Tay-Sachs Disease
Hexosaminidase A
beta-N-Acetylhexosaminidases
Lipidoses
Hexosaminidase B
G(M2) Ganglioside
Sandhoff Disease
Gangliosidoses
Gangliosidoses, GM2
Gangliosidosis, GM1
Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. (1/158)
The severe neurodegenerative disorder, Tays-Sachs disease, is caused by a beta-hexosaminidase alpha-subunit deficiency which prevents the formation of lysosomal heterodimeric alpha-beta enzyme, hexosaminidase A (HexA). No treatment is available for this fatal disease; however, gene therapy could represent a therapeutic approach. We previously have constructed and characterized, in vitro, adenoviral and retroviral vectors coding for alpha- and beta-subunits of the human beta-hexosaminidases. Here, we have determined the in vivo strategy which leads to the highest HexA activity in the maximum number of tissues in hexA -deficient knock-out mice. We demonstrated that intravenous co-administration of adenoviral vectors coding for both alpha- and beta-subunits, resulting in preferential liver transduction, was essential to obtain the most successful results. Only the supply of both subunits allowed for HexA overexpression leading to massive secretion of the enzyme in serum, and full or partial enzymatic activity restoration in all peripheral tissues tested. The enzymatic correction was likely to be due to direct cellular transduction by adenoviral vectors and/or uptake of secreted HexA by different organs. These results confirmed that the liver was the preferential target organ to deliver a large amount of secreted proteins. In addition, the need to overexpress both subunits of heterodimeric proteins in order to obtain a high level of secretion in animals defective in only one subunit is emphasized. The endogenous non-defective subunit is otherwise limiting. (+info)Sialidase-mediated depletion of GM2 ganglioside in Tay-Sachs neuroglia cells. (2/158)
Tay-Sachs disease is a severe, inherited disease of the nervous system caused by accumulation of the brain lipid GM2 ganglioside. Mouse models of Tay-Sachs disease have revealed a metabolic bypass of the genetic defect based on the more potent activity of the enzyme sialidase towards GM2. To determine whether increasing the level of sialidase would produce a similar effect in human Tay-Sachs cells, we introduced a human sialidase cDNA into neuroglia cells derived from a Tay-Sachs fetus and demonstrated a dramatic reduction in the accumulated GM2. This outcome confirmed the reversibility of GM2 accumulation and opens the way to pharmacological induction or activation of sialidase for the treatment of human Tay-Sachs disease. (+info)Structure of the GM2A gene: identification of an exon 2 nonsense mutation and a naturally occurring transcript with an in-frame deletion of exon 2. (3/158)
Deficiency of the GM2 activator protein, encoded by GM2A, results in the rare AB-variant form of GM2 gangliosidosis. Four mutations have been identified, but the human gene structure has been only partially characterized. We report a new patient from a Laotian deme whose cells are deficient in both GM2-activator mRNA and protein. However, reverse transcription (RT)-PCR detected some normal-sized cDNA and a smaller cDNA species, which was not seen in the RT-PCR products from normal controls. Sequencing revealed that, although the patient's normal-sized cDNA contained a single nonsense mutation in exon 2, his smaller cDNA was the result of an in-frame deletion of exon 2. Long PCR was used to amplify introns 1 and 2 from patient and normal genomic DNA, and no differences in size, in 5' and 3' end sequences, or in restriction-mapping patterns were observed. From these data we developed a set of four PCR primers that can be used to identify GM2A mutations. We use this procedure to demonstrate that the patient is likely homozygous for the nonsense mutation. Other reports have associated nonsense mutations with dramatically reduced levels of mRNA and with an increased level of skipping of the exon containing the mutation, thus reestablishing an open reading frame. However, a recent article has concluded that, in some cases, the latter observation is caused by an artifact of RT-PCR. In support of this conclusion, we demonstrate that, if the competing, normal-sized cDNA is removed from the initial RT-PCR products, from both patient and normal cells, by an exon 2-specific restriction digest; a second round of PCR produces similar amounts of exon 2-deleted cDNA. (+info)Biochemical consequences of mutations causing the GM2 gangliosidoses. (4/158)
The hydrolysis of GM2-ganglioside is unusual in its requirements for the correct synthesis, processing, and ultimate combination of three gene products. Whereas two of these proteins are the alpha- (HEXA gene) and beta- (HEXB) subunits of beta-hexosaminidase A, the third is a small glycolipid transport protein, the GM2 activator protein (GM2A), which acts as a substrate specific co-factor for the enzyme. A deficiency of any one of these proteins leads to storage of the ganglioside, primarily in the lysosomes of neuronal cells, and one of the three forms of GM2-gangliosidosis, Tay-Sachs disease, Sandhoff disease or the AB-variant form. Studies of the biochemical impact of naturally occurring mutations associated with the GM2 gangliosidoses on mRNA splicing and stability, and on the intracellular transport and stability of the affected protein have provided some general insights into these complex cellular mechanisms. However, such studies have revealed little in the way of structure-function information on the proteins. It appears that the detrimental effect of most mutations is not specifically on functional elements of the protein, but rather on the proteins' overall folding and/or intracellular transport. The few exceptions to this generalization are missense mutations at two codons in HEXA, causing the unique biochemical phenotype known as the B1-variant, and one codon in both the HEXB and GM2A genes. Biochemical characterization of these mutations has led to the localization of functional residues and/or domains within each of the encoded proteins. (+info)Isoenzymes of N-acetyl-beta-hexosaminidase. (5/158)
Biological significance, structure and posttranslational processing of N-acetyl-beta-hexosaminidase isoenzymes are described. Clinical application of N-acetyl-beta-hexosaminidase is also reviewed. (+info)Primer system for single cell detection of double mutation for Tay-Sachs disease. (6/158)
PURPOSE: Nearly 100% of infantile Tay-Sachs disease is produced by two mutations occurring in the alpha chain of the lysosomal enzyme beta-N-acetylhexosaminidase (HEXA) in the Ashkenazi Jewish population. Although others have described primer systems used to amplify both sites simultaneously, few discuss the allele dropout problems inherent in this test. Our goal was to construct a more robust test enabling stronger signal generation for single cell preimplantation genetic diagnosis and to investigate the occurrence of allele dropout. METHODS: New nested primers were designed to optimize detection of both major Tay-Sachs mutations. Four hundred fifty-seven single cells, including normal cells and those carrying mutations of either the 4bp insertion exon 11 or splice-site intron 12 defects, were used to screen a new primer system. RESULTS: Based on PCR amplified product analysis, total efficiency of amplification was 85.3%, (390/457). The allele dropout rate for the 4bp insertion mutation in exon 11 and splice-site mutation in intron 12 was 4.8% and 5.8%, respectively. CONCLUSIONS: Multiple mutation detection and analysis within the Tay-Sachs disease gene (HEXA) is possible using single cells for clinical preimplantation genetic diagnosis. Alternative PCR primers and conditions offer various methods for developing systems compatible to specific program requirements. (+info)Gangliosides as modulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. (7/158)
Pyramidal cells initiate the formation of dendritic arbors in a prolific burst of neurite outgrowth during early cortical development. Although morphologically mature pyramidal neurons do not normally sprout additional primary dendrites, the discovery of ectopic dendritogenesis in neuronal storage diseases has revealed that these cells do retain this ability under appropriate stimulation. The capacity for renewal of dendritogenesis has been found to exhibit a species gradient with human > cat, dog, sheep > mouse. A consistent metabolic feature of ectopic dendrite-bearing pyramidal neurons is a heightened intracellular expression of GM2 ganglioside. Elevated expression of this same glycosphingolipid has also been found to correlate with normal dendritogenesis. Immature neurons in developing cat and ferret cortex exhibit high levels of GM2 ganglioside immunoreactivity coincident with normal dendritic sprouting and a similar relationship has now been shown for human cortical development. Ultrastructural studies of all three species revealed GM2 localized to vesicles in a manner consistent with Golgi synthesis and exocytic trafficking to the somatic-dendritic plasmalemma. We propose that GM2 ganglioside functions in glycosphingolipid-enriched microdomains (lipid rafts) in the plasmalemma to promote dendritic initiation through modulation of specific membrane proteins and/or their associated second messenger cascades. (+info)Screening for genetic disorders among Jews: how should the Tay-Sachs screening program be continued? (8/158)
The screening program in Israel for Tay-Sachs disease has proven very successful, giving Jewish couples a choice not to have affected children. The technology of carrier detection is now possible in several other severe genetic diseases that are relatively frequent among Jews. Due to the current confusion, a policy is needed to determine how the TSD screening program should be continued in the Israeli Jewish population. We propose that such a screening program include only mutations agreed by consensus as causing a disease severe enough to warrant the possibility of therapeutic abortion. We also propose that general screening include only mutations that are relatively frequent, taking into account the carrier frequencies in the Israeli Jewish population. (+info)Yarrowia is a genus of fungi that belongs to the family of Dipodascaceae. It is a type of yeast that is often found in various environments, including plants, soil, and water. One species, Yarrowia lipolytica, has gained attention in biotechnology applications due to its ability to break down fats and oils, produce organic acids, and express heterologous proteins. It's also known to be an opportunistic pathogen in humans, causing rare but serious infections in individuals with weakened immune systems.
Tay-Sachs Disease is a rare, inherited autosomal recessive disorder that affects the nervous system's functioning. It results from the deficiency of an enzyme called hexosaminidase A (Hex-A), which is necessary for breaking down gangliosides, a type of fatty substance found in nerve cells. When Hex-A is absent or insufficient, gangliosides accumulate abnormally in the nerve cells, leading to their progressive destruction and severe neurological deterioration.
The classic infantile form of Tay-Sachs Disease manifests within the first six months of life with symptoms such as loss of motor skills, seizures, paralysis, dementia, blindness, and eventually death, usually by age four. Late-onset forms of the disease also exist, which may present in childhood or adulthood with milder symptoms.
Tay-Sachs Disease is more prevalent among individuals of Ashkenazi Jewish, French Canadian, and Cajun descent. Genetic counseling and prenatal testing are recommended for couples at risk of passing on the disease.
Hexosaminidase A is an enzyme that is responsible for breaking down certain complex molecules in the body, specifically gangliosides. This enzyme is composed of two subunits, alpha and beta, which are encoded by the genes HEXA and HEXB, respectively.
Deficiency or mutation in the HEXA gene can lead to a genetic disorder called Tay-Sachs disease, which is characterized by an accumulation of gangliosides in the nerve cells, leading to progressive neurological degeneration. The function of hexosaminidase A is to break down these gangliosides into simpler molecules that can be eliminated from the body. Without sufficient levels of this enzyme, the gangliosides build up and cause damage to the nervous system.
Beta-N-Acetylhexosaminidases are a group of enzymes that play a role in the breakdown and recycling of complex carbohydrates in the body. Specifically, they help to break down gangliosides, which are a type of molecule found in cell membranes.
There are several different isoforms of beta-N-Acetylhexosaminidases, including A, B, and S. These isoforms are formed by different combinations of subunits, which can affect their activity and substrate specificity.
Mutations in the genes that encode for these enzymes can lead to a variety of genetic disorders, including Tay-Sachs disease and Sandhoff disease. These conditions are characterized by an accumulation of gangliosides in the brain, which can cause progressive neurological deterioration and death.
Treatment for these conditions typically involves managing symptoms and providing supportive care, as there is currently no cure. Enzyme replacement therapy has been explored as a potential treatment option, but its effectiveness varies depending on the specific disorder and the age of the patient.
Lipidoses are a group of genetic disorders characterized by abnormal accumulation of lipids (fats or fat-like substances) in various tissues and cells of the body due to defects in lipid metabolism. These disorders include conditions such as Gaucher's disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, and Wolman disease, among others. The accumulation of lipids can lead to progressive damage in multiple organs, resulting in a range of symptoms and health complications. Early diagnosis and management are essential for improving the quality of life and prognosis of affected individuals.
Hexosaminidase B is a type of enzyme that is involved in the breakdown of complex lipids called gangliosides in the body. These enzymes are found in lysosomes, which are structures inside cells that break down and recycle various materials.
Hexosaminidase B specifically helps to break down a particular type of ganglioside called GM2 ganglioside, which is abundant in the nervous system. Mutations in the gene that provides instructions for making this enzyme can lead to a condition called Tay-Sachs disease, which is characterized by the accumulation of GM2 gangliosides in the nerve cells, leading to progressive neurological deterioration.
In summary, Hexosaminidase B is an essential enzyme for breaking down certain types of lipids in the body, and its deficiency can lead to serious health consequences.
Sandhoff disease is a rare inherited disorder that affects the nervous system. It's a type of GM2 gangliosidosis, which is a group of conditions characterized by the body's inability to break down certain fats (lipids) called gangliosides.
In Sandhoff disease, deficiencies in the enzymes hexosaminidase A and B lead to an accumulation of GM2 ganglioside in various cells, particularly in nerve cells of the brain. This accumulation results in progressive damage to the nervous system.
The symptoms of Sandhoff disease typically appear between 6 months and 2 years of age and can include developmental delay, seizures, an exaggerated startle response, muscle weakness, loss of motor skills, and vision and hearing loss. The condition is often fatal by around age 3. It's caused by mutations in the HEXB gene, and it's inherited in an autosomal recessive manner, meaning an individual must inherit two copies of the mutated gene (one from each parent) to develop the disease.
Gangliosidoses are a group of inherited metabolic disorders caused by the accumulation of certain complex lipids called gangliosides in the brain and nervous system. This buildup is due to a deficiency of specific enzymes needed to break down these substances. The three main types of gangliosidoses are:
1. Type 1 - Infantile Neurovisceral or Tay-Sachs Disease: Characterized by the absence of the enzyme hexosaminidase A, leading to severe neurological symptoms such as muscle weakness, blindness, and developmental delay in early infancy, with rapid progression and death usually occurring before age 4.
2. Type 2 - Juvenile or Subacute GM1 Gangliosidosis: Caused by a deficiency of the enzyme beta-galactosidase, resulting in progressive neurological symptoms such as motor and cognitive decline, beginning between ages 6 months and 2 years. Affected individuals may survive into adolescence or early adulthood.
3. Type 3 - Adult or Chronic GM1 Gangliosidosis: Characterized by a deficiency of beta-galactosidase, leading to milder neurological symptoms that appear in late childhood, adolescence, or even adulthood. The progression is slower compared to the other types, and life expectancy varies widely.
Gangliosidoses are autosomal recessive disorders, meaning an individual must inherit two copies of the defective gene (one from each parent) to develop the condition.
GM2 gangliosidoses are a group of inherited metabolic disorders caused by the accumulation of harmful amounts of GM2 gangliosides in the body's cells, particularly in the nerve cells of the brain. There are three main types of GM2 gangliosidoses: Tay-Sachs disease, Sandhoff disease, and AB variant of GM2 gangliosidosis. These conditions are characterized by progressive neurological degeneration, which can lead to severe physical and mental disabilities, and ultimately death in childhood or early adulthood.
The underlying cause of GM2 gangliosides is a deficiency in the enzyme hexosaminidase A (Tay-Sachs and AB variant) or both hexosaminidase A and B (Sandhoff disease), which are responsible for breaking down GM2 gangliosides. Without sufficient enzyme activity, GM2 gangliosides accumulate in the lysosomes of cells, leading to cell dysfunction and death.
Symptoms of GM2 gangliosidoses can vary depending on the specific type and severity of the disorder, but often include developmental delay, muscle weakness, loss of motor skills, seizures, blindness, and dementia. There is currently no cure for GM2 gangliosidoses, and treatment is focused on managing symptoms and improving quality of life.
GM1 gangliosidosis is a rare inherited lysosomal storage disorder caused by the deficiency of an enzyme called β-galactosidase. This enzyme is responsible for breaking down certain complex fats (gangliosides) in the body. When this enzyme is lacking or not working properly, these gangliosides accumulate in various cells, particularly in nerve cells of the brain, leading to progressive neurological deterioration.
The condition can present at different ages and with varying severity, depending on the amount of functional β-galactosidase enzyme activity. The three main types of GM1 gangliosidosis are:
1. Early infantile (type I): This is the most severe form, with symptoms appearing within the first few months of life. Infants may appear normal at birth but then develop rapidly progressing neurological problems such as developmental delay, muscle weakness, seizures, and cherry-red spots in the eyes. Life expectancy is typically less than 2 years.
2. Late infantile/juvenile (type II): Symptoms begin between ages 1 and 3 years or later in childhood. Affected individuals may have developmental delay, motor difficulties, muscle weakness, and cognitive decline. Some individuals with this form may also develop corneal clouding and bone abnormalities.
3. Adult/chronic (type III): This is the least severe form of GM1 gangliosidosis, with symptoms appearing in late childhood, adolescence, or adulthood. Symptoms can include neurological problems such as muscle weakness, tremors, and difficulties with coordination and speech.
Currently, there is no cure for GM1 gangliosidosis, and treatment is primarily supportive to manage symptoms and improve quality of life.
 Tay-Sachs disease
Tay-Sachs disease
History of Tay-Sachs disease
Prevention of Tay-Sachs disease
Societal and cultural aspects of Tay-Sachs disease
Howard Ronald Kaback
Hyperacusis
Pseudodeficiency alleles
Glycosphingolipid
Compound heterozygosity
Michael Kaback
Health among the Amish
Frameshift mutation
Enzyme
GM2 gangliosidoses
Jacob sheep
Hexosaminidase
Medical genetics of Jews
Ernest Beutler
GM2 (ganglioside)
Alder-Reilly anomaly
Human genetics
CHB HEX N-terminal domain
Simple Mendelian genetics in humans
Sio Gene Therapies
Sandhoff disease
Oligogenic inheritance
James Samuel Risien Russell
Carrier testing
Károly Schaffer
Sheldon Schuster
Tay-Sachs disease - Wikipedia
 Tay-Sachs Disease | MedlinePlus
Tay-Sachs Disease | MedlinePlus
 About Tay-Sachs Disease
About Tay-Sachs Disease
 Tay-Sachs disease | Sparrow
Tay-Sachs disease | Sparrow
 Tay-Sachs Disease: Genetics and More - 23andMe
Tay-Sachs Disease: Genetics and More - 23andMe
 Irish Americans warned about Tay-Sachs disease striking community | IrishCentral.com
Irish Americans warned about Tay-Sachs disease striking community | IrishCentral.com
Acción y Cura para Tay-Sachs - ACTAYS - Rare Disease Day 2024
 Tay-Sachs Disease
Summary Report | CureHunter
Tay-Sachs Disease
Summary Report | CureHunter
 WBUR reports on early results for gene therapy trial for two young children with Tay-Sachs disease
WBUR reports on early results for gene therapy trial for two young children with Tay-Sachs disease
 Novel Vector Design and Hexosaminidase Variant Enabling Self-Complementary Adeno-Associated Virus for the Treatment of Tay...
Novel Vector Design and Hexosaminidase Variant Enabling Self-Complementary Adeno-Associated Virus for the Treatment of Tay...
 GM2 Gangliosidoses: Introduction And Epidemiology, Tay-Sachs Disease - GM2 Gangliosidosis Type I, Type III, Chronic, And B1...
GM2 Gangliosidoses: Introduction And Epidemiology, Tay-Sachs Disease - GM2 Gangliosidosis Type I, Type III, Chronic, And B1...
RealChoice: New test may allow early, better treatment of Tay-Sachs and other metabolic diseases
 Tay-Sachs Disease and Sandhoff Disease - Pediatrics - MSD Manual Professional Edition
Tay-Sachs Disease and Sandhoff Disease - Pediatrics - MSD Manual Professional Edition
 Tay-Sachs And Other Lipid Storage Diseases
Tay-Sachs And Other Lipid Storage Diseases
Tay-Sachs disease
Results: Tay-Sachs Disease
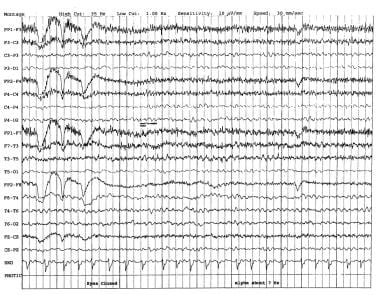
EEG in Dementia and Encephalopathy: Overview, Dementia, Vascular Dementia
 Tay-Sachs disease | Hereditary Ocular Diseases
Tay-Sachs disease | Hereditary Ocular Diseases
 Tay-Sachs Disease - Online Pathology Lecture | Lecturio
Tay-Sachs Disease - Online Pathology Lecture | Lecturio
 Carrier Screening for Genetic Conditions | ACOG
Carrier Screening for Genetic Conditions | ACOG
 Tay-Sachs Disease Solicitors | Medical Negligence Compensation | Australia
Tay-Sachs Disease Solicitors | Medical Negligence Compensation | Australia
 Tay-Sachs Disease - Symptoms, Signs & Treatment | Fitness Republic
Tay-Sachs Disease - Symptoms, Signs & Treatment | Fitness Republic
 Tay-Sachs Disease : Systemic Conditions : The Eyes Have It
Tay-Sachs Disease : Systemic Conditions : The Eyes Have It
10 Symptoms Of Tay-Sachs Disease - Healthy Info Daily
 Tay-Sachs disease in an Arab child.<...
Tay-Sachs disease in an Arab child.<...
 UCSF Tay-Sachs Disease Clinical Trials for 2022 - San Francisco Bay Area
UCSF Tay-Sachs Disease Clinical Trials for 2022 - San Francisco Bay Area
Biomedical research at UMass Chan Medical School
Tay-Sachs Disease Drugs Market Size, Share, Growth and Report, 2030 - America News Hour
 Tay-sachs Disease :: National Tay-Sachs & Allied Diseases Association of Delaware Valley (NTSAD-DV)
Tay-sachs Disease :: National Tay-Sachs & Allied Diseases Association of Delaware Valley (NTSAD-DV)
 Current page
Current page
National Tay-Sachs3
- The study is a joint project of Einstein and the National Tay-Sachs & Allied Diseases Association of Delaware Valley. (irishcentral.com)
- National Tay-Sachs & Allied Diseases Association (NTSAD) leads the worldwide fight to treat and cure Tay-Sachs, Canavan, GM1, and Sandhoff diseases by driving research, forging collaboration, and fostering community. (ntsad.org)
- She is also immediate past president of National Tay-Sachs and Allied Disease, the patient disease group. (seattlechildrens.org)
Forms of Tay-Sachs d3
- There are three forms of Tay-Sachs disease: infantile, juvenile and late onset/adult. (sparrow.org)
- There are milder forms of Tay-Sachs disease that do not develop until a child reaches early or young adulthood. (healthyinfodaily.com)
- This trial studies how the disease progresses in patients with GM1 and GM2 forms of Tay-Sachs Disease. (ucsf.edu)
Enzyme26
- Tay-Sachs disease is caused by a genetic mutation in the HEXA gene on chromosome 15, which codes a subunit of the hexosaminidase enzyme known as hexosaminidase A. It is inherited in an autosomal recessive manner. (wikipedia.org)
- medical citation needed] Tay-Sachs results from mutations in the HEXA gene on chromosome 15, which encodes the alpha-subunit of beta-N-acetylhexosaminidase A, a lysosomal enzyme. (wikipedia.org)
- Tay-Sachs is caused by the absence of a vital enzyme called hexosaminidase-A (Hex-A). Without Hex-A, a fatty substance, or lipid, called GM2 ganglioside accumulates abnormally in cells, especially in the nerve cells of the brain. (genome.gov)
- Tay-Sachs disease results from defects in a gene on chromosome 15 that codes for production of the enzyme Hex-A. We all have two copies of this gene. (genome.gov)
- Scientists are exploring enzyme replacement therapy to provide the Hex-A that is lacking in babies with Tay-Sachs. (genome.gov)
- The genetic change that causes Tay-Sachs disease results in a deficiency of the enzyme beta-hexosaminidase A. This enzyme is required to break down the fatty substance GM2 ganglioside. (sparrow.org)
- Severity and age of onset of the disease relates to how much enzyme is still produced. (sparrow.org)
- A drop of blood and an enzyme analysis can lead to early, precise diagnosis of Tay-Sachs and other devastating metabolic diseases. (blogspot.com)
- Diagnosis of Tay-Sachs disease is clinical and can be confirmed by DNA analysis and/or enzyme assay. (msdmanuals.com)
- Gene therapy or enzyme replacement therapy research may eventually lead to a cure or treatment to slow the progression of Tay-Sachs disease. (ahdubai.com)
- Tay-Sachs disease (TSD) is a mutation in the Hex-A gene that causes the body to have very low levels or none at all of the Hex-A enzyme. (fitnessrepublic.com)
- The babies with classic Tay-Sachs do not have the Hex-A enzyme, the reason why the progress of the disease is very quick. (fitnessrepublic.com)
- Just like with the juvenile form, the individual with late-onset Tay-Sachs disease produces little amount of the Hex-A enzyme. (fitnessrepublic.com)
- The cause of Tay Sachs disease is the lack of the enzyme hexosaminidase A, which is a protein found in the nervous system that breaks down gangliosides in nervous tissue. (medneg.com.au)
- There is no specific treatment for Tay Sachs disease, including no enzyme replacement yet available. (medneg.com.au)
- Children with Tay-Sachs disease lack a vital enzyme, hexosaminidase A (Hex-A). Hex-A is needed for the body to break down a fatty waste substance found in brain cells. (tay-sachs.org)
- A child who inherits two Tay-Sachs genes (one from each parent) produces no functional Hex-A enzyme and is certain to develop Tay-Sachs disease. (tay-sachs.org)
- (Encyclopedia) Tay-Sachs disease tāˈ-săksˈ, rare hereditary disease caused by a genetic mutation that leaves the body unable to produce an enzyme necessary for fat metabolism in nerve cells, producing central ne. (factmonster.com)
- Other rare deficiencies of the hex-A enzyme are sometimes included under the umbrella of Tay-Sachs disease. (girlstalkinsmack.com)
- Gaucher disease is caused by a deficiency of the enzyme glucocerebrosidase. (nih.gov)
- Only an individual with two genes for a defective enzyme will actually show the recessive trait, such as an inherited disease or condition, blue eyes, or a recessive peapod shape. (visionlearning.com)
- The concept revolves around using virus vectors, or "trucks", to get past the blood brain barrier, delivering Hex-A, the enzyme lacking in Tay-Sachs patients. (curetay-sachs.org)
- Children with Tay-Sachs lack an enzyme responsible for breaking down specific chemicals in the nerve cells of the brain. (npr.org)
- Classically, lysosomal storage diseases encompassed only enzyme deficiencies of the lysosomal hydrolases. (medscape.com)
- Enzyme replacement therapy (ERT) appears safe and effective for peripheral manifestations in patients with Gaucher disease types I and III, Fabry disease, mucopolysaccharidosis I (Hurler, Hurler-Scheie, and Scheie syndromes), mucopolysaccharidosis II (Hunter syndrome), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), and Pompe disease. (medscape.com)
- This has led to active clinical trials evaluating the safety and efficacy of intrathecal enzyme delivery in several lysosomal storage diseases (see www.ClinicalTrials.gov ). (medscape.com)
Family history of Tay-Sachs d2
- If you have a family history of Tay-Sachs disease or if you're a member of a high-risk group and plan to have children, health care providers strongly recommend genetic testing and genetic counseling. (sparrow.org)
- the fact that there is no family history of Tay-Sachs disease does not lower an individual's risk of being a carrier. (tay-sachs.org)
Infantile9
- The most common form is infantile Tay-Sachs disease, which becomes apparent around the age of three to six months of age, with the baby losing the ability to turn over, sit, or crawl. (wikipedia.org)
- Post-infantile Tay-Sachs was often misdiagnosed as another neurological disorder, such as Friedreich's ataxia. (wikipedia.org)
- Kenny's son Danny, now two years old, was diagnosed with infantile Tay-Sachs when he was just six months old. (irishcentral.com)
- In their infantile, acute forms, these diseases rapidly progress with mental and psychomotor deterioration resulting in death by approximately 4 years of age. (nih.gov)
- Generally, infantile cases have no Hex-A present the reason why the disease progresses very fast. (fitnessrepublic.com)
- For adolescents and adults, Tay-Sachs disease may rarely manifest and with less severity of symptoms than those in the infantile form. (fitnessrepublic.com)
- The Juvenile form and the Late-onset form or Chronic Tay-Sachs symptoms are rare and the severity of the symptoms tends to be milder as compared to the symptoms of the Infantile or Classic form. (fitnessrepublic.com)
- Tay Sachs disease comes in infantile forms, juvenile forms and adult forms, depending on when the symptoms begin to form. (medneg.com.au)
- Type 2 (acute infantile neuropathic Gaucher disease) typically begins within three months of birth. (nih.gov)
Cure for Tay-Sachs d3
- There is no cure for Tay-Sachs disease, but some treatments can help in managing symptoms. (ahdubai.com)
- Unfortunately, there is no treatment and no cure for Tay-Sachs disease at this time. (girlstalkinsmack.com)
- Your donation will be used to support research to find a cure for Tay-Sachs disease. (curetay-sachs.org)
Sandhoff Disease2
- Research into potential therapies for lysosomal storage diseases such as Tay-Sachs, Sandhoff disease and GM1 gangliosidosis at UMass Medical School and Auburn University has led to significant advances in the field. (umassmed.edu)
- Mutations in the α-subunit (encoded by HEXA) lead to Tay-Sachs disease (TSD), whereas mutations in the β-subunit (encoded by HEXB) lead to Sandhoff disease (SD). (nih.gov)
Late-onset9
- Less commonly, the disease may occur in later childhood or adulthood (juvenile or late-onset). (wikipedia.org)
- A rare form of this disease, known as Adult-Onset or Late-Onset Tay-Sachs disease, usually has its first symptoms during the 30s or 40s. (wikipedia.org)
- In contrast to the other forms, late-onset Tay-Sachs disease is usually not fatal as the effects can stop progressing. (wikipedia.org)
- Symptoms of late-onset Tay-Sachs - which typically begin to be seen in adolescence or early adulthood - include speech and swallowing difficulties, unsteadiness of gait, spasticity, cognitive decline, and psychiatric illness, particularly a schizophrenia-like psychosis. (wikipedia.org)
- Late-onset Tay-Sachs patients may become fully wheelchair-using. (wikipedia.org)
- A much rarer form of Tay-Sachs, Late-Onset Tay-Sachs disease, affects adults and causes neurological and intellectual impairment. (genome.gov)
- Rarely, some adults have a late-onset form of Tay-Sachs disease which is often less severe than forms that begin in childhood. (sparrow.org)
- Those inflicted with the Late-onset or Chronic form of Tay-Sachs show symptoms by the age of 10. (fitnessrepublic.com)
- Late onset Tay Sachs disease, when the disease occurs first in adulthood, is not common at all. (medneg.com.au)
Neurodegenerative4
- The fatal neurodegenerative disease, prevalent when marriage within community is common, affects one in 50 Irish. (irishcentral.com)
- The Irish American community is being warned about the risks and realities of Tay-Sachs disease, a fatal neurodegenerative disease for which an estimated one in 50 Irish and Irish Americans are carriers. (irishcentral.com)
- Tay-Sachs is a rapidly progressive and fatal pediatric neurodegenerative genetic disorder that has a median life expectancy of approximately three to four years. (umassmed.edu)
- Metachromatic leukodystrophy (MLD) is part of a larger group of inherited lysosomal storage diseases, some of which are progressive and neurodegenerative disorders (MLD included). (medscape.com)
HEXA7
- A blood test can be used to identify carriers of the HEXA gene change that causes Tay-Sachs disease. (sparrow.org)
- This test can examine the HEXA gene to identify whether there are changes that indicate Tay-Sachs disease. (sparrow.org)
- Tay-Sachs disease is caused by variants (differences) in the HEXA gene. (23andme.com)
- 23andMe tests for four genetic variants in the HEXA gene linked to Tay-Sachs disease and is most relevant for people of Ashkenazi Jewish and Cajun descent. (23andme.com)
- The Tay-Sachs Disease Carrier Status report is indicated for the detection of four of variants in the HEXA gene and is most relevant for people of Ashkenazi Jewish and Cajun descent. (23andme.com)
- Tay-Sachs disease is an autosomal recessive lysosomal storage disorder caused by genetic mutations in the hexosaminidase A (HEXA) gene, leading to progressive neurodegeneration. (lecturio.com)
- Therapeutic Candidate or Device Autologous hematopoietic stem cells transduced with a HexA/HexB expressing lentiviral vector Indication Tay-Sachs disease Therapeutic Mechanism The transplanted gene modified autologous hematopoietic stem cells will engraft in the bone marrow and start producing HexA/HexB expressing immune progeny. (ca.gov)
Mutations6
- Instead, you can choose to be screened for a wide range of disease mutations - more than 100 instead of just the one or two you may be 'at risk' for. (babycenter.com)
- From Genes to Genetic Diseases: What Kinds of Mutations Matter? (sciencebuddies.org)
- What kinds of mutations have to occur to cause a genetic disease? (sciencebuddies.org)
- Determine why some gene mutations cause genetic diseases, but others do not. (sciencebuddies.org)
- Sometimes only a single DNA mutation (change in the DNA sequence) can cause a person to have a devastating genetic disease , and researchers have been able to identify mutations responsible for causing thousands of different genetic diseases and conditions. (sciencebuddies.org)
- Age of onset and clinical manifestations may vary widely among patients with a given lysosomal storage disease, and significant phenotypic heterogeneity between family members carrying identical mutations has been reported. (medscape.com)
Carriers18
- Carriers of a single Tay-Sachs allele are typically normal. (wikipedia.org)
- Tay-Sachs disease is an autosomal recessive genetic disorder, meaning that when both parents are carriers, there is a 25% risk of giving birth to an affected child with each pregnancy. (wikipedia.org)
- Carriers of Tay-Sachs - people who have one copy of the inactive gene along with one copy of the active gene - are healthy. (genome.gov)
- If both parents are carriers and their child inherits the defective Hex-A gene from each of them, the child will have Tay-Sachs disease. (genome.gov)
- When both parents are carriers of the defective Tay-Sachs gene, each child has a 25 percent chance of having Tay-Sachs disease and a 50 percent chance of being a carrier. (genome.gov)
- Tay-Sachs has a 25% chance of being passed on to children when both parents are carriers of an altered gene. (irishcentral.com)
- Neither of them had any idea they could be carriers of Tay-Sachs. (irishcentral.com)
- A defective gene on chromosome 15 is the cause of the disease and it is a chromosomal recessive gene so that both parents need to be carriers of the disease in order to cause it to happen. (medneg.com.au)
- Half of their children will be carriers of the disease and twenty five percent will be completely free of the abnormal gene. (medneg.com.au)
- Carriers and those free of Tay Sachs disease will have no symptoms and will be allowed to continue their growth in utero. (medneg.com.au)
- When both parents are carriers, there is a 1 in 4 (25%) chance, with every pregnancy, of having a child with Tay-Sachs disease. (tay-sachs.org)
- Tay-Sachs carriers are found most frequently among families of eastern European Jewish descent (Ashkenazi Jews). (tay-sachs.org)
- The Tay-Sachs blood test, referred to as carrier screening, identifies Tay-Sachs carriers and non-carriers. (tay-sachs.org)
- Couples who are both carriers of the same disease will want to explore their many options for a healthy family. (tay-sachs.org)
- Carriers don't usually have any symptoms of the disease. (babycenter.com)
- If both you and your partner are carriers of a disorder like cystic fibrosis, sickle cell disease, or Tay-Sachs disease, your child will have a 1 in 4 chance of inheriting one defective gene from each of you and being born with the disease. (babycenter.com)
- While Tay-Sachs was once thought to be a disease linked to Ashkenazi Jews, new research shows that this disease knows no ethnicity and in fact, 1 in 50 Irish Americans are carriers of Tay-Sachs, with further links to Creole and French Canadian populations. (curetay-sachs.org)
- Examples cited include prenatal screening for congenital malformations and chromosomal abnormalities, neonatal screening for phenylketonuria, hypothyroidism and sickle-cell disease, and population screening for carriers of inherited diseases such as the haemoglobin disorders and Tay-Sachs disease. (who.int)
Bernard Sachs3
- and American neurologist Bernard Sachs, who described in 1887 the cellular changes and noted an increased rate of disease in Ashkenazi Jews. (wikipedia.org)
- Then, it was also named after Bernard Sachs, an American neurologist from New York in 1896, who did a continuous study and after several years provided the first description of the cellular changes, that is the extreme swelling of neurons in patients with Tay-Sachs disease. (fitnessrepublic.com)
- It was named after Warren Tay and Bernard Sachs who were the first to discover this genetic disorder by a red spot on the retina of the eye and changes in the cellular structure in 1881. (healthyinfodaily.com)
Hexosaminidase1
- Babies born with Tay-Sachs disease lack a protein called hexosaminidase A, or hex-A. When hex-A isn't present, substances build up and gradually destroy brain and nerve cells, until the central nervous system stops working. (girlstalkinsmack.com)
Carrier16
- A child who inherits one inactive gene is a Tay-Sachs carrier like the parent. (genome.gov)
- While anyone can be a carrier of Tay-Sachs, the incidence of the disease is significantly higher among people of eastern European (Ashkenazi) Jewish descent. (genome.gov)
- Approximately one in every 27 Jews in the United States is a carrier of the Tay-Sachs disease gene. (genome.gov)
- For people of Ashkenazi Jewish ancestry, about 1 in 30 individuals is a carrier for Tay-Sachs. (23andme.com)
- 23andMe can tell you whether you might be a carrier for Tay-Sachs disease. (23andme.com)
- 23andMe does not test for all possible genetic variants linked to Tay-Sachs disease, and individuals who have zero variants detected still have a chance of being a carrier for Tay-Sachs disease. (23andme.com)
- The Tay-Sachs Disease Carrier Status report* is included in the 23andMe Health + Ancestry Service. (23andme.com)
- These carrier reports are not intended to tell you anything about your risk for developing a disease in the future or anything about the health of your fetus, or your newborn child's risk of developing a particular disease later in life. (23andme.com)
- In order to gather more accurate data on Tay-Sachs among people of Irish descent, Weaver and Dr. Schneider are conducting the Irish Tay-Sachs Carrier Study, through which they offer free genetic testing to people with at least three grandparents of Irish descent. (irishcentral.com)
- It is an uncommon disease in the general population but, in Ashkenazi Jews, the carrier status is one in every 27 individuals, making it more likely to occur in that population. (medneg.com.au)
- A person with only one Tay-Sachs gene is perfectly healthy, but is a Tay-Sachs carrier. (tay-sachs.org)
- When only one parent is a carrier, there is no chance the child will have Tay-Sachs disease. (tay-sachs.org)
- There is a 2 in 4 (50%) chance, with every pregnancy, of having a child who is a Tay-Sachs carrier. (tay-sachs.org)
- How do you know if you're a Tay-Sachs carrier? (tay-sachs.org)
- a Tay-Sachs carrier has one normal gene for hex-A and one Tay-Sachs gene. (girlstalkinsmack.com)
- Risk factors include having a family member with the inherited disorder or who's a known carrier, or being part of an ethnic group at increased risk for the disease. (babycenter.com)
Fatal5
- Tay-Sachs disease is a fatal genetic disorder that results in progressive destruction of the nervous system. (genome.gov)
- The disease is progressively causing damages to the nerve cells which is unfortunately at all times fatal. (fitnessrepublic.com)
- Tay Sachs disease is a genetic disease of the nervous system that can be fatal. (medneg.com.au)
- Tay-Sachs disease is a severe genetic disease of the nervous system that is nearly always fatal, usually by three to four years of age. (jrank.org)
- Ronan's diagnosis that day was Tay-Sachs disease, a genetic and degenerative condition that is always fatal. (npr.org)
Symptoms13
- The disease is classified into several forms, which are differentiated based on the onset age of neurological symptoms. (wikipedia.org)
- Treatment involves managing the symptoms of the disease. (genome.gov)
- In the most common and severe form of Tay-Sachs disease, signs and symptoms start to show up at about 3 to 6 months of age. (sparrow.org)
- If you or your child has any of the signs or symptoms that may indicate Tay-Sachs disease, or if you have concerns about your child's development, schedule an appointment with your health care provider. (sparrow.org)
- To confirm that your child has Tay-Sachs disease, your health care provider will ask about symptoms and any family hereditary disorders, and also do a physical exam. (sparrow.org)
- Symptoms of Tay-Sachs disease typically develop during infancy. (23andme.com)
- Babies born with Tay-Sachs disease appear normal at birth, and symptoms of the disease do not appear until the infants are about four to six months of age when they begin to lose previously attained skills, such as sitting up or rolling over. (irishcentral.com)
- To confirm that your baby has Tay-Sachs disease, your doctor will ask you about the child's symptoms and any hereditary family disorders and will order a diagnostic blood test. (ahdubai.com)
- The symptoms appear the same as those of the Classic form but the development of the disease is much slower. (fitnessrepublic.com)
- In order to establish the diagnosis, the physician may gather information regarding the child's symptoms and correlate it to family history tackling the family hereditary disorders and may require a diagnostic blood analysis in order to confirm the presence of Tay-Sachs disease. (fitnessrepublic.com)
- Symptoms of Tay Sachs disease include being deaf and eventually being blind. (medneg.com.au)
- It is characterized by slowly progressive yet milder neurologic symptoms compared to type 2 Gaucher disease. (nih.gov)
- Accumulated data indicate that hematopoietic stem cell transplantation may be effective under optimal conditions in preventing the progression of central nervous system symptoms in neuronopathic forms of lysosomal storage diseases, including some of the mucopolysaccharidoses, oligosaccharidoses, sphingolipidoses, and lipidoses. (medscape.com)
Autosomal1
- Niemann-Pick disease is a group of autosomal recessive disorders caused by an accumulation of fat and cholesterol in cells of the liver, spleen, bone marrow, lungs, and, in some instances, brain. (nih.gov)
Lysosomal8
- Disorders in which intracellular material that cannot be metabolized is stored in lysosomes are called lysosomal storage diseases. (nih.gov)
- Lysosomal storage diseases describe a heterogeneous group of dozens of rare inherited disorders characterized by the accumulation of undigested or partially digested macromolecules, which ultimately results in cellular dysfunction and clinical abnormalities. (medscape.com)
- More recently, the concept of lysosomal storage disease has been expanded to include deficiencies or defects in proteins necessary for the normal post-translational modification of lysosomal enzymes (which themselves are often glycoproteins), activator proteins, or proteins important for proper intracellular trafficking between the lysosome and other intracellular compartments. (medscape.com)
- More than 50 lysosomal storage diseases have been described, some of which are discussed in this article. (medscape.com)
- Lysosomal storage diseases are generally classified by the accumulated substrate and include the sphingolipidoses, oligosaccharidoses, mucolipidoses, mucopolysaccharidoses (MPSs), lipoprotein storage disorders, lysosomal transport defects, neuronal ceroid lipofuscinoses and others. (medscape.com)
- Thus far, ERT has been largely unsuccessful in improving central nervous system manifestations of the lysosomal storage diseases, putatively due to difficulty in penetrating the blood-brain barrier. (medscape.com)
- In general, transplantation yields the best results when performed early in the course of the disease (ie, in an asymptomatic affected sibling of a child with a lysosomal storage disorder), in centers with experience in performing transplantations to treat inherited metabolic disorders, and in patients healthy enough to tolerate the conditioning and transplantation regimen. (medscape.com)
- The availability of both ERT and hematopoietic stem cell transplantation has prompted ongoing consideration of newborn screening efforts to diagnose lysosomal storage diseases. (medscape.com)
Warren Tay1
- Initially, it was named after Warren Tay, a British ophthalmologist who described the cherry-red spot on the retina of the eye of the patient in 1881. (fitnessrepublic.com)
Lipid1
- Lipid storage diseases (also known as lipidoses) are a group of inherited metabolic disorders in which harmful amounts of fatty materials (lipids) accumulate in various cells and tissues in the body. (nih.gov)
Ashkenazi Jews1
- It is a rare disease that is found in all populations, but it is particularly prevalent in Ashkenazi Jews of Eastern European origin. (jrank.org)
Cystic2
- Some of the more common disorders screened for include cystic fibrosis , sickle cell disease , thalassemia, and Tay-Sachs disease, but there are more than 100 others that can be tested for. (babycenter.com)
- Cystic fibrosis , a disease that causes mucus buildup in the lungs and other organs, making it hard to breathe. (medlineplus.gov)
Defective3
- The catabolism of which of the following is defective in Tay-Sachs disease? (lecturio.com)
- Tay-Sachs is an inherited disease that only occurs when both parents carry a Tay-Sachs gene and each parent transmits the defective gene to their child. (tay-sachs.org)
- Here's how it works: These disorders are recessive, which means that a baby must inherit a defective gene from each parent to have the disease. (babycenter.com)
Infants4
- Infants with Tay-Sachs disease appear to develop normally for the first six months after birth. (wikipedia.org)
- Infants affected by Tay-Sachs disease lose their motor skills like sitting, crawling, and turning over. (fitnessrepublic.com)
- Infants affected with Tay-Sachs disease mostly begin in the fetal stage - before the birth, when the nerve damage started. (fitnessrepublic.com)
- There can be intrauterine testing for Tay Sachs disease with abortion of the infants known to have the disease. (medneg.com.au)
Canavan2
- Prenatal diagnosis early in pregnancy will reveal if the fetus has Tay-Sachs or Canavan disease. (tay-sachs.org)
- In either case, if the fetus is affected with Tay-Sachs or Canavan, couples may elect to have a therapeutic abortion. (tay-sachs.org)
Genetics2
- Until the 1970s and 1980s, when the disease's molecular genetics became known, the juvenile and adult forms of the disease were not always recognized as variants of Tay-Sachs disease. (wikipedia.org)
- Amybeth Weaver, a licensed genetic counselor with Einstein Healthcare Network, and Dr. Adele Schneider, head of the clinical genetics program at Einstein Medical Center , are campaigning fiercely to spread knowledge about Tay-Sachs within the Irish American community. (irishcentral.com)
Progression2
- There is a rapid progression of the disease and because of this, the child may only live until early childhood that is only about 4 to 5 years old. (fitnessrepublic.com)
- In general, young patients have the most rapidly progressive disease, whereas patients with adult onset MLD experience a more chronic and insidious progression of disease. (medscape.com)
Sphingolipidosis1
- Tay-Sachs disease is a type of GM2 gangliosidosis and sphingolipidosis. (wikipedia.org)
Deterioration2
- Children with Tay-Sachs disease start missing developmental milestones after age 6 months and develop progressive cognitive and motor deterioration resulting in seizures, intellectual disability, paralysis, and death by age 5 years. (msdmanuals.com)
- All forms of the disease involve a progressive deterioration of motor and neurocognitive function. (medscape.com)
Diagnosis3
- It is almost indistinguishable from Tay-Sachs disease in course, diagnosis, and management, except that there is visceral involvement (hepatomegaly and bone change) and no ethnic association. (msdmanuals.com)
- After Nathan's genetic testing came back, we finally got a diagnosis - Nathan had Tay-Sachs Disease. (curetay-sachs.org)
- ABSTRACT We reviewed the medical and economic burden of thalassaemia major with emphasis on prenatal diagnosis for disease prevention as the most economic health care policy approach. (who.int)
Rarer1
- Juvenile Tay-Sachs disease is rarer than other forms of Tay-Sachs, and usually is initially seen in children between two and ten years old. (wikipedia.org)
Sickle cell di1
- Sickle cell disease , a disorder of the red blood cells. (medlineplus.gov)
Typically1
- Children with this form of Tay-Sachs disease typically live only a few years. (sparrow.org)
Gangliosidosis1
- In 2018, Axovant Gene Therapies licensed exclusive worldwide rights from UMass Medical School for the development and commercialization of gene therapy programs for GM1 gangliosidosis and GM2 gangliosidosis, including Tay-Sachs and Sandhoff diseases. (umassmed.edu)
Chronic9
- Therapeutic Candidate or Device Hematopoietic stem and progenitor cells collected from X-CGD patients modified with a highly regulated lentiviral vector Indication X-linked Chronic Granulomatous Disease Therapeutic Mechanism Lentiviral vector (LV) modification of autologous hematopoietic stem and progenitor cells (HSPCs) to restore physiologic gp91phox expression. (ca.gov)
- How Are Chronic Kidney Disease and High Potassium Related? (healthline.com)
- Chronic kidney disease is the gradual loss of kidney function. (healthline.com)
- Your kidneys may not be able to process excess potassium if you have chronic kidney disease. (healthline.com)
- Here's how to manage your potassium levels if you have or are at risk of developing chronic kidney disease. (healthline.com)
- Chronic kidney disease increases your risk of high blood potassium levels, known as hyperkalemia. (healthline.com)
- It's important to monitor your potassium intake if you have chronic kidney disease. (healthline.com)
- Chronic kidney disease can reduce your kidney's ability to eliminate extra potassium in your bloodstream. (healthline.com)
- If you have chronic kidney disease, your doctor may recommend limiting high potassium fruits and vegetables to reduce your risk of hyperkalemia. (healthline.com)
Babies2
- Bone marrow transplantation has been attempted also, but to date has not been successful in reversing or slowing damage to the central nervous system in babies with Tay-Sachs. (genome.gov)
- The most common form of the disease affects babies, who appear healthy at birth and seem to develop normally for the first few months of life. (girlstalkinsmack.com)
NTsAD1
- Get the latest news from NTSAD about our Community, the research that provides hope, and what is happening in the world of rare disease. (ntsad.org)
Children11
- Even with the best of care, children with Tay-Sachs disease usually die by age 4. (medlineplus.gov)
- They do not have Tay-Sachs disease but they may pass on the faulty gene to their children. (genome.gov)
- Less commonly, some children have the juvenile form of Tay-Sachs disease and may live into their teen years. (sparrow.org)
- Two young children with Tay-Sachs disease were treated safely at UMass Memorial Medical Center with a gene therapy developed at UMass Medical School and, in one case, the child's condition has stabilized, according to an interview published by WBUR with Terence R. Flotte, MD. (umassmed.edu)
- Children who have Tay-Sachs disease are at high risk of lung infections that cause breathing problems and frequently accumulate mucus in their lungs. (ahdubai.com)
- A total of twenty five percent of their children will get the disease if they procreate. (medneg.com.au)
- Seizures are possible at any age and children who have the disease do not grow very well. (medneg.com.au)
- Gradually, Tay-Sachs children lose motor skills and mental functions. (tay-sachs.org)
- Tay-Sachs children usually die by age five. (tay-sachs.org)
- Most children with Tay-Sachs die by the age of 5. (medlineplus.gov)
- Children with Tay-Sachs lose skills and motor function and ultimately die by the age of 3-5. (curetay-sachs.org)
Variants1
- Tay-Sachs disease (TSD) and its variants are caused by absence or defects of the alpha subunit of Hex A. (medscape.com)
Disorder6
- Tay-Sachs disease is a genetic disorder that results in the destruction of nerve cells in the brain and spinal cord. (wikipedia.org)
- Tay-Sachs disease is a rare genetic disorder passed from parents to child. (sparrow.org)
- Tay-Sachs disease is a rare genetic disorder. (23andme.com)
- Tay-Sachs disease (TSD) is a rare congenital disorder of the central nervous system - the brain and spinal cord. (fitnessrepublic.com)
- Tay-Sachs Disease is a genetic disorder that affects the nervous system. (ucsf.edu)
- Tay-Sachs disease , a disorder that causes fatty proteins to build up in the brain. (medlineplus.gov)
Juvenile form1
- The juvenile form of Tay-Sachs disease is less common. (sparrow.org)
Affects2
- Tay-Sachs Disease is a rare inherited gene that affects the spinal cord and kills off the nerves cells around the brain. (healthyinfodaily.com)
- The disease affects males and females equally. (nih.gov)
Child's1
- While performing a careful eye exam of your child, the doctor may see a cherry-red spot in the back of the child's eyes, which is a sign of the disease. (ahdubai.com)
Disorders1
- Tay-Sachs disease is one of the most common Jewish genetic disorders. (girlstalkinsmack.com)
Research4
- Donor support has accelerated research in cancer, heart disease, ALS, Alzheimer's disease, diabetes, AIDS, multiple sclerosis, RNA interference (gene silencing) and stem cell research, and represent some of the most innovative and medically promising work being done at UMass Chan today. (umassmed.edu)
- There is no treatment, but research aimed at treating the disease by blocking synthesis of the affected molecules has been ongoing since the late 1990s. (jrank.org)
- There is a very promising line of research currently being conducted by the Tay-Sachs Gene Therapy Consortium, a group of scientists, doctors, professors, and geneticists. (curetay-sachs.org)
- We have a plan that begins with keeping our son as healthy as possible, and raising enough money to push Tay-Sachs gene therapy research from animal to human trials. (curetay-sachs.org)
Progresses3
- As the disease progresses, development slows and muscles begin to weaken. (sparrow.org)
- As the disease progresses, the child loses muscle control. (ahdubai.com)
- As the disease progresses, your child may benefit from physical therapy to help keep joints flexible and maintain as much ability to move (range of motion) as possible. (ahdubai.com)
Treatment6
- The treatment of Tay-Sachs disease is supportive in nature. (wikipedia.org)
- There is no cure or effective treatment for Tay-Sachs disease. (genome.gov)
- Currently, there is no cure for this disease, and treatment is aimed at improving the patient's quality of life. (lecturio.com)
- A comprehensive and highly informative account of genetic diseases in Europe and the technologies and services now available for treatment and prevention. (who.int)
- Details range from estimated numbers of Europeans suffering from specific genetic diseases to the average annual costs, per patient, of treatment, from advice on the safety and reliability of screening tests to a point-by-point account of deficiencies in most existing services. (who.int)
- Facts and figures are used to indicate the magnitude of the public health problem posed by these diseases, the costs and outcome of treatment, the future number of people likely to be effected, and the urgent need for rational planning of services based on both projected needs and the development of new screening tools. (who.int)
Descent2
- Tay-Sachs disease is most common in people of Ashkenazi Jewish, Cajun, and French Canadian descent. (23andme.com)
- Because of this it was at one point believed to be a disease primarily affecting people of Jewish descent, but the volume of cases among those of Irish descent has proven that Tay-Sachs is found in groups where marrying within the community is common. (irishcentral.com)
Childhood2
- As for the childhood form of Tay-Sachs, there is no cure. (genome.gov)
- These technologies are increasingly utilized to identify genetic causes of rare, mysterious diseases, particularly childhood conditions . (cdc.gov)
Pregnancy3
- The tests return in a few days to weeks so that the infant with double markers for Tay Sachs disease can be aborted before the pregnancy continues to completion. (medneg.com.au)
- Given the impact of pregnancy on CNS diseases, it is crucial for healthcare providers and patients alike to be aware of these potential effects. (bvsalud.org)
- This paper aims to review the available evidence on the impact of pregnancy on CNS diseases, including mental health conditions, from both the clinical and biomolecular perspectives. (bvsalud.org)