ins 22bp) were identified in this cohort. Mutation 1082-1083 ins C, which had been reported in the French VP population, was shared by five apparently unrelated patients of this study. CONCLUSION: The novel PPOX gene mutations are apparently unique to the Swiss population. Both clinical and biochemical presentations varied considerably even among those patients who carried an identical mutation, which does not favour the existence of a genotype-phenotype correlation in VP. (+info)montalcino, A zebrafish model for variegate porphyria. (4/11)
(+info)Genetic and biochemical studies in Argentinean patients with variegate porphyria. (5/11)
(+info)Hepatocellular carcinoma in variegate porphyria: a serious complication. (6/11)
(+info)Structural insight into human variegate porphyria disease. (7/11)
(+info)Digenic inheritance of mutations in the coproporphyrinogen oxidase and protoporphyrinogen oxidase genes in a unique type of porphyria. (8/11)
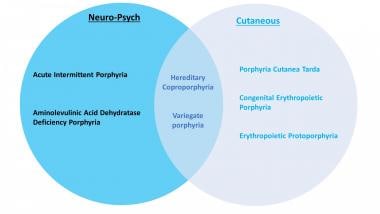
(+info)Porphyrias are a group of rare genetic disorders that affect the production of heme, a component in hemoglobin that carries oxygen in the blood. The diseases are caused by mutations in the genes involved in the production of heme, leading to the buildup of porphyrins or their precursors in the body. These substances can be toxic and can cause various symptoms depending on the specific type of porphyria. Symptoms may include abdominal pain, neurological problems, and skin issues. Porphyrias are typically divided into two categories: acute porphyrias, which affect the nervous system, and cutaneous porphyrias, which primarily affect the skin.
Hepatic porphyrias are a group of rare genetic disorders that affect the production of heme in the liver. Heme is a crucial component of hemoglobin, the protein in red blood cells that carries oxygen throughout the body. In hepatic porphyrias, there is a buildup of porphyrins or porphyrin precursors, which are toxic and can cause a variety of symptoms.
The four types of hepatic porphyrias are:
1. Acute Intermittent Porphyria (AIP): This is the most common type of hepatic porphyria. It is characterized by attacks of abdominal pain, nausea, vomiting, constipation, and neurological symptoms such as muscle weakness, seizures, and mental changes.
2. Variegate Porphyria (VP): This type of porphyria is more common in South Africa but can occur worldwide. It is characterized by skin symptoms such as blistering and scarring after exposure to sunlight, as well as acute attacks similar to those seen in AIP.
3. Hereditary Coproporphyria (HCP): This type of porphyria is similar to VP, but the symptoms are usually less severe. It can cause both skin symptoms and acute attacks.
4. ALA Dehydratase Deficiency Porphyria (ADDP): This is the rarest type of hepatic porphyria. It is characterized by severe neurological symptoms and is often diagnosed in infancy or early childhood.
The diagnosis of hepatic porphyrias typically involves measuring the levels of porphyrins and their precursors in the urine, blood, or stool during an attack or between attacks. Treatment may include avoiding trigger factors such as certain medications, alcohol, and smoking, as well as providing supportive care during acute attacks. In some cases, medication to reduce porphyrin production or prevent attacks may be necessary.
Acute Intermittent Porphyria (AIP) is a rare inherited metabolic disorder that affects the production of heme, a component in hemoglobin. This condition is part of a group of disorders known as the porphyrias, which are caused by genetic mutations that result in enzyme deficiencies needed to produce heme.
In AIP, specifically, there is a deficiency in the enzyme porphobilinogen deaminase (PBGD). This leads to the buildup of porphyrin precursors, particularly porphobilinogen and delta-aminolevulinic acid (ALA), in the body. These substances are toxic and can cause acute attacks when they accumulate in high concentrations.
Acute attacks are characterized by severe abdominal pain, nausea, vomiting, constipation or diarrhea, muscle weakness, seizures, and mental changes such as confusion, hallucinations, or anxiety. These symptoms can be triggered by certain factors like drugs, alcohol, hormonal changes, infections, or stress.
It is essential to differentiate AIP from other medical conditions that may present with similar symptoms, as the treatment strategies differ significantly. Diagnosis typically involves measuring porphyrin precursors in urine, especially during an acute attack, and can be confirmed by genetic testing for the PBGD gene mutation.
Treatment of AIP primarily focuses on managing acute attacks with intravenous heme preparations, which help to reduce the production of toxic porphyrin precursors. In addition, providing supportive care such as hydration, pain management, and addressing any triggers or complications is crucial. Long-term management includes avoiding identified triggers, monitoring for early signs of acute attacks, and implementing a low-purine diet in some cases.
Erythropoietic Porphyria (EP) is a rare inherited disorder of the heme biosynthesis pathway, specifically caused by a deficiency of the enzyme uroporphyrinogen III synthase. This results in the accumulation of porphyrin precursors, particularly uroporphyrin I and coproporphyrin I, in erythrocytes (red blood cells), bone marrow, and other tissues. The accumulation of these porphyrins leads to photosensitivity, hemolysis, and iron overload.
The symptoms of EP typically appear in childhood or early adulthood and include severe skin fragility and blistering, particularly on sun-exposed areas, which can result in scarring, disfigurement, and increased susceptibility to infection. Other features may include anemia due to hemolysis, iron overload, and splenomegaly (enlarged spleen).
The diagnosis of EP is based on clinical symptoms, laboratory tests measuring porphyrin levels in blood and urine, and genetic testing to confirm the presence of pathogenic variants in the UROS gene. Treatment for EP includes avoidance of sunlight exposure, use of sun-protective measures, and management of anemia with blood transfusions or erythropoietin injections. In some cases, bone marrow transplantation may be considered as a curative treatment option.
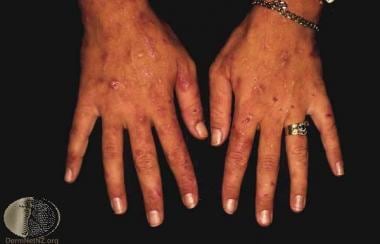
Variegate Porphyria (VP) is a rare inherited metabolic disorder that affects the production of heme, a component in hemoglobin. It is one of the types of porphyrias, which are caused by genetic mutations that result in deficiencies of enzymes needed to synthesize heme.
In variegate porphyria, the deficient enzyme is protoporphyrinogen oxidase (PPOX). This leads to the accumulation of porphyrins and their precursors, particularly coproporphyrin III and protoporphyrin, in the body. These substances can cause neurological symptoms when they are excreted in urine and exposed to light.
Variegate porphyria is characterized by both cutaneous (skin) and neurovisceral (neurological) manifestations. Cutaneous symptoms include skin sensitivity to sunlight, blistering, scarring, and fragility. Neurovisceral symptoms can include abdominal pain, nausea, vomiting, constipation, muscle weakness, seizures, and mental changes such as anxiety, hallucinations, or confusion.
The severity of variegate porphyria can vary widely between individuals, even among family members who carry the same genetic mutation. Symptoms may be triggered by certain medications, hormonal changes, alcohol consumption, infections, or other factors that increase heme synthesis. Diagnosis typically involves measuring porphyrin levels in blood and urine, as well as genetic testing for the PPOX gene mutation. Treatment usually focuses on managing symptoms, avoiding triggers, and providing supportive care during acute attacks.
Porphyria Cutanea Tarda (PCT) is a type of porphyria, a group of rare genetic disorders that affect the production of heme, a component in hemoglobin. PCT is primarily an acquired disorder, although it can have a hereditary component as well.
In PCT, there is a dysfunction in the enzyme uroporphyrinogen decarboxylase (UROD), which leads to the accumulation of porphyrins and porphyrin precursors in the skin. This buildup causes the characteristic symptoms of PCT, which include:
* Blisters, particularly on sun-exposed areas such as the hands and face
* Fragile, thin skin that tears easily
* Scarring
* Hypertrichosis (abnormal hair growth)
* Changes in skin color, including redness, increased pigmentation, or loss of pigment
PCT is typically triggered by factors such as alcohol consumption, estrogen use, hepatitis C infection, and exposure to certain chemicals. Treatment often involves addressing these triggers, along with the use of phlebotomy (removal of blood) or low-dose hydroxychloroquine to reduce porphyrin levels in the body.
It's important to note that PCT is a complex disorder and its diagnosis and management should be done by healthcare professionals with experience in managing porphyrias.
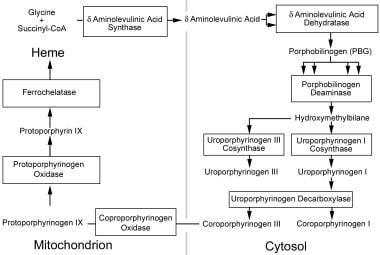
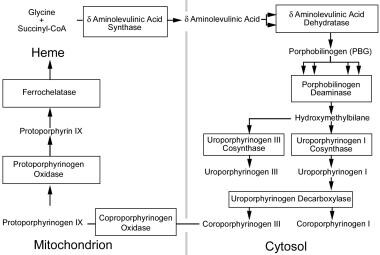
Hydroxymethylbilane Synthase (HMBS) is an enzyme that plays a crucial role in the metabolic pathway known as heme biosynthesis. Heme is an essential component of various proteins, including hemoglobin, which is responsible for oxygen transport in the blood.
The HMBS enzyme catalyzes the conversion of aminolevulinic acid (ALA) and glycine into a linear tetrapyrrole intermediate called hydroxymethylbilane. This reaction is the third step in the heme biosynthesis pathway, and it takes place in the mitochondria of cells.
Deficiencies in HMBS can lead to a rare genetic disorder called acute intermittent porphyria (AIP), which is characterized by neurovisceral attacks and neurological symptoms such as abdominal pain, vomiting, hypertension, tachycardia, and mental disturbances.
Porphobilinogen (PBG) is a bioactive compound that plays a crucial role in the biosynthesis pathway of heme, which is an essential component of hemoglobin and other hemoproteins. It is a porphyrin precursor and is synthesized from aminolevulinic acid (ALA) by the enzyme ALA dehydratase in the second step of heme biosynthesis.
In medical terms, abnormal accumulation or increased levels of PBG in the body can indicate an underlying disorder in heme biosynthesis, such as acute intermittent porphyria (AIP), variegate porphyria (VP), or hereditary coproporphyria (HCP). These disorders are known as porphyrias and are characterized by the buildup of porphyrin precursors in various tissues, leading to neurological and gastrointestinal symptoms.
Therefore, measuring PBG levels in urine or blood can help diagnose and monitor these conditions.
Uroporphyrinogen decarboxylase is a vital enzyme in the biosynthetic pathway of heme, which is a crucial component of hemoglobin in red blood cells. This enzyme is responsible for catalyzing the decarboxylation of uroporphyrinogen III, a colorless porphyrinogen, to produce coproporphyrinogen III, a brownish-red porphyrinogen.
The reaction involves the sequential removal of four carboxyl groups from the four acetic acid side chains of uroporphyrinogen III, resulting in the formation of coproporphyrinogen III. This enzyme's activity is critical for the normal biosynthesis of heme, and any defects or deficiencies in its function can lead to various porphyrias, a group of metabolic disorders characterized by the accumulation of porphyrins and their precursors in the body.
The gene responsible for encoding uroporphyrinogen decarboxylase is UROD, located on chromosome 1p34.1. Mutations in this gene can lead to a deficiency in the enzyme's activity, causing an autosomal recessive disorder known as congenital erythropoietic porphyria (CEP), also referred to as Günther's disease. This condition is characterized by severe photosensitivity, hemolytic anemia, and scarring or thickening of the skin.
Protoporphyrinogen Oxidase (PPO) is a mitochondrial enzyme that plays a crucial role in the heme biosynthesis pathway. It catalyzes the oxidation of protoporphyrinogen IX to protporphyrin IX, which is the penultimate step in the production of heme. This enzyme is the target of certain herbicides, such as those containing the active ingredient diphenyl ether, and genetic deficiencies in PPO can lead to a rare genetic disorder called Protoporphyria.
Porphyrins are complex organic compounds that contain four pyrrole rings joined together by methine bridges (=CH-). They play a crucial role in the biochemistry of many organisms, as they form the core structure of various heme proteins and other metalloproteins. Some examples of these proteins include hemoglobin, myoglobin, cytochromes, and catalases, which are involved in essential processes such as oxygen transport, electron transfer, and oxidative metabolism.
In the human body, porphyrins are synthesized through a series of enzymatic reactions known as the heme biosynthesis pathway. Disruptions in this pathway can lead to an accumulation of porphyrins or their precursors, resulting in various medical conditions called porphyrias. These disorders can manifest as neurological symptoms, skin lesions, and gastrointestinal issues, depending on the specific type of porphyria and the site of enzyme deficiency.
It is important to note that while porphyrins are essential for life, their accumulation in excessive amounts or at inappropriate locations can result in pathological conditions. Therefore, understanding the regulation and function of porphyrin metabolism is crucial for diagnosing and managing porphyrias and other related disorders.
Uroporphyrinogen III Synthase is a crucial enzyme in the biosynthetic pathway of heme and chlorophyll. This enzyme, specifically classified under EC 4.2.1.75, catalyzes the conversion of coproporphyrinogen III to protoporphyrinogen IX, which is a key step in the synthesis of heme.
The reaction it facilitates is:
Coproporphyrinogen III + reduced ferredoxin → Protoporphyrinogen IX + oxidized ferredoxin + CO2
Deficiency or malfunctioning of this enzyme can lead to a rare genetic disorder known as "congenital erythropoietic porphyria" (CEP), also known as Günther's disease, which is characterized by severe photosensitivity and related symptoms.
Uroporphyrins are porphyrin derivatives that contain four carboxylic acid groups. They are intermediates in the biosynthesis of heme, which is a component of hemoglobin and other hemoproteins. Uroporphyrinogen I and III are precursors to uroporphyrin I and III, respectively, through the action of uroporphyrinogen decarboxylase.
Uroporphyrin I and III differ in the position of acetate and propionate side chains on the porphyrin ring. Uroporphyrins are usually elevated in the urine of patients with certain inherited metabolic disorders, such as acute intermittent porphyria, variegate porphyria, and hereditary coproporphyria, due to enzyme deficiencies in the heme biosynthetic pathway.
The measurement of uroporphyrins in urine or other body fluids can be helpful in diagnosing and monitoring these disorders.
Hepatoerythropoietic porphyria (HEP) is a rare inherited metabolic disorder that affects the production of heme, a component in hemoglobin. It is a subtype of porphyria known as "erythropoietic porphyria," which primarily affects the bone marrow and erythroid cells.
In HEP, there are deficiencies in the activity of two enzymes involved in heme biosynthesis: uroporphyrinogen III synthase (UROS) and coproporphyrinogen oxidase (CPOX). This double enzyme deficiency leads to the accumulation of porphyrin precursors, particularly uroporphyrinogen I and coproporphyrinogen I, in erythrocytes, plasma, and tissues.
The main clinical manifestations of HEP include severe cutaneous photosensitivity, blistering, scarring, and hypertrichosis (excessive hair growth) on sun-exposed areas. Other features may include hemolytic anemia, splenomegaly, and liver dysfunction. The condition typically presents in infancy or early childhood, and it can be associated with significant morbidity and mortality if not properly managed.
Diagnosis of HEP is based on the detection of elevated levels of porphyrin precursors in plasma, erythrocytes, and stool, as well as genetic testing to confirm mutations in the UROS and CPOX genes. Treatment involves avoidance of sunlight exposure, use of sun-protective measures, and management of anemia with blood transfusions or other therapies. In some cases, hematopoietic stem cell transplantation may be considered as a curative treatment option.
Coproporphyrins are porphyrin molecules that contain four carboxylic acid groups (four propionic side chains and two acetic side chains). They are intermediates in the biosynthesis of heme, which is a component of hemoglobin and other hemoproteins. Coproporphyrins can be further metabolized to form protoporphyrins, which are converted into heme by the enzyme ferrochelatase.
Coproporphyrins can be excreted in urine and feces, and their levels can be measured in clinical testing. Elevated coproporphyrin levels in urine or feces may indicate the presence of certain medical conditions, such as lead poisoning, porphyrias, or liver dysfunction.
There are two types of coproporphyrins, coproporphyrin I and coproporphyrin III, which differ in the arrangement of their side chains. Coproporphyrin III is the form that is normally produced in the body, while coproporphyrin I is a byproduct of abnormal porphyrin metabolism.
5-Aminolevulinate synthase (ALAS) is an enzyme that catalyzes the first step in heme biosynthesis, a metabolic pathway that produces heme, a porphyrin ring with an iron atom at its center. Heme is a crucial component of hemoglobin, cytochromes, and other important molecules in the body.
ALAS exists in two forms: ALAS1 and ALAS2. ALAS1 is expressed in all tissues, while ALAS2 is primarily expressed in erythroid cells (precursors to red blood cells). The reaction catalyzed by ALAS involves the condensation of glycine and succinyl-CoA to form 5-aminolevulinate.
Deficiencies or mutations in the ALAS2 gene can lead to a rare genetic disorder called X-linked sideroblastic anemia, which is characterized by abnormal red blood cell maturation and iron overload in mitochondria.
Hereditary coproporphyria (HCP) is a rare inherited disorder of the heme biosynthesis pathway, which is the process by which your body produces heme. Heme is a crucial component of various proteins, including hemoglobin, which carries oxygen in red blood cells.
In HCP, there is a deficiency of an enzyme called coproporphyrinogen oxidase. This enzyme is essential for converting coproporphyrinogen III to protoporphyrin IX in the heme biosynthesis pathway. As a result, coproporphyrinogen III accumulates and gets converted to coproporphyrin, which is excreted in urine and stool in abnormally high amounts.
The symptoms of HCP can be diverse and may include both neurological and gastrointestinal manifestations. Neurological symptoms might include abdominal pain, muscle weakness, numbness, tingling, seizures, and psychiatric disturbances. Gastrointestinal symptoms could encompass nausea, vomiting, constipation, or diarrhea. These symptoms are typically triggered by certain factors such as infections, drugs, hormonal changes, or alcohol consumption.
HCP is usually inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the disease-causing gene from a parent with the disorder. However, some cases may result from de novo mutations, which means the mutation occurs spontaneously without a family history of the condition.
Diagnosis of HCP is usually made through measuring porphyrin levels and their precursors in urine, stool, and blood during an acute attack or between attacks. Genetic testing can confirm the diagnosis by identifying mutations in the CPOX gene, which encodes coproporphyrinogen oxidase.
Treatment for HCP typically involves avoiding triggers, providing supportive care during acute attacks, and using medications to manage symptoms. In some cases, heme arginate or hemine may be given to help decrease porphyrin precursor production. Preventive measures such as avoidance of potential triggers, adequate hydration, and a balanced diet are essential in managing HCP.
Uroporphyrinogens are organic compounds that are intermediate products in the synthesis of heme, which is a crucial component of hemoglobin and other important molecules in the body. Specifically, uroporphyrinogens are tetrapyrroles, which means they contain four pyrrole rings linked together. They have eight carboxylic acid side chains and two propionic acid side chains.
There are two types of uroporphyrinogens: Type I and Type III. Uroporphyrinogen III is the precursor to heme, while uroporphyrinogen I is a dead-end metabolite that is not used in heme synthesis. Defects in the enzymes involved in heme biosynthesis can lead to various porphyrias, which are genetic disorders characterized by the accumulation of porphyrins and their precursors in the body.
Aminolevulinic acid (ALA) is a naturally occurring compound in the human body and is a key precursor in the biosynthesis of heme, which is a component of hemoglobin in red blood cells. It is also used as a photosensitizer in dermatology for the treatment of certain types of skin conditions such as actinic keratosis and basal cell carcinoma.
In medical terms, ALA is classified as an α-keto acid and a porphyrin precursor. It is synthesized in the mitochondria from glycine and succinyl-CoA in a reaction catalyzed by the enzyme aminolevulinic acid synthase. After its synthesis, ALA is transported to the cytosol where it undergoes further metabolism to form porphyrins, which are then used for heme biosynthesis in the mitochondria.
In dermatology, topical application of ALA followed by exposure to a specific wavelength of light can lead to the production of reactive oxygen species that destroy abnormal cells in the skin while leaving healthy cells unharmed. This makes it an effective treatment for precancerous and cancerous lesions on the skin.
It is important to note that ALA can cause photosensitivity, which means that patients who have undergone ALA-based treatments should avoid exposure to sunlight or other sources of bright light for a period of time after the treatment to prevent adverse reactions.
Porphyrinogens are organic compounds that are the precursors to porphyrins, which are ring-shaped molecules found in many important biological molecules such as hemoglobin and cytochromes. Porphyrinogens are themselves derived from the condensation of four pyrrole molecules, and they undergo further reactions to form porphyrins.
In particular, porphyrinogens are intermediates in the biosynthesis of heme, which is a complex organic ring-shaped molecule that contains iron and plays a critical role in oxygen transport and storage in the body. Abnormalities in heme biosynthesis can lead to various medical conditions known as porphyrias, which are characterized by the accumulation of porphyrinogens and other intermediates in this pathway. These conditions can cause a range of symptoms, including neurological problems, skin sensitivity to light, and abdominal pain.
Levulinic acid is not specifically a medical term, but it is a chemical compound with the formula C5H8O2. It is a white crystalline solid that is used in the production of various chemicals and materials. However, I can provide you with some general information about levulinic acid:
Levulinic acid is a saturated carboxylic acid, which means it contains a carboxyl group (-COOH) and is fully saturated with hydrogen atoms. It is an alpha-beta unsaturated carboxylic acid due to the presence of a carbon-carbon double bond (C=C) between the second and third carbon atoms in its structure.
Levulinic acid can be found naturally in small amounts in various fruits, such as apples and grapes, and is also present in some fermented foods like beer and wine. It can be produced industrially from biomass sources, such as cellulose or lignocellulosic materials, through a process called acid hydrolysis.
In the medical field, levulinic acid may have potential applications as an antimicrobial agent due to its ability to inhibit the growth of certain bacteria and fungi. It is also used in the synthesis of pharmaceuticals and other chemical products. However, it is not a substance that is typically directly associated with medical treatment or diagnosis.
Hexachlorobenzene (HCB) is not typically included in standard medical textbooks or resources as it's more of a environmental and industrial chemical. However, I can provide you with an definition from a toxicological perspective:
Hexachlorobenzene (C6Cl6) is an organic compound that consists of a benzene ring with six chlorine atoms attached to it. It is a persistent organic pollutant, which means it does not break down easily and can accumulate in the environment and living organisms. HCB has been used as a pesticide, fungicide, and chemical intermediate in various industrial processes. Exposure to this compound can lead to several health issues, including skin lesions, damage to the nervous system, and impaired immune function. It's also considered a possible human carcinogen by some agencies. Long-term environmental exposure to HCB is of particular concern due to its bioaccumulation in the food chain and potential adverse effects on human health and the environment.
Porphobilinogen Synthase (also known as PBGD or hydroxymethylbilane synthase) is an enzyme that catalyzes the second step in the heme biosynthesis pathway. This enzyme is responsible for converting two molecules of porphobilinogen into a linear tetrapyrrole called hydroxymethylbilane, which is then converted into uroporphyrinogen III by uroporphyrinogen III synthase.
Deficiency in Porphobilinogen Synthase can lead to a rare genetic disorder known as acute intermittent porphyria (AIP), which is characterized by the accumulation of porphobilinogen and other precursors in the heme biosynthesis pathway, resulting in neurovisceral symptoms such as abdominal pain, vomiting, neuropathy, and psychiatric disturbances.
Ammonia-lyases are a class of enzymes that catalyze the removal of an amino group from a substrate, releasing ammonia in the process. These enzymes play important roles in various biological pathways, including the biosynthesis and degradation of various metabolites such as amino acids, carbohydrates, and aromatic compounds.
The reaction catalyzed by ammonia-lyases typically involves the conversion of an alkyl or aryl group to a carbon-carbon double bond through the elimination of an amine group. This reaction is often reversible, allowing the enzyme to also catalyze the addition of an amino group to a double bond.
Ammonia-lyases are classified based on the type of substrate they act upon and the mechanism of the reaction they catalyze. Some examples of ammonia-lyases include aspartate ammonia-lyase, which catalyzes the conversion of aspartate to fumarate, and tyrosine ammonia-lyase, which converts tyrosine to p-coumaric acid.
These enzymes are important in both plant and animal metabolism and have potential applications in biotechnology and industrial processes.
Flavoproteins are a type of protein molecule that contain noncovalently bound flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) as cofactors. These flavin cofactors play a crucial role in redox reactions, acting as electron carriers in various metabolic pathways such as cellular respiration and oxidative phosphorylation. Flavoproteins are involved in several biological processes, including the breakdown of fatty acids, amino acids, and carbohydrates, as well as the synthesis of steroids and other lipids. They can also function as enzymes that catalyze various redox reactions, such as oxidases, dehydrogenases, and reductases. Flavoproteins are widely distributed in nature and found in many organisms, from bacteria to humans.
Ferrochelatase is a medical/biochemical term that refers to an enzyme called Fe-chelatase or heme synthase. This enzyme plays a crucial role in the biosynthesis of heme, which is a vital component of hemoglobin, cytochromes, and other important biological molecules.
Ferrochelatase functions by catalyzing the insertion of ferrous iron (Fe2+) into protoporphyrin IX, the final step in heme biosynthesis. This enzyme is located within the inner mitochondrial membrane of cells and is widely expressed in various tissues, with particularly high levels found in erythroid precursor cells, liver, and brain.
Defects or mutations in the ferrochelatase gene can lead to a rare genetic disorder called erythropoietic protoporphyria (EPP), which is characterized by an accumulation of protoporphyrin IX in red blood cells, plasma, and other tissues. This accumulation results in photosensitivity, skin lesions, and potential complications such as liver dysfunction and gallstones.
Heme is not a medical term per se, but it is a term used in the field of medicine and biology. Heme is a prosthetic group found in hemoproteins, which are proteins that contain a heme iron complex. This complex plays a crucial role in various biological processes, including oxygen transport (in hemoglobin), electron transfer (in cytochromes), and chemical catalysis (in peroxidases and catalases).
The heme group consists of an organic component called a porphyrin ring, which binds to a central iron atom. The iron atom can bind or release electrons, making it essential for redox reactions in the body. Heme is also vital for the formation of hemoglobin and myoglobin, proteins responsible for oxygen transport and storage in the blood and muscles, respectively.
In summary, heme is a complex organic-inorganic structure that plays a critical role in several biological processes, particularly in electron transfer and oxygen transport.
Griseofulvin is an antifungal medication used to treat various fungal infections, including those affecting the skin, hair, and nails. It works by inhibiting the growth of fungi, particularly dermatophytes, which cause these infections. Griseofulvin can be obtained through a prescription and is available in oral (by mouth) and topical (on the skin) forms.
The primary mechanism of action for griseofulvin involves binding to tubulin, a protein necessary for fungal cell division. This interaction disrupts the formation of microtubules, which are crucial for the fungal cell's structural integrity and growth. As a result, the fungi cannot grow and multiply, allowing the infected tissue to heal and the infection to resolve.
Common side effects associated with griseofulvin use include gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea), headache, dizziness, and skin rashes. It is essential to follow the prescribing physician's instructions carefully when taking griseofulvin, as improper usage may lead to reduced effectiveness or increased risk of side effects.
It is important to note that griseofulvin has limited use in modern medicine due to the development of newer and more effective antifungal agents. However, it remains a valuable option for specific fungal infections, particularly those resistant to other treatments.
Coproporphyrinogen Oxidase is a mitochondrial enzyme that plays a crucial role in the biosynthesis of heme, which is an essential component of hemoglobin and other hemoproteins. This enzyme catalyzes the oxidative decarboxylation of coproporphyrinogen III to protoporphyrinogen IX, a key step in the heme biosynthetic pathway.
Deficiency or dysfunction of Coproporphyrinogen Oxidase can lead to a rare genetic disorder known as Hereditary Coproporphyria (HCP), which is characterized by the accumulation of coproporphyrinogen III and its derivative, coproporphyrin, in various tissues and body fluids. This accumulation can result in a range of symptoms, including abdominal pain, neurological disturbances, and skin manifestations.
Coproporphyrinogens are intermediates in the biosynthesis of heme, a complex molecule that is essential for various biological processes including oxygen transport and cellular respiration. There are two types of coproporphyrinogens: Coproporphyrinogen I and Coproporphyrinogen III.
Coproporphyrinogen I is an intermediate in the biosynthesis of siroheme, a porphyrin-like molecule that functions as a cofactor for enzymes involved in sulfur and nitrogen metabolism. It is produced from uroporphyrinogen III through the action of coproporphyrinogen oxidase.
Coproporphyrinogen III, on the other hand, is an intermediate in the biosynthesis of heme. It is produced from protoporphyrinogen IX through the action of coproporphyrinogen oxidase and then converted to protoporphyrin IX by the enzyme coproporphyrinogen III decarboxylase. Protoporphyrin IX is then converted to heme by the addition of iron in a reaction catalyzed by ferrochelatase.
Abnormal accumulation of coproporphyrinogens can occur due to various genetic and acquired disorders that affect enzymes involved in heme biosynthesis, leading to the accumulation of porphyrins and their precursors in tissues and bodily fluids. These conditions are known as porphyrias and can present with a variety of symptoms including neuropsychiatric manifestations, skin lesions, and gastrointestinal disturbances.
I apologize, but I am unable to find a medical definition for "dicarbethoxydihydrocollidine." It is possible that this term does not have a specific meaning within the field of medicine. A more general search indicates that dicarbethoxydihydrocollidine may be a chemical compound used in research or industry, but it does not appear to have direct relevance to medical diagnosis, treatment, or health. If you have further questions or need information on a related medical topic, I would be happy to help you with those.
Skin diseases, also known as dermatological conditions, refer to any medical condition that affects the skin, which is the largest organ of the human body. These diseases can affect the skin's function, appearance, or overall health. They can be caused by various factors, including genetics, infections, allergies, environmental factors, and aging.
Skin diseases can present in many different forms, such as rashes, blisters, sores, discolorations, growths, or changes in texture. Some common examples of skin diseases include acne, eczema, psoriasis, dermatitis, fungal infections, viral infections, bacterial infections, and skin cancer.
The symptoms and severity of skin diseases can vary widely depending on the specific condition and individual factors. Some skin diseases are mild and can be treated with over-the-counter medications or topical creams, while others may require more intensive treatments such as prescription medications, light therapy, or even surgery.
It is important to seek medical attention if you experience any unusual or persistent changes in your skin, as some skin diseases can be serious or indicative of other underlying health conditions. A dermatologist is a medical doctor who specializes in the diagnosis and treatment of skin diseases.
Oxidoreductases acting on CH-CH group donors are a class of enzymes within the larger group of oxidoreductases, which are responsible for catalyzing oxidation-reduction reactions. Specifically, this subclass of enzymes acts upon donors containing a carbon-carbon (CH-CH) bond, where one atom or group of atoms is oxidized and another is reduced during the reaction process. These enzymes play crucial roles in various metabolic pathways, including the breakdown and synthesis of carbohydrates, lipids, and amino acids.
The reactions catalyzed by these enzymes involve the transfer of electrons and hydrogen atoms between the donor and an acceptor molecule. This process often results in the formation or cleavage of carbon-carbon bonds, making them essential for numerous biological processes. The systematic name for this class of enzymes is typically structured as "donor:acceptor oxidoreductase," where donor and acceptor represent the molecules involved in the electron transfer process.
Examples of enzymes that fall under this category include:
1. Aldehyde dehydrogenases (EC 1.2.1.3): These enzymes catalyze the oxidation of aldehydes to carboxylic acids, using NAD+ as an electron acceptor.
2. Dihydrodiol dehydrogenase (EC 1.3.1.14): This enzyme is responsible for the oxidation of dihydrodiols to catechols in the biodegradation of aromatic compounds.
3. Succinate dehydrogenase (EC 1.3.5.1): A key enzyme in the citric acid cycle, succinate dehydrogenase catalyzes the oxidation of succinate to fumarate and reduces FAD to FADH2.
4. Xylose reductase (EC 1.1.1.307): This enzyme is involved in the metabolism of pentoses, where it reduces xylose to xylitol using NADPH as a cofactor.
Erythropoietic Protoporphyria (EPP) is a rare inherited disorder of porphyrin metabolism. It results from a deficiency in the ferrochelatase enzyme, which normally catalyzes the insertion of iron into protoporphyrin to form heme. This deficiency leads to an accumulation of protoporphyrin, particularly in red blood cells and plasma.
The accumulated protoporphyrin is sensitive to light, particularly wavelengths between 400-410 nm (blue light). When exposed to this light, the protoporphyrin molecules absorb the light energy and transfer it to molecular oxygen, leading to the formation of highly reactive singlet oxygen. This reaction causes oxidative damage to surrounding tissues, resulting in the symptoms of EPP.
The main symptom is severe, painful burn-like reactions on exposed skin after sunlight exposure, often accompanied by swelling and itching. These symptoms can occur within minutes of sun exposure and can last for several days. Chronic skin changes such as scarring and milia can also occur over time.
EPP is usually diagnosed through the measurement of porphyrins in the blood or stool, and genetic testing can confirm the diagnosis. Treatment typically involves avoiding sunlight exposure, using sun protection measures, and in some cases, oral beta-carotene or cysteine supplements to reduce symptoms. In severe cases, heme arginate or afamelanotide may be used.
Photosensitivity disorders refer to conditions that cause an abnormal reaction to sunlight or artificial light. This reaction can take the form of various skin changes, such as rashes, inflammation, or pigmentation, and in some cases, it can also lead to systemic symptoms like fatigue, fever, or joint pain.
The two main types of photosensitivity disorders are:
1. Phototoxic reactions: These occur when a substance (such as certain medications, chemicals, or plants) absorbs light energy and transfers it to skin cells, causing damage and inflammation. The reaction typically appears within 24 hours of exposure to the light source and can resemble a sunburn.
2. Photoallergic reactions: These occur when the immune system responds to the combination of light and a particular substance, leading to an allergic response. The reaction may not appear until several days after initial exposure and can cause redness, itching, and blistering.
It is important for individuals with photosensitivity disorders to avoid excessive sun exposure, wear protective clothing, and use broad-spectrum sunscreens with a high SPF rating to minimize the risk of phototoxic or photoallergic reactions.
Bloodletting is a medical procedure that was commonly used in the past to balance the four humors of the body, which were believed to be blood, phlegm, black bile, and yellow bile. The procedure involved withdrawing blood from a patient through various methods such as venesection (making an incision in a vein), leeches, or cupping.
The theory behind bloodletting was that if one humor became overabundant, it could cause disease or illness. By removing some of the excess humor, practitioners believed they could restore balance and promote healing. Bloodletting was used to treat a wide variety of conditions, including fever, inflammation, and pain.
While bloodletting is no longer practiced in modern medicine, it was once a common treatment for many different ailments. The practice dates back to ancient times and was used by various cultures throughout history, including the Greeks, Romans, Egyptians, and Chinese. However, its effectiveness as a medical treatment has been called into question, and it is now considered an outdated and potentially harmful procedure.
Protoporphyrins are organic compounds that are the immediate precursors to heme in the porphyrin synthesis pathway. They are composed of a porphyrin ring, which is a large, complex ring made up of four pyrrole rings joined together, with an acetate and a propionate side chain at each pyrrole. Protoporphyrins are commonly found in nature and are important components of many biological systems, including hemoglobin, the protein in red blood cells that carries oxygen throughout the body.
There are several different types of protoporphyrins, including protoporphyrin IX, which is the most common form found in humans and other animals. Protoporphyrins can be measured in the blood or other tissues as a way to diagnose or monitor certain medical conditions, such as lead poisoning or porphyrias, which are rare genetic disorders that affect the production of heme. Elevated levels of protoporphyrins in the blood or tissues can indicate the presence of these conditions and may require further evaluation and treatment.
Allylisopropylacetamide is not a term that has a widely accepted or established medical definition. It is a chemical compound with the formula (CH₂CHCH₂)N(C=O)CH(CH₃)₂, and it may have various chemical or industrial uses, but it is not a term that is commonly used in medical contexts.
If you have any specific questions about this compound or its potential uses or effects, I would recommend consulting with a relevant expert, such as a chemist or toxicologist, who can provide more detailed and accurate information based on their expertise and knowledge of the subject.
In medical terms, "precipitating factors" refer to specific events, actions, or circumstances that trigger the onset of a disease, symptom, or crisis in an individual who is already vulnerable due to pre-existing conditions. These factors can vary depending on the particular health issue, and they may include things like physical stress, emotional stress, environmental triggers, or changes in medication.
For example, in the context of a heart condition, precipitating factors might include strenuous exercise, exposure to extreme temperatures, or the use of certain drugs that increase heart rate or blood pressure. In mental health, precipitating factors for a depressive episode could include significant life changes such as the loss of a loved one, financial difficulties, or a major life transition.
Identifying and managing precipitating factors is an important aspect of preventative healthcare and disease management, as it can help individuals reduce their risk of experiencing negative health outcomes.
Hemochromatosis is a medical condition characterized by excessive absorption and accumulation of iron in the body, resulting in damage to various organs. It's often referred to as "iron overload" disorder. There are two main types: primary (hereditary) and secondary (acquired). Primary hemochromatosis is caused by genetic mutations that lead to increased intestinal iron absorption, while secondary hemochromatosis can be the result of various conditions such as multiple blood transfusions, chronic liver disease, or certain types of anemia.
In both cases, the excess iron gets stored in body tissues, particularly in the liver, heart, and pancreas, which can cause organ damage and lead to complications like cirrhosis, liver failure, diabetes, heart problems, and skin discoloration. Early diagnosis and treatment through regular phlebotomy (blood removal) or chelation therapy can help manage the condition and prevent severe complications.
Erythrocytes, also known as red blood cells (RBCs), are the most common type of blood cell in circulating blood in mammals. They are responsible for transporting oxygen from the lungs to the body's tissues and carbon dioxide from the tissues to the lungs.
Erythrocytes are formed in the bone marrow and have a biconcave shape, which allows them to fold and bend easily as they pass through narrow blood vessels. They do not have a nucleus or mitochondria, which makes them more flexible but also limits their ability to reproduce or repair themselves.
In humans, erythrocytes are typically disc-shaped and measure about 7 micrometers in diameter. They contain the protein hemoglobin, which binds to oxygen and gives blood its red color. The lifespan of an erythrocyte is approximately 120 days, after which it is broken down in the liver and spleen.
Abnormalities in erythrocyte count or function can lead to various medical conditions, such as anemia, polycythemia, and sickle cell disease.
Liver diseases refer to a wide range of conditions that affect the normal functioning of the liver. The liver is a vital organ responsible for various critical functions such as detoxification, protein synthesis, and production of biochemicals necessary for digestion.
Liver diseases can be categorized into acute and chronic forms. Acute liver disease comes on rapidly and can be caused by factors like viral infections (hepatitis A, B, C, D, E), drug-induced liver injury, or exposure to toxic substances. Chronic liver disease develops slowly over time, often due to long-term exposure to harmful agents or inherent disorders of the liver.
Common examples of liver diseases include hepatitis, cirrhosis (scarring of the liver tissue), fatty liver disease, alcoholic liver disease, autoimmune liver diseases, genetic/hereditary liver disorders (like Wilson's disease and hemochromatosis), and liver cancers. Symptoms may vary widely depending on the type and stage of the disease but could include jaundice, abdominal pain, fatigue, loss of appetite, nausea, and weight loss.
Early diagnosis and treatment are essential to prevent progression and potential complications associated with liver diseases.
Carboxy-lyases are a class of enzymes that catalyze the removal of a carboxyl group from a substrate, often releasing carbon dioxide in the process. These enzymes play important roles in various metabolic pathways, such as the biosynthesis and degradation of amino acids, sugars, and other organic compounds.
Carboxy-lyases are classified under EC number 4.2 in the Enzyme Commission (EC) system. They can be further divided into several subclasses based on their specific mechanisms and substrates. For example, some carboxy-lyases require a cofactor such as biotin or thiamine pyrophosphate to facilitate the decarboxylation reaction, while others do not.
Examples of carboxy-lyases include:
1. Pyruvate decarboxylase: This enzyme catalyzes the conversion of pyruvate to acetaldehyde and carbon dioxide during fermentation in yeast and other organisms.
2. Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO): This enzyme is essential for photosynthesis in plants and some bacteria, as it catalyzes the fixation of carbon dioxide into an organic molecule during the Calvin cycle.
3. Phosphoenolpyruvate carboxylase: Found in plants, algae, and some bacteria, this enzyme plays a role in anaplerotic reactions that replenish intermediates in the citric acid cycle. It catalyzes the conversion of phosphoenolpyruvate to oxaloacetate and inorganic phosphate.
4. Aspartate transcarbamylase: This enzyme is involved in the biosynthesis of pyrimidines, a class of nucleotides. It catalyzes the transfer of a carboxyl group from carbamoyl aspartate to carbamoyl phosphate, forming cytidine triphosphate (CTP) and fumarate.
5. Urocanase: Found in animals, this enzyme is involved in histidine catabolism. It catalyzes the conversion of urocanate to formiminoglutamate and ammonia.
 Variegate porphyria
Variegate porphyria
Porphyria
Polymorphous light eruption
Protoporphyrinogen oxidase
Zuytdorp
Carrot or Stick
Acute intermittent porphyria
Hives
Alcohol and cancer
Hereditary coproporphyria
Hepatocellular carcinoma
Hepatic porphyria
Heme
VP (disambiguation)
Afrikaners
Psychomotor agitation
Barbiturate
List of MeSH codes (C06)
Prince William of Gloucester
Dysuria
Syndrome of inappropriate antidiuretic hormone secretion
List of MeSH codes (C18)
List of MeSH codes (C16)
List of diseases (V)
List of MeSH codes (C17)
Chromosome 1
List of moths of Nepal (Sphingidae)
List of skin conditions
List of OMIM disorder codes
RNA interference
Variegate porphyria - Wikipedia
 Variegate Porphyria: Background, Pathophysiology, Etiology
Variegate Porphyria: Background, Pathophysiology, Etiology
 A plasma porphyrin fluorescence marker for variegate porphyria
A plasma porphyrin fluorescence marker for variegate porphyria
Variegate porphyria
Variegate Porphyria: Background, Pathophysiology, Etiology
Variegate Porphyria Differential Diagnoses
Variegate Porphyria: Background, Pathophysiology, Epidemiology
Safe use of perampanel in a carrier of variegate porphyria | Practical Neurology
 Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
Porphyria Overview: Practice Essentials, Background, Pathophysiology
 A 25-Hour Fast Among Quiescent Hereditary Coproporphyria and Variegate Porphyria Patients is Associated With a Low Risk of...
A 25-Hour Fast Among Quiescent Hereditary Coproporphyria and Variegate Porphyria Patients is Associated With a Low Risk of...
 A boy with blistering of sun-exposed skin and finger shortening: the first case of Variegate Porphyria with a novel mutation in...
A boy with blistering of sun-exposed skin and finger shortening: the first case of Variegate Porphyria with a novel mutation in...
 Porphyria cutanea tarda - Symptoms, diagnosis and treatment | BMJ Best Practice US
Porphyria cutanea tarda - Symptoms, diagnosis and treatment | BMJ Best Practice US
 Natural Pedia Com | NaturalNews - NaturalPedia
Natural Pedia Com | NaturalNews - NaturalPedia
 8-MOP (Methoxsalen): Uses, Dosage, Side Effects, Interactions, Warning
8-MOP (Methoxsalen): Uses, Dosage, Side Effects, Interactions, Warning
 Science.NaturalNews.com - Conduct powerful scientific research in mere seconds for your book, blog, website article or news...
Science.NaturalNews.com - Conduct powerful scientific research in mere seconds for your book, blog, website article or news...
Volume 81, Issue | Acta Dermato-Venereologica
 Elevated iron levels and erosions on the hands - Clinical Advisor
Elevated iron levels and erosions on the hands - Clinical Advisor
 DailyMed - CARBAMAZEPINE- carbamazepine tablet, extended release
DailyMed - CARBAMAZEPINE- carbamazepine tablet, extended release
 Patient Perspective on Acute Intermittent Porphyria with Frequent Attacks: A Disease with Intermittent and Chronic...
Patient Perspective on Acute Intermittent Porphyria with Frequent Attacks: A Disease with Intermittent and Chronic...
 Genetic Testing for the Healthy | Harvard Medical School
Genetic Testing for the Healthy | Harvard Medical School
 WikiGenes - Europe
WikiGenes - Europe
 Chapter 186. Other Bullous Diseases | The Color Atlas of Family Medicine, 2e | AccessMedicine | McGraw Hill Medical
Chapter 186. Other Bullous Diseases | The Color Atlas of Family Medicine, 2e | AccessMedicine | McGraw Hill Medical
 Lidocaine Toxicity - Liposuction.com - Liposuction.com
Lidocaine Toxicity - Liposuction.com - Liposuction.com
 Genetic Testing & Counseling | Alnylam Act® | Guide for HCPs
Genetic Testing & Counseling | Alnylam Act® | Guide for HCPs
 Acute Porphyrias - Endocrine and Metabolic Disorders - MSD Manual Professional Edition
Acute Porphyrias - Endocrine and Metabolic Disorders - MSD Manual Professional Edition
 Porphyrias | Choose the Right Test
Porphyrias | Choose the Right Test
Specific PHGKB|Rare Diseases PHGKB|PHGKB
Cutanea26
- Twelve patients with porphyria cutanea tarda, eight patients with erythropoietic protoporphyria, one patient with congenital erythropoietic porphyria, two patients with acute intermittent porphyria, and four patients with hereditary coproporphyria, whose plasma specimens were similarly examined, had plasma fluorescence characteristics that were different from those of the patients with variegate porphyria. (nih.gov)
- Porphyrias with blistering cutaneous features include porphyria cutanea tarda (PCT) and hepatoerythropoietic porphyria . (logicalimages.com)
- Porphyria cutanea tarda (PCT) - Lacks acute systemic findings, is more easily provoked, and presents with severe cutaneous findings. (logicalimages.com)
- Misdiagnosis of symptomatic variegate porphyria as porphyria cutanea tarda may lead to inappropriate treatment with phlebotomy or antimalarial therapies that are ineffective. (medscape.com)
- Aminolevulinic acid dehydrase (ALAD) porphyria and acute intermittent porphyria (AIP) cause predominately neurovisceral symptoms, whereas congenital erythropoietic porphyria (CEP), porphyria cutanea tarda (PCT), and erythropoietic porphyria (EP) cause mostly cutaneous symptoms. (medscape.com)
- Phlebotomy and apheresis can remove excessive iron in patients with porphyria cutanea tarda (PCT). (medscape.com)
- Acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and the familial form of porphyria cutanea tarda (PCT) follow an autosomal dominant inheritance pattern with low penetration. (medscape.com)
- Cutaneous signs often result from photosensitivity (eg, skin fragility and blistering in porphyria cutanea tarda). (medscape.com)
- Porphyria cutanea tarda presents with blistering and crusted skin lesions on the back of hands and other sun-exposed areas of the body. (bmj.com)
- Porphyria cutanea tarda (PCT) is a blistering cutaneous condition caused by a substantial deficiency of hepatic uroporphyrinogen decarboxylase, the fifth enzyme in the heme biosynthetic pathway. (bmj.com)
- The nonacute porphyrias include porphyria cutanea tarda (PCT), erythropoietic protoporphyria, congenital erythropoietic porphyria, and hepatoerythropoietic porphyria. (clinicaladvisor.com)
- Porphyria cutanea tarda is a porphyria that has no extracutaneous manifestations ( Figures 186-1 , 186-2 , and 186-3 ). (mhmedical.com)
- Porphyria cutanea tarda in a middle-aged woman. (mhmedical.com)
- Porphyria cutanea tarda in a man with hepatitis C. ( Courtesy of the University of Texas Health Sciences Center, Division of Dermatology . (mhmedical.com)
- Porphyria cutanea tarda in a man with hepatitis C and alcohol abuse. (mhmedical.com)
- Work-up showed elevated porphyrins in the urine (which fluoresced orange-red under a Wood lamp) and the patient was diagnosed with porphyria cutanea tarda. (mhmedical.com)
- Porphyria cutanea tarda (PCT) occurs mostly in middle-aged adults (typically 30 to 50 years of age) and is rare in children. (mhmedical.com)
- Individuals with any of the cutaneous porphyrias, which include porphyria cutanea tarda (PCT), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and XLP, can experience photosensitivity as a result of sun exposure, which can manifest with either blisters and scarring or immediate redness and pain. (arupconsult.com)
- It has the same clinical and histologic features as porphyria cutanea tarda (PCT) but does not cause biochemical porphyrin abnormalities. (arupconsult.com)
- Gunn GB, Anderson KE, Patel AJ, Gallegos J, Hallberg C, Sood G, Hatch SS, Sanguineti, G: Severe radiation therapy-related soft tissue toxicity in a patient with porphyria cutanea tarda: case report and review of the literature. (porphyria.org)
- Grady JJ, Lee C, Anderson KE, Associations among behavior-related susceptibility factors in porphyria cutanea tarda. (porphyria.org)
- If you have a history of hepatic porphyrias (acute intermittent porphyria, variegate porphyria, porphyria, cutanea tarda). (pharmeasy.in)
- The nonacute type includes the porphyria cutanea tarda and erythropoietic porphyrias. (medscape.com)
- Porphyria is not a single disease but a group of nine disorders: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), δ-aminolevulinic acid dehydratase deficiency porphyria (ADP), porphyria cutanea tarda (PCT), hepatoerythropoietic porphyria (HEP), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and X-linked protoporphyria (XLP). (qxmd.com)
- Among people with porphyria cutanea tarda (PCT) and chronic hepatitis C infection, treatment with modern antiviral medicines has been found to cure the infection and resolve PCT symptoms, according to a new study. (porphyrianews.com)
- The Mount Sinai Genetic Testing Laboratory in New York City is proud to announce availability of DNA testing for seven Porphyrias, including Acute Intermittent Porphyria (AIP), Hereditary Coproporphyria (HCP), Variegate Porphyria (VP), familial Porphyria Cutanea Tarda (f-PCT), Hepatoerythropoietic Porphyria (HEP), Erythropoietic Protoporphyria (EPP) and Congenital Erythropoietic Porphyria (CEP). (porphyriafoundation.org)
Hereditary19
- Porphyrias that can have both blistering cutaneous features and acute neurovisceral attacks include hereditary coproporphyria and variegate porphyria (VP). (logicalimages.com)
- Two porphyrias overlap these categories and can cause both neurovisceral and cutaneous symptoms, namely hereditary coproporphyria (HCP) and variegate porphyria (VP). (medscape.com)
- Porphyria is the common term for a group of syndromes, largely hereditary, that result from defects in porphyrins (the enzymes involved in heme synthesis). (medscape.com)
- A 25-Hour Fast Among Quiescent Hereditary Coproporphyria and Variegate Porphyria Patients is Associated With a Low Risk of Complications. (bvsalud.org)
- Despite this, some Jewish AHP patients -mainly hereditary coproporphyria (HCP) and variegate porphyria (VP) patients -fast for 25 consecutive hours during the traditional Jewish holy day known as Yom Kippur. (bvsalud.org)
- The acute porphyrias include acute intermittent porphyria, variegate porphyria, hereditary coproporphyria, and ALA-D-deficiency porphyria. (clinicaladvisor.com)
- Also included in the differential are the other porphyrias that present with blistering (congenital erythropoietic porphyria, hepatoerythropoietic porphyria, variegate porphyria, and hereditary coproporphyria). (clinicaladvisor.com)
- Alnylam Act® is a sponsored, no-charge, third-party genetic testing and counseling program for patients with a family history or suspected diagnosis of hereditary ATTR (hATTR) amyloidosis, acute hepatic porphyria, or primary hyperoxaluria type 1. (alnylam.com)
- All four types of AHP-acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and ALAD-deficient porphyria (ADP)-are characterized by acute, potentially life-threatening attacks and in some patients, chronic debilitating symptoms that negatively impact patients' quality of life. (alnylam.com)
- Patients with variegate porphyria and hereditary coproporphyria, with or without neurovisceral symptoms, may develop bullous eruptions especially on the hands, forearms, face, neck, or other areas of the skin exposed to sunlight. (msdmanuals.com)
- Exposure to sunlight precipitates cutaneous symptoms in variegate porphyria and rarely also in hereditary coproporphyria. (msdmanuals.com)
- The acute porphyrias (also referred to as acute hepatic porphyrias), which include acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP), are characterized by neurovisceral attacks that can cause neurologic damage and death if not treated promptly. (arupconsult.com)
- Lead poisoning and hereditary tyrosinemia type I can cause neuropathies similar to those of acute intermittent porphyria (AIP), as well as elevated porphyrins and aminolevulinic acid (ALA). Conditions with similar presentations to AIP but without elevated porphobilinogen (PBG) excretions include Guillain-Barré syndrome and seizures. (arupconsult.com)
- The four disorders are ALA dehydratase deficiency porphyria, acute intermittent porphyria, hereditary coproporphyria, and variegate porphyria. (ashpublications.org)
- Other conditions that clinically and biochemically may mimic acute porphyria include lead poisoning and hereditary tyrosinemia type I. The diagnosis of one of these acute porphyric syndromes should be considered in many patients with otherwise unexplained abdominal pain, severe constipation, systemic arterial hypertension, or other characteristic symptoms. (ashpublications.org)
- A woman with a transient episode of severe photosensitivity showed a biochemical porphyrin profile suggestive of hereditary coproporphyria (HCP), whereas some of her relatives had a profile that was suggestive of variegate porphyria (VP). (maastrichtuniversity.nl)
- Acute type includes the acute intermittent porphyria, variegate porphyria, and hereditary coproporphyria. (medscape.com)
- Plasma porphobilinogen (PBG) and aminolevulinic acid (ALA) are elevated during the symptomatic phase of the acute porphyrias: acute intermittent porphyria (AIP), hereditary coproporphyria, and variegate porphyria. (mayocliniclabs.com)
- Increased porphobilinogen (PBG) in urine is pathognomonic of an attack or crisis of acute porphyria acute intermittent porphyria, variegate porphyria, hereditary coproporphyria the absence of increased urinary PBG in a suspected attack excludes the diagnosis. (blallab.com)
Congenital4
- VP, also known as porphyria variegata, mixed porphyria, congenital cutaneous hepatic porphyria, and South African porphyria, is a blistering disorder caused by an autosomal dominantly inherited deficiency in protoporphyrinogen oxidase, a cytoplasmic enzyme involved in heme biosynthesis. (logicalimages.com)
- Demonstration of elevated porphyrins in plasma (particularly for congenital erythropoietic porphyria [CEP]), urine, and stool is very useful for diagnosis of the porphyrias. (medscape.com)
- Aminolevulinic acid dehydratase deficiency porphyria (ADP), congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), and hepatoerythropoietic porphyria (HEP) are autosomal recessive. (medscape.com)
- Seen in Inherited disorders like Acute intermittent porphyria, Congenital erythropoietic porphyria and Acquired disorders like Due to toxic agents such as heavy metals, chemicals, acute alcoholism and cirrhosis. (blallab.com)
Hepatic porphyrias6
- Acute hepatic porphyrias (AHPs) are a family of rare metabolic disorders each caused by a deficiency in one of four enzymes responsible for heme synthesis in the liver. (springer.com)
- What are potential complications for patients with acute hepatic porphyrias? (arupconsult.com)
- Patients with acute hepatic porphyrias are at greater risk for hepatic fibrosis or cirrhosis, as well as hepatocellular carcinoma (HCC). (arupconsult.com)
- The acute or inducible hepatic porphyrias comprise four inherited disorders of heme biosynthesis. (ashpublications.org)
- Diagnosis of the specific Porphyria can be difficult because the three acute Hepatic Porphyrias (AIP, HCP, VP) typically have similar acute symptoms, biochemical findings, and responses to treatment. (porphyriafoundation.org)
- For patients with symptoms of an acute Porphyria, but without a specific diagnosis, we offer a 'triple test,' which includes DNA testing for the three major acute hepatic Porphyrias (AIP, HCP, and VP). (porphyriafoundation.org)
Deficiency8
- Porphyrias with only neurovisceral symptoms without skin findings include acute intermittent porphyria and delta-aminolevulinic acid (ALA) dehydratase deficiency porphyria. (logicalimages.com)
- Porphyria is a predominantly inherited metabolic disorder resulting from a deficiency of an enzyme in the heme production pathway and overproduction of toxic heme precursors. (medscape.com)
- Acute porphyrias result from deficiency of certain enzymes in the heme biosynthetic pathway, resulting in accumulation of heme precursors that cause intermittent attacks of abdominal pain and neurologic symptoms. (msdmanuals.com)
- Accumulation of the porphyrin precursors porphobilinogen (PBG) and delta- aminolevulinic acid (ALA), or in the case of ALAD-deficiency porphyria, ALA alone, results. (msdmanuals.com)
- Each of the porphyrias is due to the deficiency of a specific enzyme involved in heme synthesis. (ashpublications.org)
- Each porphyria results from overproduction of heme precursors secondary to partial deficiency or, in XLP, increased activity of one of the enzymes of heme biosynthesis. (qxmd.com)
- This schematic diagram of biochemical abnormality shows the sites of enzymatic defects of the various porphyrias on the left side of the diagram and the dual enzyme abnormality of Chester porphyria (deficiency of porphobilinogen deaminase [PBGD] and protoporphyrinogen oxidase) on the right. (medscape.com)
- An isolated elevation of ALA may be due to the very rare ALA dehydratase deficiency porphyria (ADP) or more commonly, a secondary inhibition of ALA dehydratase. (mayocliniclabs.com)
American Porphyria Foundation3
- The American Porphyria Foundation provides a Drug Safety Database Search that provides information about the interaction of specific drugs in patients with porphyria. (logicalimages.com)
- current information should be sought from online databases such as www.drugs-porphyria.org and the American Porphyria Foundation . (msdmanuals.com)
- The testing program was developed with a grant from the American Porphyria Foundation. (porphyriafoundation.org)
Symptoms23
- Variegate porphyria, also known by several other names, is an autosomal dominant porphyria that can have acute (severe but usually not long-lasting) symptoms along with symptoms that affect the skin. (wikipedia.org)
- When symptoms occur, they can include acute attacks (similar to acute intermittent porphyria) or skin damage. (wikipedia.org)
- citation needed] Rarely, the signs and symptoms of variegate porphyria can begin in infancy or early childhood. (wikipedia.org)
- Systemic stress, such as liver disease or cholelithiasis, has been noted to precipitate cutaneous symptoms in patients with underlying variegate porphyria. (medscape.com)
- Porphyrias are divided into two types according to the predominant symptoms: (1) the neurovisceral or acute porphyrias, with abdominal pain, neuropathy, autonomic instability, and psychosis, and (2) the cutaneous porphyrias, with symptoms of photosensitive lesions on the skin. (medscape.com)
- Enzyme-inducing medications induce hepatic heme synthesis, which can exacerbate porphyria symptoms, or provoke acute attacks. (bmj.com)
- Depending on the specific enzyme affected, porphyria may manifest clinically in an acute or non-acute manner, and the signs and symptoms may be predominantly neurovisceral, psychiatric, cutaneous, or some combination of those. (medscape.com)
- The inductive coding approach targeted textual data related to acute intermittent porphyria attack symptoms, chronic symptoms, and the impact of the disease. (springer.com)
- In this study population of acute intermittent porphyria with frequent attacks, most patients had symptoms during and between attacks. (springer.com)
- Acute intermittent porphyria (AIP) is a rare, often mis/underdiagnosed, inherited metabolic disease characterized by acute potentially life-threatening attacks and in some patients, chronic debilitating multi-systemic symptoms and manifestations that negatively impact patients' daily functioning and quality of life. (springer.com)
- Symptoms and signs of acute porphyrias involve the nervous system, abdomen, or both (neurovisceral). (msdmanuals.com)
- Signs and symptoms of porphyrias are variable and nonspecific. (arupconsult.com)
- Porphyrias are generally classified as either acute or cutaneous, but some types can have overlapping symptoms, which can complicate diagnosis. (arupconsult.com)
- Diagnostic testing for porphyrias should be performed in individuals who present with severe, diffuse neuropathic abdominal pain and accompanying symptoms and in individuals with cutaneous photosensitivity. (arupconsult.com)
- Acute porphyria is often called the 'little imitator' because its typical symptoms-acute abdominal pain, rapid heart rate, increased blood pressure, nausea, vomiting, constipation or diarrhea, dizziness and fatigue, weakness and sometimes paralysis-are shared with many other more common conditions. (porphyriafoundation.org)
- In Robert Dawson's case, the diagnosis may be a little bit trickier than usual because until recently he was not too strongly affected by neurological symptoms like severe abdominal pain that occur in acute porphyria and are seen so much in Acute Intermittent Porphyria (the most common of the acute porphyrias). (porphyriafoundation.org)
- This medication is used to treat the symptoms that occur with certain blood disorders (porphyrias). (webmd.com)
- This medication may relieve symptoms such as pain, high blood pressure , rapid heartbeat, or mental changes that may occur during an acute attack of porphyria. (webmd.com)
- The researchers noted the nonspecific symptoms of porphyria attacks may make the diagnosis difficult. (porphyrianews.com)
- People with inactive porphyria who choose to fast for a day as part of their religious observances generally don't see any unusual porphyria symptoms, a recent study reported. (porphyrianews.com)
- Many of the symptoms of erythropoietic protoporphyria (EPP), a form of porphyria, may not be reported fully or at all, and this may make it harder to diagnose the rare disease, according to a new U.S. study. (porphyrianews.com)
- Acute hepatic porphyria (AHP) is a family of rare genetic diseases characterized by potentially life-threatening attacks and, for some people, chronic debilitating symptoms that negatively impact daily functioning and quality of life. (jacksoncountysentinel.net)
- Moreover, activated charcoal may worsen symptoms in individuals with variegate porphyria, a rare genetic disease affecting the skin, gut and nervous system. (foodpharmacy.blog)
Attacks14
- An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. (medscape.com)
- Mild attacks of variegate porphyria may resolve within a few to several days with conservative management. (medscape.com)
- Acute attacks of variegate porphyria can be life-threatening. (medscape.com)
- Urine porphyrin studies are the mainstay in the diagnosis of acute porphyria attacks. (medscape.com)
- Avoidance of sunlight is the key in preventing attacks of cutaneous porphyrias. (medscape.com)
- In patients with acute hepatic porphyria (AHP), prolonged fasting is a known trigger of AHP attacks. (bvsalud.org)
- The acute porphyrias are characterized by potentially life-threatening neurologic attacks that do not occur in the nonacute porphyrias. (clinicaladvisor.com)
- Patients with acute intermittent porphyria may experience acute debilitating neurovisceral attacks that require frequent hospitalizations and negatively impact quality of life. (springer.com)
- Although clinical aspects of acute intermittent porphyria attacks have been documented, the experience of patients is not well known, particularly for those more severely affected patients who experience frequent attacks. (springer.com)
- The aim of the present study was to qualitatively characterize the experience of patients with acute intermittent porphyria who have frequent attacks, as well as the impact of the disease on daily living. (springer.com)
- Patients with acute intermittent porphyria who experience frequent attacks were recruited and took part in 2-h qualitative one-on-one interviews with a semi-structured guide. (springer.com)
- The treatment of choice for all but mild attacks of the acute porphyrias is intravenous hemin therapy, which should be started as soon as possible. (ashpublications.org)
- because these two types, HCP, and VP, are documented to not have urinary elevations in ALA/ PBG during life threatening attacks (Hematin treatment of Acute Porphyria, Peterson, 1976) (Janus, 2017). (porphyriaalliance.org)
- Acute attacks usually require hospitalisation and treatment by a porphyria specialist. (porphyria-australia.org)
Type of Porphyria1
- Precisely which of these chemicals builds up depends on the type of porphyria. (qxmd.com)
Mutation14
- An additional aggravating mutation affecting variegate porphyria can be found at 6p21.3 on the HFE gene. (wikipedia.org)
- Variegate porphyria arises from autosomal dominant inheritance of a gene mutation encoding a defective protoporphyrinogen oxidase enzyme. (medscape.com)
- Four South African children manifesting variegate porphyria were compound heterozygotes with the founder mutation on one allele, but a different mutation apparently encoding an enzyme with enough residual activity to enable survival, on the other. (medscape.com)
- [ 18 ] A haplotype study of Argentinean patients with variegate porphyria suggested a founder mutation within its population as well. (medscape.com)
- Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. (medscape.com)
- A R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria. (medscape.com)
- van Serooskerken AM, Ernst M, Bladergroen RS, Wolff C, Floderus Y, Harper P. A recurrent mutation in variegate porphyria patients from Chile and Sweden: Evidence for a common genetic background? (medscape.com)
- Frank J, Alta VM, Ahmad W, Lam H, Wolff C, Christiano AM. Identification of a Founder Mutation in the Protoporphyrinogen Oxidase Gene in Variegate Porphyria Patients from Chile. (medscape.com)
- A Chinese woman with acute intermittent porphyria (AIP) accompanied by brain lesions was found to have a new mutation in the HMBS gene, a case report described. (porphyrianews.com)
- Demystification of Chester porphyria: a nonsense mutation in the porphobilinogen deaminase gene. (medscape.com)
- If a mutation (or change) in the DNA sequence is found in a specific Porphyria-causing gene, the diagnosis of that Porphyria is confirmed. (porphyriafoundation.org)
- Once a mutation has been identified, DNA analysis can then be performed on other family members to determine if they have inherited that Porphyria, thus allowing identification of individuals who can be counseled about appropriate management in order to avoid or minimize disease complications. (porphyriafoundation.org)
- Each Porphyria is caused by a mutation in the DNA sequence of a specific gene. (porphyriafoundation.org)
- This means, for example, that if a patient has been given the diagnosis of AIP and no AIP gene mutation is identified, it is possible that the patient has a different acute Porphyria. (porphyriafoundation.org)
Symptomatic4
- Acute symptomatic seizures occur in 10-20% of patients with acute intermittent porphyria in relapse. (bmj.com)
- Porphyria might be the cause of chronic symptomatic epilepsy (if so, this would be rare or frequently undiagnosed) or there might be a chance association, given that epilepsy is common. (bmj.com)
- A normal urine PBG result has a sensitivity of almost 100% (ie, rules out) in the diagnosis of porphyria in acutely symptomatic patients. (medscape.com)
- This test is an alternative for the evaluation of a suspected acute porphyria when a urine specimen cannot be obtained during a symptomatic episode. (mayocliniclabs.com)
Management of the acute porphyrias2
- For explanation of diagnosis and management of the acute porphyrias and the acute manifestations of porphyrias with both neurovisceral and cutaneous components, please refer to the companion article Porphyria, Acute. (medscape.com)
- Downregulation of ALA synthase-1 by avoidance or removal of inducing drugs and chemicals by nutritional means (high carbohydrate intakes) and by administration of exogenous heme remains the cornerstone of management of the acute porphyrias. (ashpublications.org)
Erythropoietic porphyria1
- The t wo current classifications of porphyria are: acute hepatic porphyria (AHP), and erythropoietic porphyria (EP). (porphyriaalliance.org)
Patients with variegate porphyria1
- Ten patients with variegate porphyria were uniformly found to have distinctive plasma porphyrin fluorescence wavelength maxima in saline-diluted plasma specimens. (nih.gov)
Protoporphyria3
- Porphyrias with nonblistering cutaneous features include erythropoietic protoporphyria and X-linked protoporphyria . (logicalimages.com)
- [ 2 ] There is also an X-linked dominant inherited porphyria called X-linked protoporphyria (XLP). (medscape.com)
- The porphyrias are caused by loss (or gain, in the case of X-linked erythropoietic protoporphyria [XLP]) of specific enzyme functions in the heme biosynthesis pathway. (arupconsult.com)
Homozygous1
- Homozygous variegate porphyria in South Africa: genotypic analysis in two cases. (medscape.com)
Types of porphyrias2
- [ 1 ] There are at least 8 different types of porphyrias. (medscape.com)
- The other enzymes, when deficient, give rise to the types of porphyrias shown in Tables 1 Table 1A , Table 1B and 2 . (ashpublications.org)
Photosensitivity2
- Beta-carotene is a pigment found in various green and yellow fruits and vegetables and can decrease the severity of photosensitivity reactions in patients with porphyria. (medscape.com)
- Photosensitivity is seen (as with variegate porphyria). (mhmedical.com)
Enzyme2
- The activity of this enzyme is reduced by 50 percent in most people with variegate porphyria. (wikipedia.org)
- Variegate porphyria (VP) is an inherited disorder of porphyrin-heme metabolism arising from mutations of the gene encoding the enzyme protoporphyrinogen oxidase. (medscape.com)
Heme synthesis2
- Acute intermittent porphyria is a rare metabolic disorder that affects heme synthesis. (springer.com)
- Overview of Porphyrias Porphyrias are rare disorders in which there are defects in the pathway of heme synthesis due to genetic or acquired deficiencies of enzymes of the heme biosynthetic pathway. (msdmanuals.com)
Protoporphyrinogen oxidase gene1
- To date, there have been 184 different mutations in the protoporphyrinogen oxidase gene that results in variegate porphyria. (medscape.com)
Porphyrins4
- Inherited as an autosomal dominant trait, variegate porphyria is biochemically characterized by accumulations of the photosensitizing porphyrins, protoporphyrin and coproporphyrin. (medscape.com)
- The mutations that underlie porphyria result in accumulation and increased excretion of porphyrins and their precursors. (medscape.com)
- Diagnosis is established by finding substantial increases in porphyrins in urine or plasma and excluding other blistering cutaneous porphyrias. (bmj.com)
- Porphyria diseases are a group of metabolic disorders caused by abnormal functioning of heme biosynthesis enzymes and characterized by excessive accumulation and excretion of porphyrins and their precursors. (qxmd.com)
Clinical8
- A 2006 clinical, biochemical and mutational study of eight Swiss variegate porphyria patients and their families found four novel PPOX gene mutations believed to be unique to the Swiss population. (wikipedia.org)
- The clinical and biochemical features of variegate porphyria: an analysis of 300 cases studied at Groote Schuur Hospital, Cape Town. (medscape.com)
- von und zu Fraunberg M, Timonen K, Mustajoki P, Kauppinen R. Clinical and biochemical characteristics and genotype-phenotype correlation in Finnish variegate porphyria patients. (medscape.com)
- Clinical classification of the different porphyrias. (medscape.com)
- Medicine Central , im.unboundmedicine.com/medicine/view/5-Minute-Clinical-Consult/816140/all/Porphyria. (unboundmedicine.com)
- This unit summarizes the current knowledge on the classification, clinical features, etiology, pathogenesis, and genetics of porphyria diseases. (qxmd.com)
- The case of a teenager in Nepal who was diagnosed with porphyria following a detailed series of clinical tests - part of a "diagnostic conundrum" - was described in a recent report. (porphyrianews.com)
- Qadiri MR, Church SE, McColl KE, Moore MR, Youngs GR. Chester porphyria: a clinical study of a new form of acute porphyria. (medscape.com)
Mutations3
- citation needed] Mutations in the PPOX gene cause variegate porphyria. (wikipedia.org)
- As shown in Figure 1 , there is no porphyria associated with a defect in ALA synthase-1, but mutations of the X-linked ALA synthase-2 (the erythroid form) are causative for X-linked sideroblastic anemia. (ashpublications.org)
- In the Porphyrias, there are no common mutations so the entire gene must be sequenced in each new family. (porphyriafoundation.org)
People with porphyria1
- It is, therefore, important to determine the correct dose and safety of use of certain drugs in people with porphyria. (bmj.com)
Diagnosis of porphyrias1
- What is the role of skin biopsy in the diagnosis of porphyrias? (arupconsult.com)
Metabolic disorders1
- Porphyrias are a group of metabolic disorders, usually genetic in origin, secondary to deficiencies of various enzymes involved in the heme biosynthetic pathways. (medscape.com)
Variegata1
- porphyria variegata" at Dorland's Medical Dictionary Frank J, Christiano AM (1998). (wikipedia.org)
Porphyrin3
- An outline of the porphyrin pathway reveals the pathophysiological mechanisms that cause porphyria. (medscape.com)
- To assess for cutaneous porphyria, the plasma porphyrin level should be measured, using fluorescence emission spectroscopy. (medscape.com)
- The porphyrias are a family of illnesses caused by various metabolic derangements in the metabolism of porphyrin, the chemical backbone of hemoglobin. (mhmedical.com)
Differential Diagnoses1
- What are the differential diagnoses for porphyrias? (arupconsult.com)
Chester Porphyria5
- There are 2 and possibly many more known sub types of porphyria so far, HEP, a dual PCT, and Chester Porphyria a VP/ AIP hybrid. (porphyriaalliance.org)
- Currently, no cure exists for Chester porphyria. (medscape.com)
- Patients with Chester porphyria are at risk of an acute attack of porphyria at the time of surgery. (medscape.com)
- Crimlisk H. Dobson's complaint: the story of the Chester porphyria. (medscape.com)
- Chester porphyria: biochemical studies of a new form of acute porphyria. (medscape.com)
Porphobilinogen deaminase1
- Meissner PN, Day RS, Moore MR, Disler PB, Harley E. Protoporphyrinogen oxidase and porphobilinogen deaminase in variegate porphyria. (medscape.com)
Biochemical1
- The current porphyria literature is very exhaustive and a brief overview of porphyria diseases is essential in order for the reader to better appreciate the relevance of this area of research prior to undertaking biochemical diagnostics procedures. (qxmd.com)
Prevalence1
- In South Africa, the prevalence of variegate porphyria is approximately 1 in 300. (wikipedia.org)
Manifestations5
- Bonkowsky HL, Schady W. Neurologic manifestations of acute porphyria. (medscape.com)
- Acute porphyrias: pathogenesis of neurological manifestations. (medscape.com)
- Only the cutaneous manifestations of the porphyrias are considered in this article. (medscape.com)
- In these patients, acute intermittent porphyria appears to have acute exacerbations as well as chronic day-to-day manifestations, and is not just intermittent as its name implies. (springer.com)
- Whereas the other porphyrias (acute intermittent porphyria and variegate porphyria) are associated with well-known systemic manifestations (abdominal pain, peripheral neuropathy, and pulmonary complications), PCT has no extracutaneous manifestations. (mhmedical.com)
Genetic3
- A subtype of porphyria, variegate porphyria is an uncommon genetic disorder. (naturalpedia.com)
- Another patient was found to have a genetic variant associated with variegate porphyria, which finally explained the patient's and family members' mysterious rashes and sun sensitivity. (harvard.edu)
- In contrast, DNA testing is the most accurate and reliable method for determining if a person has a specific Porphyria and is considered the 'gold standard' for the diagnosis of genetic disorders. (porphyriafoundation.org)
Abdominal1
- AIP is an acute porphyria known to cause abdominal pain, gastrointestinal issues, and neurological changes. (porphyria-australia.org)
Severe1
- An early diagnosis and treatment for acute intermittent porphyria (AIP) might have prevented severe complications in a 21-year-old woman, according to a U.K. case report. (porphyrianews.com)
Gene3
- citation needed] Variegate porphyria is inherited in an autosomal dominant pattern, which means the defective gene is located on an autosome, and inheriting one copy of the defective gene from an affected parent is sufficient to cause the disorder. (wikipedia.org)
- Of the 44 FDA approvals of new drugs, 8 were for 6 single-gene diseases: DMD, beta thalassemia, cystic fibrosis, a form of amyloidosis, and two each for sickle cell disease and porphyria. (plos.org)
- Thus, the diagnosis of a specific Porphyria determines what gene to test. (porphyriafoundation.org)
Fluorescence emission1
- Plasma fluorescence emission that is maximal at 626 +/- 1 nm is a diagnostic marker for variegate porphyria. (nih.gov)
Diseases4
- Porphyrias are a group of diseases resulting from defects / dysfunction in enzymes involved in heme biosynthesis. (logicalimages.com)
- The cutaneous porphyrias are dermatologic diseases that may or may not involve the liver and nervous system. (medscape.com)
- The porphyrias are metabolic diseases caused by deficiencies in enzymes involved in the heme biosynthetic pathway. (clinicaladvisor.com)
- With the exception of acquired PCT, the porphyrias are inherited diseases. (clinicaladvisor.com)