Lamin Type A
Aging, Premature
Farnesyltranstransferase
Nuclear Lamina
Werner Syndrome
Prenylation
Protein Precursors
Protein Prenylation
Bone Demineralization, Pathologic
Fibroblasts
Nuclear Envelope
Nuclear Proteins
Lamins
Protein Modification, Translational
Lamin Type B
Contracture
Lipodystrophy
Maxillofacial Abnormalities
Cell Aging
Cockayne Syndrome
Cell Nucleus
Aging
Bone Diseases, Developmental
Phenotype
Mutation
Alkyl and Aryl Transferases
Progeria
Heterochromatin
Cells, Cultured
Membrane Proteins
Skin
Disease Models, Animal
Enzyme Inhibitors
DNA Repair
Heterozygote
Mitosis
DNA Damage
Bone and Bones
Mice, Transgenic
Mice, Knockout
Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. (1/254)
Progeroid type Ehlers-Danlos (E-D) syndrome was reported to be caused by defects in galactosyltransferase I (EC 2.4.1.133), which is involved in the synthesis of common linkage regions of proteoglycans. Recently, we isolated cDNA of the galactosyltransferase I (XGalT-1) (Okajima, T., Yoshida, K., Kondo, T., and Furukawa, K. (1999) J. Biol. Chem. 274, 22915-22918). Therefore, we analyzed mutations in this gene of a patient with progeroid type E-D syndrome by reverse transcription polymerase chain reaction and direct sequencing. Two changes of G and T to A and C at 186 and 206, respectively, were detected. Then, we determined the genomic DNA sequences encompassing the A186D and L206P mutations, revealing that the unaffected parents and two siblings were heterozygous for either one of the two different mutations and normal, while the patient had both of two different mutant genes. Enzymatic functions of cDNA clones of XGalT-1 containing the individual mutations were examined, elucidating that L206P clone completely lost the activity, while A186D retained approximately 50% or 10% of the activity when analyzed with extracts from cDNA transfectant cells or recombinant soluble enzymes, respectively. Moreover, L206P enzyme showed diffuse staining in the cytoplasm of transfectant cells, while the wild type or A186D clones showed Golgi pattern. These results indicated that the mutations in XGalT-1 were at least one of main molecular basis for progeroid type E-D syndrome. (+info)The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. (2/254)
The limited life span of normal human cells represents a substantial obstacle for biochemical analysis, genetic manipulation and genetic screens. To overcome this technical barrier, immortal human cell lines are often derived from tumors or produced by transformation with viral oncogenes such as SV40 large T antigen. Cell lines produced by these approaches are invariably transformed, genomically unstable and display cellular properties that differ from their normal counterpart. It was recently shown that the ectopic expression of hTERT, encoding the catalytic subunit of human telomerase, can extend the life span of normal human cells without causing cellular transformation and genomic instability. In the present study, we have used hTERT to extend the life span of normal human skin fibroblasts derived from patients afflicted with syndromes of genomic instability and/or premature aging. Our results show that hTERT efficiently extends the life span without altering the characteristic phenotypic properties of the cells. Thus, the ectopic expression of telomerase represents a major improvement over the use of viral oncogenes for the establishment of human cell lines. (+info)Mitotic misregulation and human aging. (3/254)
Messenger RNA levels were measured in actively dividing fibroblasts isolated from young, middle-age, and old-age humans and humans with progeria, a rare genetic disorder characterized by accelerated aging. Genes whose expression is associated with age-related phenotypes and diseases were identified. The data also suggest that an underlying mechanism of the aging process involves increasing errors in the mitotic machinery of dividing cells in the postreproductive stage of life. We propose that this dysfunction leads to chromosomal pathologies that result in misregulation of genes involved in the aging process. (+info)Horizontal transmission of Candida parapsilosis candidemia in a neonatal intensive care unit. (4/254)
This report describes the nosocomial acquisition of Candida parapsilosis candidemia by one of the six premature newborns housed in the same room of a neonatal intensive care unit at the Ospedale Santa Chiara, Pisa, Italy. The infant had progeria, a disorder characterized by retarded physical development and progressive senile degeneration. The infant, who was not found to harbor C. parapsilosis at the time of his admission to the intensive care unit, had exhibited symptomatic conjunctivitis before the onset of a severe bloodstream infection. In order to evaluate the source of infection and the route of transmission, two independent molecular typing methods were used to determine the genetic relatedness among the isolates recovered from the newborn, the inanimate hospital environment, hospital personnel, topically and intravenously administered medicaments, and indwelling catheters. Among the isolates collected, only those recovered from the hands of two nurses attending the newborns and from both the conjunctiva and the blood of the infected infant were genetically indistinguishable. Since C. parapsilosis was never recovered from indwelling catheters or from any of the drugs administered to the newborn, we concluded that (i) horizontal transmission of C. parapsilosis occurred through direct interaction between nurses and the newborn and (ii) the conjunctiva was the site through which C. parapsilosis entered the bloodstream. This finding highlights the possibility that a previous C. parapsilosis colonization and/or infection of other body sites may be a predisposing condition for subsequent C. parapsilosis hematogenous dissemination in severely ill newborns. (+info)Telomere length predicts replicative capacity of human fibroblasts. (5/254)
When human fibroblasts from different donors are grown in vitro, only a small fraction of the variation in their finite replicative capacity is explained by the chronological age of the donor. Because we had previously shown that telomeres, the terminal guanine-rich sequences of chromosomes, shorten throughout the life-span of cultured cells, we wished to determine whether variation in initial telomere length would account for the unexplained variation in replicative capacity. Analysis of cells from 31 donors (aged 0-93 yr) indicated relatively weak correlations between proliferative ability and donor age (m = -0.2 doubling per yr; r = -0.42; P = 0.02) and between telomeric DNA and donor age (m = -15 base pairs per yr; r = -0.43; P = 0.02). However, there was a striking correlation, valid over the entire age range of the donors, between replicative capacity and initial telomere length (m = 10 doublings per kilobase pair; r = 0.76; P = 0.004), indicating that cell strains with shorter telomeres underwent significantly fewer doublings than those with longer telomeres. These observations suggest that telomere length is a biomarker of somatic cell aging in humans and are consistent with a causal role for telomere loss in this process. We also found that fibroblasts from Hutchinson-Gilford progeria donors had short telomeres, consistent with their reduced division potential in vitro. In contrast, telomeres from sperm DNA did not decrease with age of the donor, suggesting that a mechanism for maintaining telomere length, such as telomerase expression, may be active in germ-line tissue. (+info)Lack of peroxisomal catalase causes a progeric phenotype in Caenorhabditis elegans. (6/254)
Studies using the nematode Caenorhabditis elegans as a model system to investigate the aging process have implicated the insulin/insulin-like growth factor-I signaling pathway in the regulation of organismal longevity through its action on a subset of target genes. These targets can be classified into genes that shorten or extend life-span upon their induction. Genes that shorten life-span include a variety of stress response genes, among them genes encoding catalases; however, no evidence directly implicates catalases in the aging process of nematodes or other organisms. Using genetic mutants, we show that lack of peroxisomal catalase CTL-2 causes a progeric phenotype in C. elegans. Lack of peroxisomal catalase also affects the developmental program of C. elegans, since Deltactl-2 mutants exhibit decreased egg laying capacity. In contrast, lack of cytosolic catalase CTL-1 has no effect on either nematode aging or egg laying capacity. The Deltactl-2 mutation also shortens the maximum life-span of the long lived Deltaclk-1 mutant and accelerates the onset of its egg laying period. The more rapid aging of Deltactl-2 worms is apparently not due to increased carbonylation of the major C. elegans proteins, although altered peroxisome morphology in the Deltactl-2 mutant suggests that changes in peroxisomal function, including increased production of reactive oxygen species, underlie the progeric phenotype of the Deltactl-2 mutant. Our findings support an important role for peroxisomal catalase in both the development and aging of C. elegans and suggest the utility of the Deltactl-2 mutant as a convenient model for the study of aging and the human diseases acatalasemia and hypocatalasemia. (+info)Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. (7/254)
Hutchinson-Gilford progeria syndrome (HGPS) is a premature aging disorder, commonly caused by a point mutation in the lamin A gene that results in a protein lacking 50 aa near the C terminus, denoted LADelta50. Here we show by light and electron microscopy that HGPS is associated with significant changes in nuclear shape, including lobulation of the nuclear envelope, thickening of the nuclear lamina, loss of peripheral heterochromatin, and clustering of nuclear pores. These structural defects worsen as HGPS cells age in culture, and their severity correlates with an apparent increase in LADelta50. Introduction of LADelta50 into normal cells by transfection or protein injection induces the same changes. We hypothesize that these alterations in nuclear structure are due to a concentration-dependent dominant-negative effect of LADelta50, leading to the disruption of lamin-related functions ranging from the maintenance of nuclear shape to regulation of gene expression and DNA replication. (+info)Laminopathies and atherosclerosis. (8/254)
Laminopathies are genetic diseases that encompass a wide spectrum of phenotypes with diverse tissue pathologies and result mainly from mutations in the LMNA gene encoding nuclear lamin A/C. Some laminopathies affect the cardiovascular system, and a few (namely, Dunnigan-type familial partial lipodystrophy [FPLD2] and Hutchinson-Gilford progeria syndrome [HGPS]) feature atherosclerosis as a key component. The premature atherosclerosis of FPLD2 is probably related to characteristic proatherogenic metabolic disturbances such as dyslipidemia, hyperinsulinemia, hypertension, and diabetes. In contrast, the premature atherosclerosis of HGPS occurs with less exposure to metabolic proatherogenic traits and probably reflects the generalized process of accelerated aging in HGPS. Although some common polymorphisms of LMNA have been associated with traits related to atherosclerosis, the monogenic diseases FPLD2 and HGPS are more likely to provide clues about new pathways for the general process of atherosclerosis. Dunnigan-type familial partial lipodystrophy and Hutchinson-Gilford progeria syndrome are laminopathies caused by mutation in LMNA that feature atherosclerosis, which is related to proatherogenic metabolic disturbances and to the generalized process of accelerated aging, respectively. These monogenic diseases may provide clues about new pathways for atherogenesis. (+info)Lamin Type A, also known as LMNA, is a gene that provides instructions for making proteins called lamins. These proteins are part of the nuclear lamina, a network of fibers that lies just inside the nuclear envelope, which is the membrane that surrounds the cell's nucleus. The nuclear lamina helps maintain the shape and stability of the nucleus and plays a role in regulating gene expression and DNA replication.
Mutations in the LMNA gene can lead to various diseases collectively known as laminopathies, which affect different tissues and organs in the body. These conditions include Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, dilated cardiomyopathy with conduction system disease, and a type of premature aging disorder called Hutchinson-Gilford progeria syndrome. The specific symptoms and severity of these disorders depend on the particular LMNA mutation and the tissues affected.
Premature aging, also known as "accelerated aging" or "early aging," refers to the physiological process in which the body shows signs of aging at an earlier age than typically expected. This can include various symptoms such as wrinkles, graying hair, decreased energy and mobility, cognitive decline, and increased risk of chronic diseases.
The medical definition of premature aging is not well-established, as aging is a complex process influenced by a variety of genetic and environmental factors. However, certain conditions and syndromes are associated with premature aging, such as Hutchinson-Gilford progeria syndrome, Werner syndrome, and Down syndrome.
In general, the signs of premature aging may be caused by a combination of genetic predisposition, lifestyle factors (such as smoking, alcohol consumption, and poor diet), exposure to environmental toxins, and chronic stress. While some aspects of aging are inevitable, maintaining a healthy lifestyle and reducing exposure to harmful factors can help slow down the aging process and improve overall quality of life.
Farnesyltranstransferase (FTase) is an enzyme that plays a role in the post-translational modification of proteins, specifically by adding a farnesyl group to certain protein substrates. This process, known as farnesylation, is essential for the proper localization and function of many proteins, including Ras family GTPases, which are involved in signal transduction pathways that regulate cell growth, differentiation, and survival.
FTase catalyzes the transfer of a farnesyl group from farnesyl pyrophosphate (FPP) to a cysteine residue near the C-terminus of its protein substrates. This modification allows the protein to interact with membranes and other cellular structures, which is critical for their function. Inhibitors of FTase have been developed as potential therapeutic agents for cancer and other diseases associated with aberrant Ras signaling.
The cell nucleus is a membrane-bound organelle that contains most of the genetic material in eukaryotic cells. The shape of the cell nucleus can vary widely among different cell types and can be influenced by various factors, including the organization of the nuclear envelope, the distribution of chromatin (the complex of DNA, RNA, and proteins that makes up chromosomes), and the presence or absence of a nucleolus (a structure within the nucleus where ribosomal RNA is synthesized).
The shape of the cell nucleus can be described in several ways, including:
* Spherical: The nucleus has a round, ball-like shape.
* Ellipsoidal: The nucleus has an oval or ellipse-like shape.
* Irregular: The nucleus has a shape that is not easily described as spherical or ellipsoidal and may be lobed, indented, or have other irregularities.
The shape of the cell nucleus can provide important clues about the function and health of a cell. For example, certain diseases and conditions, such as cancer, can cause changes in the shape of the nucleus. In addition, some researchers have suggested that the shape of the nucleus may be related to the mechanical properties of the cell and its ability to migrate or change shape in response to its environment.
The nuclear lamina is a protein network that lies beneath the inner nuclear membrane of the nuclear envelope in eukaryotic cells. It is primarily composed of type V intermediate filament proteins called lamins, which assemble into heteropolymeric filaments and provide structural support to the nucleus. The nuclear lamina plays essential roles in various nuclear processes, including DNA replication, gene expression, and nuclear division. Additionally, it serves as an attachment site for chromatin, nuclear pore complexes, and other nuclear envelope components, helping maintain the overall organization and integrity of the nucleus.
Werner Syndrome is a rare, autosomal recessive genetic disorder characterized by the appearance of premature aging. It's often referred to as "progeria of the adult" or "adult progeria." The syndrome is caused by mutations in the WRN gene, which provides instructions for making a protein involved in repairing damaged DNA and maintaining the stability of the genetic information.
The symptoms typically begin in a person's late teens or early twenties and may include:
- Short stature
- Premature graying and loss of hair
- Skin changes, such as scleroderma (a thickening and hardening of the skin) and ulcers
- Voice changes
- Type 2 diabetes
- Cataracts
- Atherosclerosis (the buildup of fats, cholesterol, and other substances in and on the artery walls)
- Increased risk of cancer
The life expectancy of individuals with Werner Syndrome is typically around 45 to 50 years. It's important to note that while there are similarities between Werner Syndrome and other forms of progeria, such as Hutchinson-Gilford Progeria Syndrome, they are distinct conditions with different genetic causes and clinical features.
Prenylation is a post-translational modification process in which a prenyl group, such as a farnesyl or geranylgeranyl group, is added to a protein covalently. This modification typically occurs at a cysteine residue within a CAAX motif (C is cysteine, A is an aliphatic amino acid, and X is any amino acid) found at the carboxyl-terminus of the protein. Prenylation plays a crucial role in membrane association, protein-protein interactions, and intracellular trafficking of proteins, particularly those involved in signal transduction pathways.
Protein precursors, also known as proproteins or prohormones, are inactive forms of proteins that undergo post-translational modification to become active. These modifications typically include cleavage of the precursor protein by specific enzymes, resulting in the release of the active protein. This process allows for the regulation and control of protein activity within the body. Protein precursors can be found in various biological processes, including the endocrine system where they serve as inactive hormones that can be converted into their active forms when needed.
Protein prenylation is a post-translational modification process in which a lipophilic group, such as a farnesyl or geranylgeranyl moiety, is covalently attached to specific cysteine residues near the carboxy-terminus of proteins. This modification plays a crucial role in membrane targeting and protein-protein interactions, particularly for proteins involved in signal transduction pathways, such as Ras family GTPases. The enzymes responsible for prenylation are called protein prenyltransferases, and their dysfunction has been implicated in various diseases, including cancer and neurodegenerative disorders.
Pathologic bone demineralization is a condition characterized by the loss of minerals, such as calcium and phosphate, from the bones. This process makes the bones more porous, weaker, and more susceptible to fractures. It can occur due to various medical conditions, including osteoporosis, hyperparathyroidism, Paget's disease of bone, and cancer that has spread to the bones (metastatic cancer).
In a healthy individual, the body constantly remodels the bones by removing old bone tissue (resorption) and replacing it with new tissue. This process is regulated by two types of cells: osteoclasts, which are responsible for bone resorption, and osteoblasts, which produce new bone tissue. In pathologic bone demineralization, there is an imbalance between the activity of these two cell types, with excessive resorption and inadequate formation of new bone tissue.
Pathologic bone demineralization can lead to a range of symptoms, including bone pain, fractures, loss of height, and a decreased ability to perform daily activities. Treatment for this condition depends on the underlying cause but may include medications that slow down bone resorption or promote bone formation, as well as lifestyle changes such as exercise and dietary modifications.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
The nuclear envelope is a complex and double-membrane structure that surrounds the eukaryotic cell's nucleus. It consists of two distinct membranes: the outer nuclear membrane, which is continuous with the endoplasmic reticulum (ER) membrane, and the inner nuclear membrane, which is closely associated with the chromatin and nuclear lamina.
The nuclear envelope serves as a selective barrier between the nucleus and the cytoplasm, controlling the exchange of materials and information between these two cellular compartments. Nuclear pore complexes (NPCs) are embedded in the nuclear envelope at sites where the inner and outer membranes fuse, forming aqueous channels that allow for the passive or active transport of molecules, such as ions, metabolites, and RNA-protein complexes.
The nuclear envelope plays essential roles in various cellular processes, including DNA replication, transcription, RNA processing, and chromosome organization. Additionally, it is dynamically regulated during the cell cycle, undergoing disassembly and reformation during mitosis to facilitate equal distribution of genetic material between daughter cells.
Nuclear proteins are a category of proteins that are primarily found in the nucleus of a eukaryotic cell. They play crucial roles in various nuclear functions, such as DNA replication, transcription, repair, and RNA processing. This group includes structural proteins like lamins, which form the nuclear lamina, and regulatory proteins, such as histones and transcription factors, that are involved in gene expression. Nuclear localization signals (NLS) often help target these proteins to the nucleus by interacting with importin proteins during active transport across the nuclear membrane.
Skin abnormalities refer to any changes in the skin that deviate from its normal structure, function, or color. These can manifest as various conditions such as lesions, growths, discolorations, or textural alterations. Examples include moles, freckles, birthmarks, rashes, hives, acne, eczema, psoriasis, rosacea, skin cancer, and many others. Some skin abnormalities may be harmless and require no treatment, while others might indicate an underlying medical condition that requires further evaluation and management.
Lamins are type V intermediate filament proteins that play a structural role in the nuclear envelope. They are the main components of the nuclear lamina, a mesh-like structure located inside the inner membrane of the nuclear envelope. Lamins are organized into homo- and heterodimers, which assemble into higher-order polymers to form the nuclear lamina. This structure provides mechanical support to the nucleus, helps maintain the shape and integrity of the nucleus, and plays a role in various nuclear processes such as DNA replication, transcription, and chromatin organization. Mutations in the genes encoding lamins have been associated with various human diseases, collectively known as laminopathies, which include muscular dystrophies, neuropathies, cardiomyopathies, and premature aging disorders.
Translational protein modification refers to the covalent alteration of a protein during or shortly after its synthesis on the ribosome. This process is an essential mechanism for regulating protein function and can have a significant impact on various aspects of protein biology, including protein stability, localization, activity, and interaction with other molecules.
During translation, as the nascent polypeptide chain emerges from the ribosome, it can be modified by enzymes that recognize specific sequences or motifs within the protein. These modifications can include the addition of chemical groups such as phosphate, acetyl, methyl, ubiquitin, or SUMO (small ubiquitin-like modifier) groups, among others.
Examples of translational protein modifications include:
1. N-terminal acetylation: The addition of an acetyl group to the alpha-amino group of the first amino acid in a polypeptide chain. This modification can affect protein stability and localization.
2. Ubiquitination: The covalent attachment of ubiquitin molecules to lysine residues within a protein, which can target it for degradation by the proteasome or regulate its activity and interactions with other proteins.
3. SUMOylation: The addition of a SUMO group to a lysine residue in a protein, which can modulate protein-protein interactions, subcellular localization, and stability.
4. Phosphorylation: The addition of a phosphate group to serine, threonine, or tyrosine residues within a protein, which can regulate enzymatic activity, protein-protein interactions, and signal transduction pathways.
Translational protein modifications play crucial roles in various cellular processes, including gene expression regulation, DNA repair, cell cycle control, stress response, and apoptosis. Dysregulation of these modifications has been implicated in numerous diseases, such as cancer, neurodegenerative disorders, and metabolic disorders.
I'm sorry for any confusion, but "Lamin Type B" is not a recognized medical term or concept in human physiology or pathology. The term "lamin" refers to proteins that are part of the nuclear lamina, a filamentous network found inside the nucleus of cells. There are three types of lamin proteins: A, B, and C.
Lamin A and Lamin C are produced from the LMNA gene, while Lamin B1 and Lamin B2 are produced from the LMNB1 and LMNB2 genes, respectively. Therefore, "Lamin Type B" is not a specific designation, but rather encompasses two distinct proteins: Lamin B1 and Lamin B2.
If you have any questions about lamins or another medical topic, please provide more context or clarify your question so I can give you a more accurate answer.
I'm sorry for any confusion, but "organelle shape" is not a medical term with a specific definition. Organelles are specialized structures within cells that have specific functions, and they can vary in shape between different types of cells and organisms. However, the shape of an organelle is typically described as part of its overall function or structure, rather than being a distinct concept with its own definition.
For example, mitochondria are often described as having a distinctive "fried egg" shape, with a round or oval outer membrane surrounding a more irregular inner membrane. Similarly, chloroplasts in plant cells have a characteristic disk or sac-like shape and contain stacks of flattened sacs called thylakoids.
In summary, while organelle shape is an important aspect of cell biology, it does not have a specific medical definition.
A contracture, in a medical context, refers to the abnormal shortening and hardening of muscles, tendons, or other tissue, which can result in limited mobility and deformity of joints. This condition can occur due to various reasons such as injury, prolonged immobilization, scarring, neurological disorders, or genetic conditions.
Contractures can cause significant impairment in daily activities and quality of life, making it difficult for individuals to perform routine tasks like dressing, bathing, or walking. Treatment options may include physical therapy, splinting, casting, medications, surgery, or a combination of these approaches, depending on the severity and underlying cause of the contracture.
Lipodystrophy is a medical condition characterized by abnormal distribution or absence of fat (adipose tissue) in the body. It can lead to metabolic complications such as insulin resistance, diabetes mellitus, high levels of fats in the blood (dyslipidemia), and liver disease. There are different types of lipodystrophy, including congenital generalized lipodystrophy, acquired generalized lipodystrophy, and partial lipodystrophy, which can affect different parts of the body and have varying symptoms and causes.
Maxillofacial abnormalities, also known as craniofacial anomalies, refer to a broad range of structural and functional disorders that affect the development of the skull, face, jaws, and related soft tissues. These abnormalities can result from genetic factors, environmental influences, or a combination of both. They can vary in severity, from minor cosmetic issues to significant impairments of vital functions such as breathing, speaking, and eating.
Examples of maxillofacial abnormalities include cleft lip and palate, craniosynostosis (premature fusion of the skull bones), hemifacial microsomia (underdevelopment of one side of the face), and various other congenital anomalies. These conditions may require multidisciplinary treatment involving surgeons, orthodontists, speech therapists, and other healthcare professionals to address both functional and aesthetic concerns.
Metalloendopeptidases are a type of enzymes that cleave peptide bonds in proteins, specifically at interior positions within the polypeptide chain. They require metal ions as cofactors for their catalytic activity, typically zinc (Zn2+) or cobalt (Co2+). These enzymes play important roles in various biological processes such as protein degradation, processing, and signaling. Examples of metalloendopeptidases include thermolysin, matrix metalloproteinases (MMPs), and neutrophil elastase.
Cellular aging, also known as cellular senescence, is a natural process that occurs as cells divide and grow older. Over time, cells accumulate damage to their DNA, proteins, and lipids due to various factors such as genetic mutations, oxidative stress, and epigenetic changes. This damage can impair the cell's ability to function properly and can lead to changes associated with aging, such as decreased tissue repair and regeneration, increased inflammation, and increased risk of age-related diseases.
Cellular aging is characterized by several features, including:
1. Shortened telomeres: Telomeres are the protective caps on the ends of chromosomes that shorten each time a cell divides. When telomeres become too short, the cell can no longer divide and becomes senescent or dies.
2. Epigenetic changes: Epigenetic modifications refer to chemical changes to DNA and histone proteins that affect gene expression without changing the underlying genetic code. As cells age, they accumulate epigenetic changes that can alter gene expression and contribute to cellular aging.
3. Oxidative stress: Reactive oxygen species (ROS) are byproducts of cellular metabolism that can damage DNA, proteins, and lipids. Accumulated ROS over time can lead to oxidative stress, which is associated with cellular aging.
4. Inflammation: Senescent cells produce pro-inflammatory cytokines, chemokines, and matrix metalloproteinases that contribute to a low-grade inflammation known as inflammaging. This chronic inflammation can lead to tissue damage and increase the risk of age-related diseases.
5. Genomic instability: DNA damage accumulates with age, leading to genomic instability and an increased risk of mutations and cancer.
Understanding cellular aging is crucial for developing interventions that can delay or prevent age-related diseases and improve healthy lifespan.
Somatotrophs are a type of cell found within the anterior pituitary gland, a small endocrine gland located at the base of the brain. These cells are responsible for producing and secreting the hormone known as somatotropin or growth hormone (GH). This hormone plays a crucial role in regulating growth, cell reproduction, and regeneration. It also helps to regulate the body's metabolism and maintain proper body composition by promoting the breakdown of fats and the synthesis of proteins. Disorders related to somatotrophs can lead to conditions such as gigantism or dwarfism, depending on whether there is an overproduction or underproduction of growth hormone.
Cockayne Syndrome is a rare genetic disorder that affects the body's ability to repair DNA. It is characterized by progressive growth failure, neurological abnormalities, and premature aging. The syndrome is typically diagnosed in childhood and is often associated with photosensitivity, meaning that affected individuals are unusually sensitive to sunlight.
Cockayne Syndrome is caused by mutations in either the ERCC6 or ERCC8 gene, which are involved in the repair of damaged DNA. There are two types of Cockayne Syndrome: Type I and Type II. Type I is the more common form and is characterized by normal development during the first year of life followed by progressive growth failure, neurological abnormalities, and premature aging. Type II is a more severe form that is apparent at birth or within the first few months of life and is associated with severe developmental delays, intellectual disability, and early death.
There is no cure for Cockayne Syndrome, and treatment is focused on managing symptoms and improving quality of life. This may include physical therapy, occupational therapy, speech therapy, and special education services. In some cases, medications may be used to treat specific symptoms such as seizures or gastrointestinal problems.
The cell nucleus is a membrane-bound organelle found in the eukaryotic cells (cells with a true nucleus). It contains most of the cell's genetic material, organized as DNA molecules in complex with proteins, RNA molecules, and histones to form chromosomes.
The primary function of the cell nucleus is to regulate and control the activities of the cell, including growth, metabolism, protein synthesis, and reproduction. It also plays a crucial role in the process of mitosis (cell division) by separating and protecting the genetic material during this process. The nuclear membrane, or nuclear envelope, surrounding the nucleus is composed of two lipid bilayers with numerous pores that allow for the selective transport of molecules between the nucleoplasm (nucleus interior) and the cytoplasm (cell exterior).
The cell nucleus is a vital structure in eukaryotic cells, and its dysfunction can lead to various diseases, including cancer and genetic disorders.
Aging is a complex, progressive and inevitable process of bodily changes over time, characterized by the accumulation of cellular damage and degenerative changes that eventually lead to increased vulnerability to disease and death. It involves various biological, genetic, environmental, and lifestyle factors that contribute to the decline in physical and mental functions. The medical field studies aging through the discipline of gerontology, which aims to understand the underlying mechanisms of aging and develop interventions to promote healthy aging and extend the human healthspan.
Developmental bone diseases are a group of medical conditions that affect the growth and development of bones. These diseases are present at birth or develop during childhood and adolescence, when bones are growing rapidly. They can result from genetic mutations, hormonal imbalances, or environmental factors such as poor nutrition.
Some examples of developmental bone diseases include:
1. Osteogenesis imperfecta (OI): Also known as brittle bone disease, OI is a genetic disorder that affects the body's production of collagen, a protein necessary for healthy bones. People with OI have fragile bones that break easily and may also experience other symptoms such as blue sclerae (whites of the eyes), hearing loss, and joint laxity.
2. Achondroplasia: This is the most common form of dwarfism, caused by a genetic mutation that affects bone growth. People with achondroplasia have short limbs and a large head relative to their body size.
3. Rickets: A condition caused by vitamin D deficiency or an inability to absorb or use vitamin D properly. This leads to weak, soft bones that can bow or bend easily, particularly in children.
4. Fibrous dysplasia: A rare bone disorder where normal bone is replaced with fibrous tissue, leading to weakened bones and deformities.
5. Scoliosis: An abnormal curvature of the spine that can develop during childhood or adolescence. While not strictly a developmental bone disease, scoliosis can be caused by various underlying conditions such as cerebral palsy, muscular dystrophy, or spina bifida.
Treatment for developmental bone diseases varies depending on the specific condition and its severity. Treatment may include medication, physical therapy, bracing, or surgery to correct deformities and improve function. Regular follow-up with a healthcare provider is essential to monitor growth, manage symptoms, and prevent complications.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
Alkyl and aryl transferases are a group of enzymes that catalyze the transfer of alkyl or aryl groups from one molecule to another. These enzymes play a role in various biological processes, including the metabolism of drugs and other xenobiotics, as well as the biosynthesis of certain natural compounds.
Alkyl transferases typically catalyze the transfer of methyl or ethyl groups, while aryl transferases transfer larger aromatic rings. These enzymes often use cofactors such as S-adenosylmethionine (SAM) or acetyl-CoA to donate the alkyl or aryl group to a recipient molecule.
Examples of alkyl and aryl transferases include:
1. Methyltransferases: enzymes that transfer methyl groups from SAM to various acceptor molecules, such as DNA, RNA, proteins, and small molecules.
2. Histone methyltransferases: enzymes that methylate specific residues on histone proteins, which can affect chromatin structure and gene expression.
3. N-acyltransferases: enzymes that transfer acetyl or other acyl groups to amino groups in proteins or small molecules.
4. O-acyltransferases: enzymes that transfer acyl groups to hydroxyl groups in lipids, steroids, and other molecules.
5. Arylsulfatases: enzymes that remove sulfate groups from aromatic rings, releasing an alcohol and sulfate.
6. Glutathione S-transferases (GSTs): enzymes that transfer the tripeptide glutathione to electrophilic centers in xenobiotics and endogenous compounds, facilitating their detoxification and excretion.
Progeria, also known as Hutchinson-Gilford Progeria Syndrome (HGPS), is a rare and fatal genetic condition characterized by the rapid aging of children. The term "progeria" comes from the Greek words "pro," meaning prematurely, and "gereas," meaning old age.
Individuals with progeria typically appear normal at birth but begin to display signs of accelerated aging within the first two years of life. These symptoms can include growth failure, loss of body fat and hair, aged-looking skin, joint stiffness, hip dislocation, and cardiovascular disease. The most common cause of death in progeria patients is heart attack or stroke due to widespread atherosclerosis (the hardening and narrowing of the arteries).
Progeria is caused by a mutation in the LMNA gene, which provides instructions for making a protein called lamin A. This protein is essential for the structure and function of the nuclear envelope, the membrane that surrounds the cell's nucleus. The mutation leads to the production of an abnormal form of lamin A called progerin, which accumulates in cells throughout the body, causing premature aging.
There is currently no cure for progeria, and treatment is focused on managing symptoms and complications. Researchers are actively studying potential treatments that could slow or reverse the effects of the disease.
Heterochromatin is a type of chromatin (the complex of DNA, RNA, and proteins that make up chromosomes) that is characterized by its tightly packed structure and reduced genetic activity. It is often densely stained with certain dyes due to its high concentration of histone proteins and other chromatin-associated proteins. Heterochromatin can be further divided into two subtypes: constitutive heterochromatin, which is consistently highly condensed and transcriptionally inactive throughout the cell cycle, and facultative heterochromatin, which can switch between a condensed, inactive state and a more relaxed, active state depending on the needs of the cell. Heterochromatin plays important roles in maintaining the stability and integrity of the genome by preventing the transcription of repetitive DNA sequences and protecting against the spread of transposable elements.
Longevity, in a medical context, refers to the condition of living for a long period of time. It is often used to describe individuals who have reached a advanced age, such as 85 years or older, and is sometimes associated with the study of aging and factors that contribute to a longer lifespan.
It's important to note that longevity can be influenced by various genetic and environmental factors, including family history, lifestyle choices, and access to quality healthcare. Some researchers are also studying the potential impact of certain medical interventions, such as stem cell therapies and caloric restriction, on lifespan and healthy aging.
"Cells, cultured" is a medical term that refers to cells that have been removed from an organism and grown in controlled laboratory conditions outside of the body. This process is called cell culture and it allows scientists to study cells in a more controlled and accessible environment than they would have inside the body. Cultured cells can be derived from a variety of sources, including tissues, organs, or fluids from humans, animals, or cell lines that have been previously established in the laboratory.
Cell culture involves several steps, including isolation of the cells from the tissue, purification and characterization of the cells, and maintenance of the cells in appropriate growth conditions. The cells are typically grown in specialized media that contain nutrients, growth factors, and other components necessary for their survival and proliferation. Cultured cells can be used for a variety of purposes, including basic research, drug development and testing, and production of biological products such as vaccines and gene therapies.
It is important to note that cultured cells may behave differently than they do in the body, and results obtained from cell culture studies may not always translate directly to human physiology or disease. Therefore, it is essential to validate findings from cell culture experiments using additional models and ultimately in clinical trials involving human subjects.
Membrane proteins are a type of protein that are embedded in the lipid bilayer of biological membranes, such as the plasma membrane of cells or the inner membrane of mitochondria. These proteins play crucial roles in various cellular processes, including:
1. Cell-cell recognition and signaling
2. Transport of molecules across the membrane (selective permeability)
3. Enzymatic reactions at the membrane surface
4. Energy transduction and conversion
5. Mechanosensation and signal transduction
Membrane proteins can be classified into two main categories: integral membrane proteins, which are permanently associated with the lipid bilayer, and peripheral membrane proteins, which are temporarily or loosely attached to the membrane surface. Integral membrane proteins can further be divided into three subcategories based on their topology:
1. Transmembrane proteins, which span the entire width of the lipid bilayer with one or more alpha-helices or beta-barrels.
2. Lipid-anchored proteins, which are covalently attached to lipids in the membrane via a glycosylphosphatidylinositol (GPI) anchor or other lipid modifications.
3. Monotopic proteins, which are partially embedded in the membrane and have one or more domains exposed to either side of the bilayer.
Membrane proteins are essential for maintaining cellular homeostasis and are targets for various therapeutic interventions, including drug development and gene therapy. However, their structural complexity and hydrophobicity make them challenging to study using traditional biochemical methods, requiring specialized techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single-particle cryo-electron microscopy (cryo-EM).
In medical terms, the skin is the largest organ of the human body. It consists of two main layers: the epidermis (outer layer) and dermis (inner layer), as well as accessory structures like hair follicles, sweat glands, and oil glands. The skin plays a crucial role in protecting us from external factors such as bacteria, viruses, and environmental hazards, while also regulating body temperature and enabling the sense of touch.
Animal disease models are specialized animals, typically rodents such as mice or rats, that have been genetically engineered or exposed to certain conditions to develop symptoms and physiological changes similar to those seen in human diseases. These models are used in medical research to study the pathophysiology of diseases, identify potential therapeutic targets, test drug efficacy and safety, and understand disease mechanisms.
The genetic modifications can include knockout or knock-in mutations, transgenic expression of specific genes, or RNA interference techniques. The animals may also be exposed to environmental factors such as chemicals, radiation, or infectious agents to induce the disease state.
Examples of animal disease models include:
1. Mouse models of cancer: Genetically engineered mice that develop various types of tumors, allowing researchers to study cancer initiation, progression, and metastasis.
2. Alzheimer's disease models: Transgenic mice expressing mutant human genes associated with Alzheimer's disease, which exhibit amyloid plaque formation and cognitive decline.
3. Diabetes models: Obese and diabetic mouse strains like the NOD (non-obese diabetic) or db/db mice, used to study the development of type 1 and type 2 diabetes, respectively.
4. Cardiovascular disease models: Atherosclerosis-prone mice, such as ApoE-deficient or LDLR-deficient mice, that develop plaque buildup in their arteries when fed a high-fat diet.
5. Inflammatory bowel disease models: Mice with genetic mutations affecting intestinal barrier function and immune response, such as IL-10 knockout or SAMP1/YitFc mice, which develop colitis.
Animal disease models are essential tools in preclinical research, but it is important to recognize their limitations. Differences between species can affect the translatability of results from animal studies to human patients. Therefore, researchers must carefully consider the choice of model and interpret findings cautiously when applying them to human diseases.
Enzyme inhibitors are substances that bind to an enzyme and decrease its activity, preventing it from catalyzing a chemical reaction in the body. They can work by several mechanisms, including blocking the active site where the substrate binds, or binding to another site on the enzyme to change its shape and prevent substrate binding. Enzyme inhibitors are often used as drugs to treat various medical conditions, such as high blood pressure, abnormal heart rhythms, and bacterial infections. They can also be found naturally in some foods and plants, and can be used in research to understand enzyme function and regulation.
DNA repair is the process by which cells identify and correct damage to the DNA molecules that encode their genome. DNA can be damaged by a variety of internal and external factors, such as radiation, chemicals, and metabolic byproducts. If left unrepaired, this damage can lead to mutations, which may in turn lead to cancer and other diseases.
There are several different mechanisms for repairing DNA damage, including:
1. Base excision repair (BER): This process repairs damage to a single base in the DNA molecule. An enzyme called a glycosylase removes the damaged base, leaving a gap that is then filled in by other enzymes.
2. Nucleotide excision repair (NER): This process repairs more severe damage, such as bulky adducts or crosslinks between the two strands of the DNA molecule. An enzyme cuts out a section of the damaged DNA, and the gap is then filled in by other enzymes.
3. Mismatch repair (MMR): This process repairs errors that occur during DNA replication, such as mismatched bases or small insertions or deletions. Specialized enzymes recognize the error and remove a section of the newly synthesized strand, which is then replaced by new nucleotides.
4. Double-strand break repair (DSBR): This process repairs breaks in both strands of the DNA molecule. There are two main pathways for DSBR: non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ directly rejoins the broken ends, while HR uses a template from a sister chromatid to repair the break.
Overall, DNA repair is a crucial process that helps maintain genome stability and prevent the development of diseases caused by genetic mutations.
A heterozygote is an individual who has inherited two different alleles (versions) of a particular gene, one from each parent. This means that the individual's genotype for that gene contains both a dominant and a recessive allele. The dominant allele will be expressed phenotypically (outwardly visible), while the recessive allele may or may not have any effect on the individual's observable traits, depending on the specific gene and its function. Heterozygotes are often represented as 'Aa', where 'A' is the dominant allele and 'a' is the recessive allele.
Mitosis is a type of cell division in which the genetic material of a single cell, called the mother cell, is equally distributed into two identical daughter cells. It's a fundamental process that occurs in multicellular organisms for growth, maintenance, and repair, as well as in unicellular organisms for reproduction.
The process of mitosis can be broken down into several stages: prophase, prometaphase, metaphase, anaphase, and telophase. During prophase, the chromosomes condense and become visible, and the nuclear envelope breaks down. In prometaphase, the nuclear membrane is completely disassembled, and the mitotic spindle fibers attach to the chromosomes at their centromeres.
During metaphase, the chromosomes align at the metaphase plate, an imaginary line equidistant from the two spindle poles. In anaphase, sister chromatids are pulled apart by the spindle fibers and move toward opposite poles of the cell. Finally, in telophase, new nuclear envelopes form around each set of chromosomes, and the chromosomes decondense and become less visible.
Mitosis is followed by cytokinesis, a process that divides the cytoplasm of the mother cell into two separate daughter cells. The result of mitosis and cytokinesis is two genetically identical cells, each with the same number and kind of chromosomes as the original parent cell.
DNA damage refers to any alteration in the structure or composition of deoxyribonucleic acid (DNA), which is the genetic material present in cells. DNA damage can result from various internal and external factors, including environmental exposures such as ultraviolet radiation, tobacco smoke, and certain chemicals, as well as normal cellular processes such as replication and oxidative metabolism.
Examples of DNA damage include base modifications, base deletions or insertions, single-strand breaks, double-strand breaks, and crosslinks between the two strands of the DNA helix. These types of damage can lead to mutations, genomic instability, and chromosomal aberrations, which can contribute to the development of diseases such as cancer, neurodegenerative disorders, and aging-related conditions.
The body has several mechanisms for repairing DNA damage, including base excision repair, nucleotide excision repair, mismatch repair, and double-strand break repair. However, if the damage is too extensive or the repair mechanisms are impaired, the cell may undergo apoptosis (programmed cell death) to prevent the propagation of potentially harmful mutations.
"Bone" is the hard, dense connective tissue that makes up the skeleton of vertebrate animals. It provides support and protection for the body's internal organs, and serves as a attachment site for muscles, tendons, and ligaments. Bone is composed of cells called osteoblasts and osteoclasts, which are responsible for bone formation and resorption, respectively, and an extracellular matrix made up of collagen fibers and mineral crystals.
Bones can be classified into two main types: compact bone and spongy bone. Compact bone is dense and hard, and makes up the outer layer of all bones and the shafts of long bones. Spongy bone is less dense and contains large spaces, and makes up the ends of long bones and the interior of flat and irregular bones.
The human body has 206 bones in total. They can be further classified into five categories based on their shape: long bones, short bones, flat bones, irregular bones, and sesamoid bones.
Transgenic mice are genetically modified rodents that have incorporated foreign DNA (exogenous DNA) into their own genome. This is typically done through the use of recombinant DNA technology, where a specific gene or genetic sequence of interest is isolated and then introduced into the mouse embryo. The resulting transgenic mice can then express the protein encoded by the foreign gene, allowing researchers to study its function in a living organism.
The process of creating transgenic mice usually involves microinjecting the exogenous DNA into the pronucleus of a fertilized egg, which is then implanted into a surrogate mother. The offspring that result from this procedure are screened for the presence of the foreign DNA, and those that carry the desired genetic modification are used to establish a transgenic mouse line.
Transgenic mice have been widely used in biomedical research to model human diseases, study gene function, and test new therapies. They provide a valuable tool for understanding complex biological processes and developing new treatments for a variety of medical conditions.
A cell line is a culture of cells that are grown in a laboratory for use in research. These cells are usually taken from a single cell or group of cells, and they are able to divide and grow continuously in the lab. Cell lines can come from many different sources, including animals, plants, and humans. They are often used in scientific research to study cellular processes, disease mechanisms, and to test new drugs or treatments. Some common types of human cell lines include HeLa cells (which come from a cancer patient named Henrietta Lacks), HEK293 cells (which come from embryonic kidney cells), and HUVEC cells (which come from umbilical vein endothelial cells). It is important to note that cell lines are not the same as primary cells, which are cells that are taken directly from a living organism and have not been grown in the lab.
A "knockout" mouse is a genetically engineered mouse in which one or more genes have been deleted or "knocked out" using molecular biology techniques. This allows researchers to study the function of specific genes and their role in various biological processes, as well as potential associations with human diseases. The mice are generated by introducing targeted DNA modifications into embryonic stem cells, which are then used to create a live animal. Knockout mice have been widely used in biomedical research to investigate gene function, disease mechanisms, and potential therapeutic targets.
Protein binding, in the context of medical and biological sciences, refers to the interaction between a protein and another molecule (known as the ligand) that results in a stable complex. This process is often reversible and can be influenced by various factors such as pH, temperature, and concentration of the involved molecules.
In clinical chemistry, protein binding is particularly important when it comes to drugs, as many of them bind to proteins (especially albumin) in the bloodstream. The degree of protein binding can affect a drug's distribution, metabolism, and excretion, which in turn influence its therapeutic effectiveness and potential side effects.
Protein-bound drugs may be less available for interaction with their target tissues, as only the unbound or "free" fraction of the drug is active. Therefore, understanding protein binding can help optimize dosing regimens and minimize adverse reactions.
 Progeria - Wikipedia
Progeria - Wikipedia
 Progeria: MedlinePlus Medical Encyclopedia
Progeria: MedlinePlus Medical Encyclopedia
 Hutchinson-Gilford Progeria: Practice Essentials, Background, Pathophysiology
Hutchinson-Gilford Progeria: Practice Essentials, Background, Pathophysiology
 CRISPR doubles lifespan of mice with rapid ageing disease progeria | New Scientist
CRISPR doubles lifespan of mice with rapid ageing disease progeria | New Scientist
 Becoming pro-geriatric by understanding progeria
Becoming pro-geriatric by understanding progeria
 Progeria «DIS Magazine
Progeria «DIS Magazine
 Homepage | The Progeria Research Foundation
Homepage | The Progeria Research Foundation
China | The Progeria Research Foundation
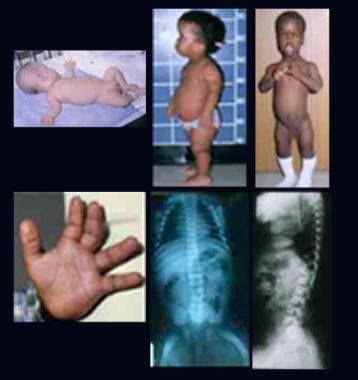
 Clinical and radiographic features of Hutchinson-Gilford progeria syndrome: A case report
Clinical and radiographic features of Hutchinson-Gilford progeria syndrome: A case report
 Hutchinson-Gilford Progeria Treatment Market Detailed Analysis, Growth Factors, Top Key Companies, Trends, Developments and...
Hutchinson-Gilford Progeria Treatment Market Detailed Analysis, Growth Factors, Top Key Companies, Trends, Developments and...
Progeria - LC Linked Data Service: Authorities and Vocabularies | Library of Congress
 Figures and data in Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria | eLife
Figures and data in Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria | eLife
 Progeria - Pediatrics - MSD Manual Professional Edition
Progeria - Pediatrics - MSD Manual Professional Edition
 Learn more about hutchinson gilford progeria syndrome | (e) Science News
Learn more about hutchinson gilford progeria syndrome | (e) Science News
 WebmedCentral.com :: The Integral Role Of The Dentist In Treating Individuals With Hutchinson-Gilford Progeria Syndrome
WebmedCentral.com :: The Integral Role Of The Dentist In Treating Individuals With Hutchinson-Gilford Progeria Syndrome
 Hutchinson-Gilford Progeria Syndrome (HGPS) - News, Articles, Whitepapers - Drug Target Review
Hutchinson-Gilford Progeria Syndrome (HGPS) - News, Articles, Whitepapers - Drug Target Review
 Progeria Symptoms - Health Hearty
Progeria Symptoms - Health Hearty
 Progeria Archives - InnoHEALTH magazine
Progeria Archives - InnoHEALTH magazine
 Micrognathia: Causes, Treatments, and When to Seek Help
Micrognathia: Causes, Treatments, and When to Seek Help
 Progeria Archives - Rabbi Jason Miller
Progeria Archives - Rabbi Jason Miller
 Progeria Forum: the discussions on Carenity
Progeria Forum: the discussions on Carenity
 Progeria - Types | Symptoms | Causes | Complications | Diagnosis | Treatment
Progeria - Types | Symptoms | Causes | Complications | Diagnosis | Treatment
 Gastrointestinal Tract Hemorrhage due to Angiodysplasia in Hutchinson Gilfort Progeria Syndrome | Aktas | Journal of Medical...
Gastrointestinal Tract Hemorrhage due to Angiodysplasia in Hutchinson Gilfort Progeria Syndrome | Aktas | Journal of Medical...
 Progeria: Children with an Elderly Body
Progeria: Children with an Elderly Body
 Progeria
Progeria
Team Zach Attack - Progeria Research Foundation - KY Chapter: 2020
 Barbara Walters Reports on Progeria- Saturday, January 19 - Global Genes
Barbara Walters Reports on Progeria- Saturday, January 19 - Global Genes
 Progeria trial offers little Enzo hope - Welcome to Team Enzo
Progeria trial offers little Enzo hope - Welcome to Team Enzo
 Why is it called Hutchinson-Gilford progeria syndrome? - Cutlergrp.com
Why is it called Hutchinson-Gilford progeria syndrome? - Cutlergrp.com
 Major Clue Could Lead to Treatment for Progeria - Face2Face Africa
Major Clue Could Lead to Treatment for Progeria - Face2Face Africa
HGPS16
- Hutchinson-Gilford progeria syndrome (HGPS) is an extremely rare hereditary disease that affects the skin, musculoskeletal system, and vasculature. (medscape.com)
- Patients with Hutchinson-Gilford progeria syndrome (HGPS) develop clinical features of accelerated aging, including accelerated atherosclerosis of the cerebral and coronary arteries. (medscape.com)
- I have never seen a case of Hutchinson-Gilford progeria syndrome (HGPS). (aad.org)
- Hutchinson-Gilford progeria syndrome (HGPS) is a rare dysmorphic syndrome characterized by several features of premature aging with clinical involvement of the skin, bones, and cardiovascular system. (wjgnet.com)
- Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare genetic disorder in which the individual displays a phenotypic expression similar to that of an aged individual. (webmedcentral.com)
- This disorder has since become known as Hutchinson-Gilford Progeria Syndrome (HGPS). (webmedcentral.com)
- Scientists have found that Hutchinson-Gilford Progeria Syndrome (HGPS) can be prevented with treatments targeting the cardiovascular system using a novel mouse model. (drugtargetreview.com)
- Researchers have uncovered new answers about why cells rapidly age in children with the rare and fatal disease, Hutchinson-Gilford Progeria Syndrome (HGPS). (drugtargetreview.com)
- First described by Jonathan Hutchinson in 1886, and then later described independently by Hastings Gilford in 1897, this genetic disorder is also known as Hutchinson-Gilford Progeria Syndrome (HGPS). (healthhearty.com)
- Hutchinson-Gilford progeria syndrome (HGPS) is the classic type of progeria caused by a mutation in the lamin A (LMNA) gene. (icliniq.com)
- Hutchinson-Gilford Progeria Syndrome (HGPS) - It is the classic type of progeria, with its onset in early childhood. (icliniq.com)
- Hutchinson Gilfort Progeria Syndrome (HGPS) is an aging disease which encounters in childhood and includes a higher risk for atherosclerosis, cerebrovascular event, stroke and coronary artery disease. (journalmc.org)
- Hutchinson-Gilfort Progeria Syndrome (HGPS) is an extremely rare genetic disorder that can be seen after birth, prematurity and a cause of aging. (journalmc.org)
- Among the different forms of progeria, the classical and most extensively studied type is the Hutchinson-Gilford progeria syndrome (HGPS), named after the two scientists (Jonathan Hutchinson in 1886 and Hastings Gilford in 1897) who independently delineated and described the syndrome. (cutlergrp.com)
- Aims Hutchinson-Gilford progeria syndrome (HGPS) is a pre-mature aging disorder caused by the mutation of the LMNA gene leading to an irreversibly farnesylated lamin A protein: progerin. (houstonmethodist.org)
- Singapore - Scientists from A*STAR's Institute of Medical Biology (IMB) have successfully established a comprehensive model of rare accelerated ageing syndrome Hutchinson-Gilford Progeria Syndrome (HGPS), thereby opening up the possibility of curing HGPS, with far-reaching implications on ageing and human health. (asiaresearchnews.com)
Syndrome46
- Progeria is a specific type of progeroid syndrome, also known as Hutchinson-Gilford syndrome. (wikipedia.org)
- citation needed] Before the late 20th century, research on progeria yielded very little information about the syndrome. (wikipedia.org)
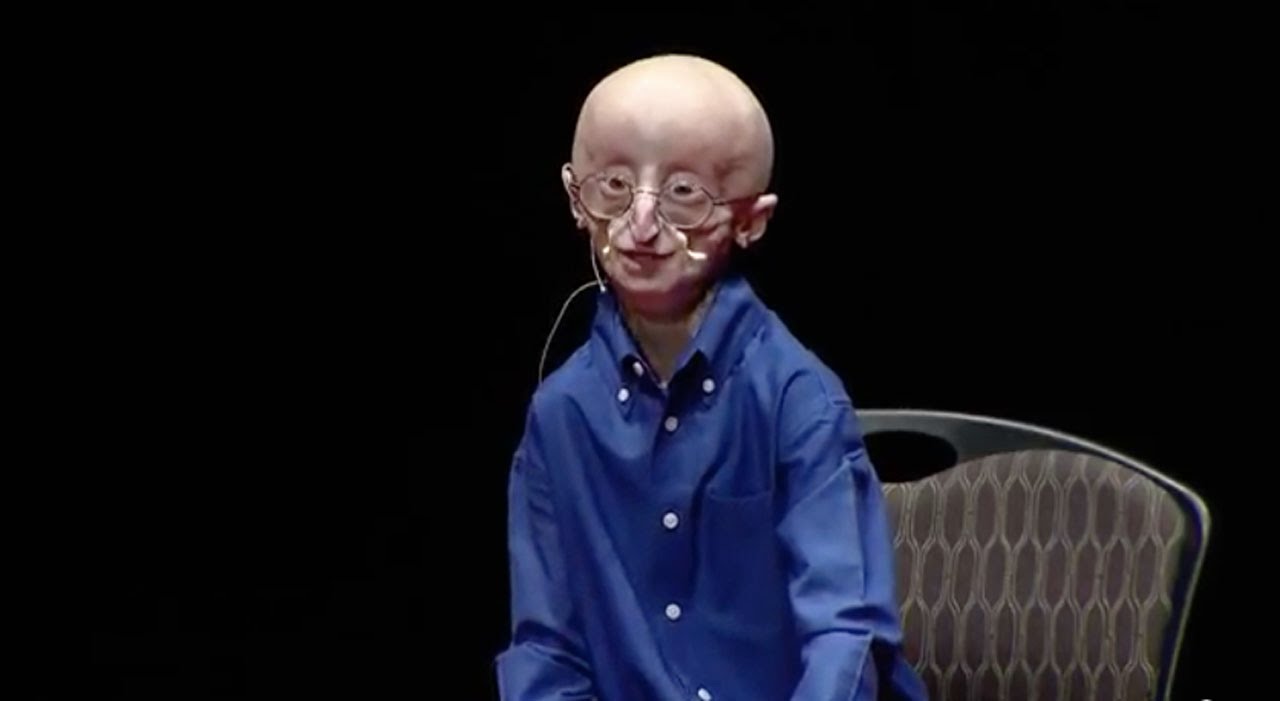
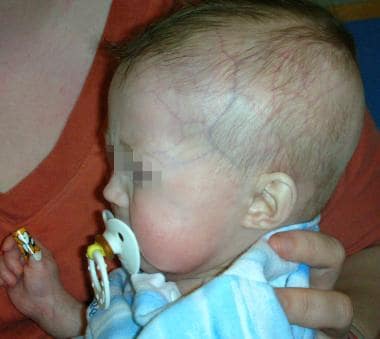
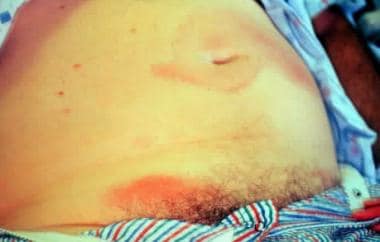
- See the image shown below depicting Hutchinson-Gilford progeria syndrome in an infant. (medscape.com)
- Early Hutchinson-Gilford progeria syndrome. (medscape.com)
- Note the alopecia, prominent scalp veins, and frontal bossing apparent in this 12-month-old infant with Hutchinson-Gilford progeria syndrome. (medscape.com)
- Hutchinson-Gilford progeria syndrome is a rare condition caused when a mutation, which probably took place in the testes or ovaries of a child's parents, results in a single DNA letter change in one of the two copies of the gene for the lamin A protein. (newscientist.com)
- A fifteen-year-old boy with Hutchinson-Gilford progeria syndrome. (aad.org)
- Hutchinson-Gilford progeria syndrome is a genetic condition characterized by the rapid, dramatic appearance of ageing in children. (medgadget.com)
- Individuals suffering from Hutchinson-Gilford progeria syndrome are known to experience severe hardening of the arteries (arteriosclerosis) since early childhood. (medgadget.com)
- In 2012, findings of the first clinical trial of the drug Lonafarnib, a farnesyltransferase inhibitor (FTI), offered a new hope for the treatment of children ailing from Hutchinson-Gilford progeria syndrome. (medgadget.com)
- In 2015, a team of scientists at the Agency for Science, Technology & Research (A*STAR) successfully established a model of Hutchinson-Gilford progeria syndrome market . (medgadget.com)
- Researchers are currently working on determining how genetic changes further lead to the significant characteristic features of Hutchinson-Gilford progeria syndrome. (medgadget.com)
- Furthermore, in March 2019, researchers at the Centro Nacional de Investigaciones Cardiovasculares (CNIC), together with Universidad de Oviedo researchers identified a new molecular mechanism involved in the premature development of atherosclerosis in mice with Hutchinson-Gilford progeria syndrome. (medgadget.com)
- Progeria is a rare syndrome of accelerated aging that manifests early in childhood and causes premature death. (msdmanuals.com)
- Scientists have identified a potential therapeutic target in the devastating genetic disease Hutchinson-Gilford progeria syndrome. (drugtargetreview.com)
- Werner Syndrome (Adult Progeria) - The symptoms usually start in teenagers, and such adults live up to 40 or 50 years of age. (icliniq.com)
- Wiedemann-Rautenstrauch Syndrome (Neonatal Progeroid Syndrome) - The type of progeria affects fetuses that are still in the womb. (icliniq.com)
- In fact, however, there a medical condition known as progeria (technical name = Hutchinson-Gilford Progeria Syndrome). (medfriendly.com)
- This disease is therefore also known as the Hutchinson-Gilford progeria syndrome. (symptoma.com)
- Enzo Cornejo enjoys playtime like any other toddler, but in August, just three weeks before his third birthday, his family was shocked to find out he suffers progeria syndrome. (teamenzoprogeria.com)
- Why is it called Hutchinson-Gilford progeria syndrome? (cutlergrp.com)
- What does Hutchinson-Gilford progeria syndrome do? (cutlergrp.com)
- Progeria (pro-JEER-e-uh), also known as Hutchinson-Gilford syndrome, is an extremely rare, progressive genetic disorder that causes children to age rapidly, starting in their first two years of life. (cutlergrp.com)
- Who discovered Hutchinson-Gilford progeria syndrome? (cutlergrp.com)
- The most severe form of the disease is Hutchinson-Gilford progeria syndrome, recognizing the efforts of Dr. Jonathan Hutchinson, who first described the disease in 1886, and Dr. Hastings Gilford who did the same in 1904. (cutlergrp.com)
- How is Hutchinson-Gilford Progeria Syndrome diagnosed? (cutlergrp.com)
- Werner syndrome is a rare progressive disorder that is characterized by the appearance of unusually accelerated aging (progeria). (cutlergrp.com)
- Williams was born with progeria, also known as Hutchinson-Gilford syndrome. (cutlergrp.com)
- When she was about three months old, Natalia Pallante, her mother, discovered that she had Hutchinson-Gilford progeria syndrome. (starsgab.com)
- Progeria syndrome is a progressive genetic disorder that causes children to age quickly. (familyhealth.today)
- It is called progeria syndrome, and this article aims to enlighten parents about this disease, its cause, symptoms and treatment option. (familyhealth.today)
- What is Progeria Syndrome? (familyhealth.today)
- Progeria syndrome, or progeria, is a rare progressive genetic disorder that causes a child's appearance to age at an accelerated rate. (familyhealth.today)
- Progeria has notable similarities with Wiedemann-Rautenstrauch syndrome and Werner syndrome. (familyhealth.today)
- How Does One Acquire Progeria Syndrome? (familyhealth.today)
- Progeria is a rare syndrome characterized by accelerated and exponential aging beginning from birth, but latent at birth. (amazonaws.com)
- Progeria is an extremely rare and highly fatal genetic premature-ageing syndrome in children, giving them a lifespan of around 14 years on average, and doesn't have a cure yet. (thewonk.in)
- Progeria, also known as Hutchinson-Gilford syndrome, is an extremely rare genetic condition that causes a child to age prematurely. (thewonk.in)
- Identifying the underlying mechanisms of this accelerated ageing syndrome progeria brings scientists one step closer to slowing down the ageing process. (asiaresearchnews.com)
- 3D-rendering of the nuclear lamina (red) and telomeres(green) in human cells expressing the mutated form of lamin A, causing the accelerated ageing syndrome progeria. (asiaresearchnews.com)
- Hutchinson-Gilford progeria syndrome (Progeria) is a rare segmental premature aging syndrome that affects 200-250 children worldwide at any one time. (rasopathiesnet.org)
- Hutchinson-Gilford progeria syndrome is a rare condition that results from just a single DNA letter mutation in one of the copies of the gene which encodes lamin A (a protein that stabilises and protects DNA). (gowinglife.com)
- Existing in humans as well as mice, Hutchinson-Gilford progeria syndrome (or progeria in its shorter form) is a severe degenerative disorder that manifests in symptoms of accelerated aging. (longevity.technology)
- This is the first time a gene editing therapy has been applied to treat progeria syndrome," says Izpisua Belmonte, a senior author of the paper [2]. (longevity.technology)
- Progeria can also refer to Hutchinson-Gilford syndrome , which is described as a lamin A gene defect and has onset early in life. (medscape.com)
- rare cancers in Werner syndrome (adult progeria). (who.int)
Atherosclerosis4
- Clinically, children with progeria develop atherosclerosis, arteriosclerosis of small vessels, and prominent adventitial fibrosis with increasing deposition of progerin within coronary arteries. (medscape.com)
- Usually, children with progeria die due to progressive atherosclerosis, which leads to heart attacks and heart failure. (healthhearty.com)
- Definition of progeria : a rare genetic disorder of childhood marked by slowed physical growth and characteristic signs (such as baldness, wrinkled skin, and atherosclerosis) of rapid aging with death usually occurring around puberty. (cutlergrp.com)
- Children with Progeria die of global, accelerated atherosclerosis at an average age of 13 years. (rasopathiesnet.org)
Jonathan Hutchinson1
- Jonathan Hutchinson and Gilford Progeria have been reported, for the first time in England in 1886. (journalmc.org)
Adult Progeria1
- Adult progeria is usually diagnosed on the basis of characteristic clinical features and typical concomitant diseases. (medscape.com)
Hutchinson-Gilford Progeria3
- Hutchinson-Gilford progeria is a rare condition known to affect at least one in around four million newborns across the world, as surveyed by the National Institutes of Health (NIH). (medgadget.com)
- For instance, in August 2019, researchers from the Houston Methodist Research Institute at the Texas Medical Center are focused on using RNA therapeutics-treatment that is focused on ribonucleic acids, a substance found in all living cells-to slow, and possibly reverse Hutchinson-Gilford Progeria. (medgadget.com)
- The research may help in designing the targeted probiotic treatments for age-related conditions, such as Hutchinson-Gilford Progeria in humans. (medgadget.com)
Cure for progeria6
- To discover treatments and the cure for Progeria and its aging-related disorders, including heart disease. (progeriaresearch.org)
- PRF is the only non-profit organization solely dedicated to finding treatments and the cure for Progeria, and is making phenomenal progress toward that goal. (progeriaresearch.org)
- As is evidenced by the film, Sam's parents weren't only passionate about finding a cure for Progeria for their own son's sake, but for all of the children throughout the world who age too quickly and end up dying as they reach their teenage years. (rabbijason.com)
- As of now, there is no cure for progeria. (icliniq.com)
- Unfortunately, there is no treatment of cure for progeria although attempts are made to reduce cardiovascular problems. (medfriendly.com)
- There is no known cure for progeria at this time. (familyhealth.today)
Diagnosis3
- Diagnosis of progeria is usually obvious by appearance but must be distinguished from segmental progerias (eg, acrogeria, metageria) and other causes of growth failure. (msdmanuals.com)
- Diagnosis of progeria is based on clinical examination of the child and can be confirmed with a genetic test. (medfriendly.com)
- A genetic test for LMNA mutations can confirm the diagnosis of progeria. (cutlergrp.com)
Progerin7
- People with progeria still have one healthy copy of the lamin A gene - the problem is the mutant progerin protein. (newscientist.com)
- Our research team has discovered a vital new way to measure progerin, the toxic protein that causes Progeria. (progeriaresearch.org)
- In children with progeria, there is an accelerated accumulation of one specific toxic protein called progerin. (face2faceafrica.com)
- This protein is called Progerin, hence the source of the name: Progeria. (amazonaws.com)
- The global expression of lamin A, and hence the aberrant protein produced in Progeria called progerin, results in a multisystem disease. (rasopathiesnet.org)
- Reasoning that any potential therapy for progeria they found would also be useful in fighting aging, the team set their sights on using gene therapy to reduce the progerin produced by unhealthy LMNA variants. (longevity.technology)
- We reasoned that progeria could be treated by CRISPR/Cas9-targeted disruption of both lamin A and progerin. (longevity.technology)
Mutation14
- A single gene mutation is responsible for progeria. (wikipedia.org)
- In 2003, the cause of progeria was discovered to be a point mutation in position 1824 of the LMNA gene, which replaces a cytosine with thymine. (wikipedia.org)
- Progeria affects many different organs in the body, and the team behind the work didn't expect that correcting the mutation in a relatively low proportion of cells - 10 to 60 per cent - would have such a big effect. (newscientist.com)
- A virus carrying the genes for the base editor was injected into the blood of 2-week-old mice - roughly equivalent to 5-year-old children, says Liu - with the progeria mutation. (newscientist.com)
- Progeria is caused by a sporadic mutation in the LMNA gene that codes for a protein (lamin A) that provides the molecular scaffolding of cell nuclei. (msdmanuals.com)
- Progeria is caused by the mutation of a single gene and occurs when a joint mutation occurs in position 1824 of the LMNA gene, wherein the component of DNA called cystosine is replaced by another pyrimidine base, thyamine. (healthhearty.com)
- Lamin A plays an important role in building the nuclear envelope, thus, when a mutation occurs in this gene, a mutant form of Lamin A protein is produced, which destabilizes the person's cells in the body, thereby causing progeria. (healthhearty.com)
- Progeria is a rare and progressive condition caused by a single genetic mutation. (icliniq.com)
- The cause of progeria itself is caused by a mutation (change) in the LMNA gene. (medfriendly.com)
- Progeria is due to a mutation of the lamin A (LMNA) gene. (familyhealth.today)
- Through this technique, scientists substitute a single DNA letter for another without damaging the actual structure of DNA, to study how changing this mutation might affect progeria-like symptoms in mice. (thewonk.in)
- Researchers found that the root cause of progeria in nearly all affected children is a single specific mutation in the LMNA gene structure of the DNA and in order to find a cure for the disease that needs to be fixed. (thewonk.in)
- In this study , researchers modified CRISPR -cas9 to correct the predominant single letter mutation responsible for progeria , as opposed to cutting the DNA and hence disabling the gene. (gowinglife.com)
- They then used a virus to deliver this editor to the cells of 2-week old mice with the human lamin A gene and progeria mutation. (gowinglife.com)
Symptoms10
- Children with progeria usually develop the first symptoms during their first few months of life. (wikipedia.org)
- At birth a child born with progeria will not exhibit any symptoms. (healthhearty.com)
- Besides these symptoms, people with progeria also experience all the other symptoms associated with the typical aging process. (healthhearty.com)
- What Are the Symptoms of Progeria? (icliniq.com)
- This instability weakens the cell structure, resulting in early and rapid aging leading to the symptoms of progeria. (icliniq.com)
- Symptoms of progeria begin to show effect within the first 2 years of life. (symptoma.com)
- In addition to the above mentioned symptoms, individuals with progeria also suffer from several health issues. (symptoma.com)
- A child with progeria usually appears normal at birth but starts to develop its signs and symptoms during the first year. (familyhealth.today)
- Before I became 5 months old, I had shown almost all the signs and symptoms of having Progeria. (amazonaws.com)
- What are the symptoms you will see in persons living with Progeria? (amazonaws.com)
Complications4
- People with progeria are treated to reduce complications resulting from different illnesses arising in their bodies. (healthhearty.com)
- What Are the Complications of Progeria? (icliniq.com)
- Children with progeria suffer from accelerated aging and most often succumb to complications associated with old age. (face2faceafrica.com)
- The usual cause of death in children affected by progeria is due to heart complications. (familyhealth.today)
Term progeria is derived2
- The term progeria is derived from the Greek word geras , meaning old age. (medscape.com)
- The term progeria is derived from a Greek term meaning "before old age. (medscape.com)
LMNA1
- Genetic testing can detect changes in the gene ( LMNA ) that causes progeria. (medlineplus.gov)
Cured progeria in mice1
- According to a recent study, scientists claimed to have cured progeria in mice with the help of gene-editing technique. (thewonk.in)
Children28
- Progeria is a rare genetic condition that produces rapid aging in children. (medlineplus.gov)
- The average lifespan of children with progeria is 14 years. (newscientist.com)
- Progeria is an ultra-rare, fatal, "rapid-aging" disease that afflicts children who, without the FDA-approved treatment lonafarnib, die of heart disease at an average age of 14.5 years . (progeriaresearch.org)
- With this discovery, scientists also found that the long-term benefit of lonafarnib for children with Progeria is greater than we thought. (progeriaresearch.org)
- Your donation helps The Progeria Research Foundation treat children with Progeria today, and cure them in the future. (progeriaresearch.org)
- Although born without any unusual signs, these children begin to show slow growth and other progeria characteristics within their first year of life. (healthhearty.com)
- Progeria is an extremely rare, incurable genetic disorder that conduces to premature aging in children. (healthhearty.com)
- An Indian family has reported to have seven children, of which five are affected with progeria. (healthhearty.com)
- This is because children with progeria have a life span of not more than 12-13 years, thus, not living long enough to reproduce. (healthhearty.com)
- However, despite the difference in appearance, children with progeria are not mentally challenged. (healthhearty.com)
- Progeria-affected children live mostly up to the age of 13, however, there are some who have lived into their 20s and 30s. (healthhearty.com)
- Children suffering from progeria have often been ostracized, since people considered it to be a bad omen. (healthhearty.com)
- It mainly occurs in children who are born with certain genetic conditions, such as trisomy 13 and progeria . (healthline.com)
- In addition to a small jaw, children with progeria may also have a slow growth rate, hair loss, and a very narrow face. (healthline.com)
- At birth, children with progeria look normal, but within the first two years of their life, they start looking older than is normal for their age. (icliniq.com)
- 134 children across 46 countries are believed to have progeria. (icliniq.com)
- But if one of the children in the family has progeria, the chances of getting this disorder is about two to three percent in the next child. (icliniq.com)
- There are only 80 known children with progeria in the entire world. (medfriendly.com)
- Ninety percent of children with progeria die from one of these two causes. (medfriendly.com)
- Progeria is a rare genetic disorder wherein children age rapidly due to genetic defect. (symptoma.com)
- The Progeria Research Foundation appreciates every effort to raise awareness about Progeria and is pleased that Barbara Walters, ABC and the public recognizes the remarkable nature of these special children and Progeria's connection to the aging process in general. (globalgenes.org)
- Progeria , a rare disease that speeds up the aging process, is estimated to affect fewer than 250 children worldwide. (globalgenes.org)
- Children with progeria generally appear normal at birth. (cutlergrp.com)
- As newborns, children with progeria usually appear normal. (cutlergrp.com)
- Progeria is a genetic disorder that affects approximately 200 children every year. (face2faceafrica.com)
- Children with progeria have a misplaced letter or base pair in their genome, resulting in an inefficient waste removal system in the cell. (face2faceafrica.com)
- Progeria is an inherited condition that causes children to grow much more rapidly than usual and leads to hair loss and slowed growth, according to Mayo Clinic. (starsgab.com)
- Children with progeria appear healthy during birth, but as they grow and by the time, they reach the age of two, their ageing rapidly accelerates making them look much older than their original age. (thewonk.in)
Gene4
- CRISPR gene editing has been used to more than double the lifespan of mice engineered to have the premature ageing disease progeria , also greatly improving their health. (newscientist.com)
- He and his colleagues have now used a CRISPR base editor to correct the single-letter change that causes almost all cases of progeria, first in skin cells taken from a person with progeria and then in mice with a human version of the lamin A gene. (newscientist.com)
- In this recent approach of researchers to cure progeria, scientists used a technique called base editing, which was originally inspired by the gene-editing technology CRISPR, to edit out a single base or one of the four letters of the DNA. (thewonk.in)
- Pairwise plots or cluster analysis of activation fold of gene expression revealed closer relationships between fibroblasts from progeria or from old individual, but not between replicative senescence fibroblasts and either models. (northwestern.edu)
Mice2
- Researchers have used a novel DNA-editing method to convert one base pair to another, increasing the lifespan of mice with progeria. (drugtargetreview.com)
- Researchers, as per their latest study, have successfully used a DNA-editing technique to extend the lifespan of mice with the help of genetic variation associated with progeria, the disease that rapidly accelerates ageing and shortens life expectancy. (thewonk.in)
Disease progeria1
- A team from the I2BC publishes in FEBS Journal an exhaustive review of the various mutations affecting the structure of type A lamins, proteins that make up the envelope of the cell nucleus (nuclear lamina ), which lead to premature aging in people with the disease ( Progeria ). (cea.fr)
Cause of progeria1
- How bad is the cause of Progeria that we have to suffer so much, and die at very young ages? (amazonaws.com)
Cases of progeria1
- About 1 in 18 million babies are born with this disorder, with around 30-40 known cases of progeria across the globe. (healthhearty.com)
Signs2
- Babies with progeria typically don't show signs when they're born, but they start showing signs of the disorder within the first 2 years of their life. (healthline.com)
- As the child ages, signs of progeria become more advanced. (medfriendly.com)
Scientists1
- But as per the findings of this new study, scientists have found the first potential base-altering treatment to extend the lifespan of progeria patients. (thewonk.in)
Clinical2
- The clinical trial results demonstrated massive improvement in weight gain, increase in bone mineral density, reduced vascular stiffness, and improved sensorineural hearing in patients with progeria. (medgadget.com)
- Dr. Gordon will discuss the status of Progeria research and lonafarnib 's successful results as a treatment option in clinical trial studies. (globalgenes.org)
Prematurely1
- Progeria is a rare genetic disorder that makes a person age prematurely. (icliniq.com)
Genetically2
- In essence, in progeria, an elderly person is present in a child's body, both physically and genetically. (medfriendly.com)
- Although genetic illnesses are often hereditary, genetically acquired progeria is rare, as most cases are due to a chance occurrence. (familyhealth.today)
Treatment6
- There is no specific treatment for progeria. (medlineplus.gov)
- Therefore, if progeria is detected in the child, it is wise to ask the doctor about the treatment plan because of the limited information on this disease. (icliniq.com)
- What Is the Treatment for Progeria? (icliniq.com)
- Physical and occupational therapy are common treatment interventions for progeria. (familyhealth.today)
- Some aspects of progeria treatment can be addressed at home. (familyhealth.today)
- Novel treatment for progeria sheds light on genetic component of aging. (longevity.technology)
Research Foundation2
- The Progeria Research Foundation is proud to be a part of the Boston Athletic Association's 128th Bank of America Boston Marathon® Official Charity Program. (progeriaresearch.org)
- According to the Progeria Research Foundation, approximately 400 youngsters have progeria. (starsgab.com)
Affects1
- Progeria is an extremely rare condition, as it affects one child in 20 million live births. (icliniq.com)
Senescence1
- Using cDNA microarray containing 384 known genes, we compared the expression profiles of three different types of aging models: replicative senescence, fibroblasts from progeria or from elderly donor. (northwestern.edu)
Rare genetic disorder1
- Progeria is a rare genetic disorder that makes a 2-year-old look like he or she is aging too fast. (icliniq.com)
Severe1
- reported severe form of Progeria in 35th gestational weeks fetus of a women in 1990 [ 3 ]. (journalmc.org)
Humans2
- Think about this: Progeria causes a faster breakdown of cells, does that not imply a faster growth of humans? (amazonaws.com)
- This bodes well for the use of this approach to treat progeria in humans and genetic diseases more broadly. (gowinglife.com)
20201
- In November 2020, the US Food and Drug Administration approved the first-ever drug for treating progeria . (newscientist.com)
Patients2
- Patients born with progeria typically live to an age of mid-teens to early twenties. (wikipedia.org)
- Researchers have found that cells in patients with progeria look old and unhealthy. (face2faceafrica.com)
Life3
- Sam Berns, the Jewish teen who lived with Progeria passed away after so many learned his story from the HBO documentary "Life According to Sam. (rabbijason.com)
- The average life expectancy of those with Progeria is 13, many of whom pass away due to strokes or heart attacks. (globalgenes.org)
- What is the Life expectancy of Progeria? (amazonaws.com)





![How to Start a Presentation [+ Examples]](https://www.prowell-tech.com/wp-content/uploads/2023/09/how-to-start-presenting.webp.jpeg)