Waardenburg Syndrome
SOXE Transcription Factors
Microphthalmia-Associated Transcription Factor
Pigmentation Disorders
Hypopigmentation
Paired Box Transcription Factors
Endothelin-3
Hirschsprung Disease
Anisocoria
Laryngomalacia
High Mobility Group Proteins
Chromosomes, Human, Pair 2
Abnormalities, Multiple
Pedigree
Neural Crest
Genes, Dominant
Melanocytes
Mutation
Transcription Factors
DNA-Binding Proteins
Phenotype
An L1 element intronic insertion in the black-eyed white (Mitf[mi-bw]) gene: the loss of a single Mitf isoform responsible for the pigmentary defect and inner ear deafness. (1/92)
Waardenburg syndrome type 2 (WS2) is an autosomal dominant disorder characterized by a combination of pigmentary and auditory abnormalities. Approximately 20% of WS2 cases are associated with mutations in the gene encoding microphthalmia-associated transcription factor (MITF). MITF plays a critical role in the development of both neural-crest-derived melanocytes and optic cup-derived retinal pigmented epithelium (RPE); the loss of a functional Mitf in mice results in complete absence of all pigment cells, which in turn induces microphthalmia and inner ear deafness. The black-eyed white Mitf mi-bw homozygous mouse normally has a pigmented RPE but lacks melanocytes essential for the pigmentation of the body and hearing. We show here that Mitf mi-bw is caused by an insertion into intron 3 of a 7.2 kb novel L1 element, L1bw, which belongs to an actively retrotransposing TF subfamily. The L1bw insertion reduces the amount of mRNAs for two Mitf isoforms, Mitf-A and Mitf-H, by affecting their overall expression levels and pre-mRNA splicing patterns, while it abolishes mRNA expression of another isoform, Mitf-M, which is specifically expressed in neural-crest-derived melanocytes. The consequence of the L1 insertion in the black-eyed white Mitf mi-bw mouse is that the developmental programme for RPE cells proceeds normally, most likely because of the presence of residual, full-length Mitf-A and Mitf-H proteins, whereas the lack of Mitf-M results in loss of the melanocyte population. The results suggest that melanocyte development depends critically on a single Mitf isoform, Mitf-M, and raise the possibility that specific mutations affecting MITF-M, the human equivalent of Mitf-M, may be responsible for a subset of WS2 conditions. (+info)Ser298 of MITF, a mutation site in Waardenburg syndrome type 2, is a phosphorylation site with functional significance. (2/92)
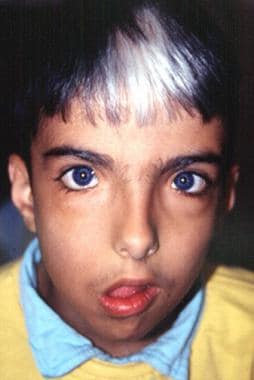
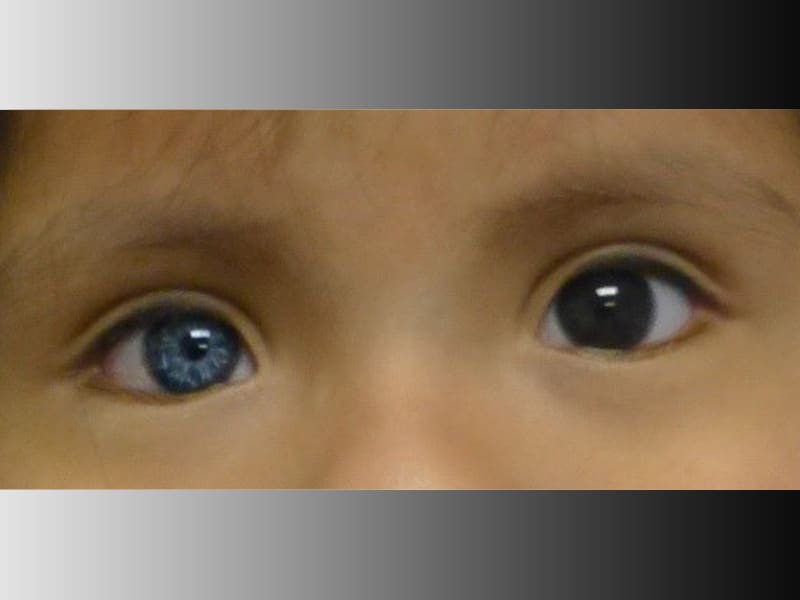
MITF (microphthalmia-associated transcription factor) is a basic-helix-loop-helix-leucine zipper (bHLHZip) factor which regulates expression of tyrosinase and other melanocytic genes via a CATGTG promoter sequence, and is involved in melanocyte differentiation. Mutations of MITF in mice or humans with Waardenburg syndrome type 2 (WS2) often severely disrupt the bHLHZip domain, suggesting the importance of this structure. Here, we show that Ser298, which locates downstream of the bHLHZip and was previously found to be mutated in individuals with WS2, plays an important role in MITF function. Glycogen synthase kinase 3 (GSK3) was found to phosphorylate Ser298 in vitro, thereby enhancing the binding of MITF to the tyrosinase promoter. The same serine was found to be phosphorylated in vivo, and expression of dominant-negative GSK3beta selectively suppressed the ability of MITF to transactivate the tyrosinase promoter. Moreover, mutation of Ser298, as found in a WS2 family, disabled phos-phorylation of MITF by GSK3beta and impaired MITF function. These findings suggest that the Ser298 is important for MITF function and is phosphorylated probably by GSK3beta. (+info)Waardenburg syndrome with anisocoria and exotropia. (3/92)
A case of Waardenburg syndrome with unusual features such as anisocoria, exotropia is reported. (+info)Structural organization of the human microphthalmia-associated transcription factor gene containing four alternative promoters. (4/92)
Microphthalmia-associated transcription factor (MITF) affects the development of many types of cells, including melanocytes and retinal pigment epithelium (RPE). MITF consists of at least three isoforms, MITF-A, MITF-H and MITF-M, differing at their amino-termini and expression patterns. Here, we characterize the structural organization of the human MITF gene. The gene contains at least four isoform-specific first exons, exons 1A, 1H, 1B and 1M in the 5' to 3' direction, each of which encodes the unique amino-terminus of a given isoform, including newly identified MITF-B. The 5'-flanking regions of these isoform-specific exons are termed promoters A, H, B and M, respectively, which showed different promoter activities, as judged by transient transfection assay. Promoter A directs the expression of a reporter gene in RPE, cervical cancer and melanoma cells, whereas promoter M is functional only in melanoma cells. Promoter H showed the significant activity in RPE and cervical cancer cells but not in melanoma cells. In contrast, the 1.7 kb 5'-flanking region of exon 1B showed no noticeable promoter activity in these cell lines. Therefore, alternative promoters provide the MITF gene with the diversity in transcriptional regulation and the capability of generating structurally different protein isoforms. (+info)Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. (5/92)
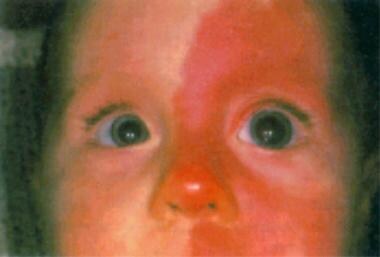
Waardenburg syndrome type 4 (WS4), also called Shah-Waardenburg syndrome, is a rare neurocristopathy that results from the absence of melanocytes and intrinsic ganglion cells of the terminal hindgut. WS4 is inherited as an autosomal recessive trait attributable to EDN3 or EDNRB mutations. It is inherited as an autosomal dominant condition when SOX10 mutations are involved. We report on three unrelated WS4 patients with growth retardation and an as-yet-unreported neurological phenotype with impairment of both the central and autonomous nervous systems and occasionally neonatal hypotonia and arthrogryposis. Each of the three patients was heterozygous for a SOX10 truncating mutation (Y313X in two patients and S251X [corrected] in one patient). The extended spectrum of the WS4 phenotype is relevant to the brain expression of SOX10 during human embryonic and fetal development. Indeed, the expression of SOX10 in human embryo was not restricted to neural-crest-derived cells but also involved fetal brain cells, most likely of glial origin. These data emphasize the important role of SOX10 in early development of both neural-crest-derived tissues, namely melanocytes, autonomic and enteric nervous systems, and glial cells of the central nervous system. (+info)Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. (6/92)
Patients with Tietz syndrome have congenital profound deafness and generalised hypopigmentation, inherited in a fully penetrant autosomal dominant fashion. The pigmentary features and complete penetrance make this syndrome distinct among syndromes with pigmentary anomalies and deafness, which characteristically have patchy depigmentation and variable penetrance. Only one family has been reported with the exact features described in the original report of this syndrome. This family was reascertained and a missense mutation was found in the basic region of the MITF gene in family members with Tietz syndrome. Mutations in other regions of this gene have been found to produce Waardenburg syndrome type 2 (WS2), which also includes pigmentary changes and hearing loss, but in contrast to Tietz syndrome, depigmentation is patchy and hearing loss is variable in WS2. (+info)Regulation of the microphthalmia-associated transcription factor gene by the Waardenburg syndrome type 4 gene, SOX10. (7/92)
The absence of melanocytes from the cochlea and epidermis is responsible of deafness and hypopigmentation, two symptoms shared by the four Waardenburg syndrome (WS) subtypes. Microphthalmia-associated transcription factor (MITF) controls melanocyte survival and differentiation. Mutations, which impair MITF function or expression, result in an abnormal melanocyte development leading to the WS2. WS1 and WS3 are caused by mutation in the gene encoding the transcription factor Pax3, which regulates MITF expression. Recently, mutations in SOX10, a gene encoding a SRY-related transcription factor, have been reported in patients with WS4. However, the molecular basis of the defective melanocyte development in these patients remained to be elucidated. In the present report, we demonstrate that Sox10 is a strong activator of the MITF promoter, and we identify a Sox10 binding site between -264 and -266 of the MITF promoter. Finally, we show that three SOX10 mutations found in WS4 abolish the transcriptional activity of the resulting Sox10 proteins toward the MITF promoter. Taken together, our observations bring new and meaningful information concerning the molecular process that leads to a defective melanocyte development in WS4 patients with SOX10 mutations. (+info)Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. (8/92)
Waardenburg syndrome (WS) is an autosomal dominant disorder with an incidence of 1 in 40 000 that manifests with sensorineural deafness and pigmentation defects. It is classified into four types depending on the presence or absence of additional symptoms. WS1 and WS3 are due to mutations in the PAX3 gene whereas some WS2 cases are associated with mutations in the microphthalmia-associated transcription factor (MITF) gene. The WS4 phenotype can result from mutations in the endothelin-B receptor gene (EDNRB), in the gene for its ligand, endothelin-3 (EDN3), or in the SOX10 gene. PAX3 has been shown to regulate MITF gene expression. The recent implication of SOX10 in WS4 prompted us to test whether this transcription factor, known to cooperate in vitro with PAX3, is also able to regulate expression from the MITF promoter. Here we show that SOX10, in synergy with PAX3, strongly activates MITF expression in transfection assays. Analyses revealed that PAX3 and SOX10 interact directly by binding to a proximal region of the MITF promoter containing binding sites for both factors. Moreover, SOX10 or PAX3 mutant proteins fail to transactivate this promoter, providing further evidence that the two genes act in concert to directly regulate expression of MITF. In situ hybridization experiments carried out in the dominant megacolon (DOM:) mouse, confirmed that SOX10 dysfunction impairs MITF: expression as well as melanocytic development and survival. These experiments, which demonstrate an interaction between three of the genes that are altered in WS, could explain the auditory-pigmentary symptoms of this disease. (+info)Waardenburg Syndrome is a genetic disorder that affects the development of melanin, a pigment responsible for hair, skin, and eye color. Named after the Dutch ophthalmologist Petrus Waardenburg who first described it in 1907, this syndrome is characterized by distinctive physical features and hearing loss.
There are four types of Waardenburg Syldrome (WS1, WS2, WS3, and WS4), each with varying degrees of symptoms. Common features include:
1. Differential coloring of the hair, skin, and eyes (poliosis, vitiligo, and heterochromia)
2. Distinctive facial features (wide-set eyes, broad nasal root, and a high arched or cleft palate)
3. Hearing loss, which can be unilateral (one-sided) or bilateral (both-sided), conductive, sensorineural, or mixed
4. Pigmentary changes in the iris, such as different colors between the eyes or within one eye
5. Sometimes, musculoskeletal abnormalities and/or developmental delays
WS1 and WS2 are more common than WS3 and WS4. The genetic causes of Waardenburg Syndrome involve mutations in several different genes associated with melanin production and transport. These include PAX3, MITF, SNAI2, EDN3, and EDNRB.
Diagnosis is typically based on clinical findings, including physical features and hearing tests. Genetic testing can confirm the diagnosis and help determine the specific type of Waardenburg Syndrome. Treatment usually involves addressing individual symptoms, such as using hearing aids or cochlear implants for hearing loss and managing any skin or eye concerns.
SOXE transcription factors are a subgroup of the SOX (SRY-related HMG box) family of proteins, which are involved in various developmental processes, including cell fate specification and differentiation. The SOXE group includes SOX8, SOX9, and SOX10, all of which contain a conserved high mobility group (HMG) box DNA-binding domain. They play crucial roles in the development of several tissues, such as the nervous system, skeletal system, and urogenital system.
SOXE transcription factors are known to regulate gene expression by binding to specific DNA sequences, often acting in combination with other transcription factors to control various cellular processes. Dysregulation of SOXE transcription factors has been implicated in several human diseases, including cancer and neurodevelopmental disorders.
The Microphthalmia-Associated Transcription Factor (MITF) is a protein that functions as a transcription factor, which means it regulates the expression of specific genes. It belongs to the basic helix-loop-helix leucine zipper (bHLH-Zip) family of transcription factors and plays crucial roles in various biological processes such as cell growth, differentiation, and survival.
MITF is particularly well-known for its role in the development and function of melanocytes, the pigment-producing cells found in the skin, eyes, and inner ear. It regulates the expression of genes involved in melanin synthesis and thus influences hair and skin color. Mutations in the MITF gene have been associated with certain eye disorders, including microphthalmia (small or underdeveloped eyes), iris coloboma (a gap or hole in the iris), and Waardenburg syndrome type 2A (an inherited disorder characterized by hearing loss and pigmentation abnormalities).
In addition to its role in melanocytes, MITF also plays a part in the development and function of other cell types, including osteoclasts (cells involved in bone resorption), mast cells (immune cells involved in allergic reactions), and retinal pigment epithelial cells (a type of cell found in the eye).
Pigmentation disorders are conditions that affect the production or distribution of melanin, the pigment responsible for the color of skin, hair, and eyes. These disorders can cause changes in the color of the skin, resulting in areas that are darker (hyperpigmentation) or lighter (hypopigmentation) than normal. Examples of pigmentation disorders include melasma, age spots, albinism, and vitiligo. The causes, symptoms, and treatments for these conditions can vary widely, so it is important to consult a healthcare provider for an accurate diagnosis and treatment plan.
Hypopigmentation is a medical term that refers to a condition where there is a decrease in the amount of pigment (melanin) in the skin, resulting in lighter patches or spots on the skin. This can occur due to various reasons such as skin injuries, certain skin disorders like vitiligo, fungal infections, burns, or as a side effect of some medical treatments like chemotherapy or radiation therapy. It is different from albinism, which is a genetic condition where the body is unable to produce melanin at all.
Paired box (PAX) transcription factors are a group of proteins that regulate gene expression during embryonic development and in some adult tissues. They are characterized by the presence of a paired box domain, a conserved DNA-binding motif that recognizes specific DNA sequences. PAX proteins play crucial roles in various developmental processes, such as the formation of the nervous system, eyes, and pancreas. Dysregulation of PAX genes has been implicated in several human diseases, including cancer.
Endothelin-3 (ET-3) is a member of the endothelin family, which are small peptides with potent vasoconstrictor properties. ET-3 is primarily produced by neurons in the central and peripheral nervous system, and it plays important roles in the development and regulation of various physiological functions, including cardiovascular function, neurotransmission, and cell proliferation.
ET-3 exerts its effects by binding to specific G protein-coupled receptors, known as endothelin A (ETA) and endothelin B (ETB) receptors. These receptors are widely distributed throughout the body, including in the cardiovascular, respiratory, gastrointestinal, and genitourinary systems.
In addition to its role as a potent vasoconstrictor, ET-3 has been implicated in various pathological conditions, such as hypertension, heart failure, pulmonary arterial hypertension, and cancer. In recent years, there has been growing interest in the potential therapeutic use of endothelin receptor antagonists to treat these conditions.
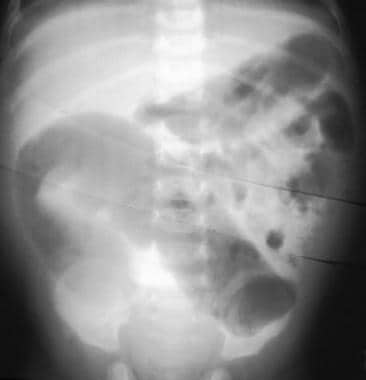
Hirschsprung disease is a gastrointestinal disorder that affects the large intestine, specifically the section known as the colon. This condition is congenital, meaning it is present at birth. It occurs due to the absence of ganglion cells (nerve cells) in the bowel's muscular wall, which are responsible for coordinating muscle contractions that move food through the digestive tract.
The affected segment of the colon cannot relax and propel the contents within it, leading to various symptoms such as constipation, intestinal obstruction, or even bowel perforation in severe cases. Common diagnostic methods include rectal suction biopsy, anorectal manometry, and contrast enema studies. Treatment typically involves surgical removal of the aganglionic segment and reattachment of the normal colon to the anus (known as a pull-through procedure).
Anisocoria is a medical term that refers to an inequality in the size of the pupils in each eye. The pupil is the black, circular opening in the center of the iris (the colored part of the eye) that allows light to enter and strike the retina. Normally, the pupils are equal in size and react similarly when exposed to light or darkness. However, in anisocoria, one pupil is larger or smaller than the other.
Anisocoria can be caused by various factors, including neurological conditions, trauma, eye diseases, or medications that affect the pupillary reflex. In some cases, anisocoria may be a normal variant and not indicative of any underlying medical condition. However, if it is a new finding or associated with other symptoms such as pain, headache, vision changes, or decreased level of consciousness, it should be evaluated by a healthcare professional to determine the cause and appropriate treatment.
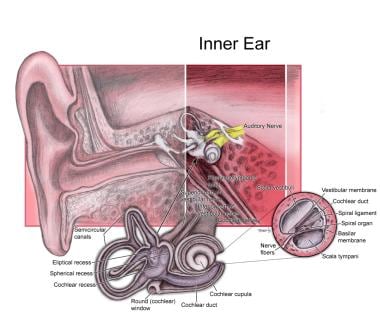
Deafness is a hearing loss that is so severe that it results in significant difficulty in understanding or comprehending speech, even when using hearing aids. It can be congenital (present at birth) or acquired later in life due to various causes such as disease, injury, infection, exposure to loud noises, or aging. Deafness can range from mild to profound and may affect one ear (unilateral) or both ears (bilateral). In some cases, deafness may be accompanied by tinnitus, which is the perception of ringing or other sounds in the ears.
Deaf individuals often use American Sign Language (ASL) or other forms of sign language to communicate. Some people with less severe hearing loss may benefit from hearing aids, cochlear implants, or other assistive listening devices. Deafness can have significant social, educational, and vocational implications, and early intervention and appropriate support services are critical for optimal development and outcomes.
Laryngomalacia is a common condition in infants characterized by soft, floppy tissues (folds) in the upper part of the windpipe (larynx) just above the vocal cords. These tissues are known as the aryepiglottic folds and the epiglottis. In laryngomalacia, these tissues are unusually soft and may prolapse or fall into the airway when an infant inhales, causing stridor (noisy breathing) or other symptoms. It's usually not a serious condition and often resolves on its own as the child grows and the tissues become stiffer. However, in some cases, it can lead to feeding difficulties, poor weight gain, or breathing problems that may require medical intervention.
Iris diseases refer to a variety of conditions that affect the iris, which is the colored part of the eye that regulates the amount of light reaching the retina by adjusting the size of the pupil. Some common iris diseases include:
1. Iritis: This is an inflammation of the iris and the adjacent tissues in the eye. It can cause pain, redness, photophobia (sensitivity to light), and blurred vision.
2. Aniridia: A congenital condition characterized by the absence or underdevelopment of the iris. This can lead to decreased visual acuity, sensitivity to light, and an increased risk of glaucoma.
3. Iris cysts: These are fluid-filled sacs that form on the iris. They are usually benign but can cause vision problems if they grow too large or interfere with the function of the eye.
4. Iris melanoma: A rare type of eye cancer that develops in the pigmented cells of the iris. It can cause symptoms such as blurred vision, floaters, and changes in the appearance of the iris.
5. Iridocorneal endothelial syndrome (ICE): A group of rare eye conditions that affect the cornea and the iris. They are characterized by the growth of abnormal tissue on the back surface of the cornea and can lead to vision loss.
It is important to seek medical attention if you experience any symptoms of iris diseases, as early diagnosis and treatment can help prevent complications and preserve your vision.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
High mobility group proteins (HMG proteins) are a family of nuclear proteins that are characterized by their ability to bind to DNA and influence its structure and function. They are named "high mobility" because of their rapid movement in gel electrophoresis. HMG proteins are involved in various nuclear processes, including chromatin remodeling, transcription regulation, and DNA repair.
There are three main classes of HMG proteins: HMGA, HMGB, and HMGN. Each class has distinct structural features and functions. For example, HMGA proteins have a unique "AT-hook" domain that allows them to bind to the minor groove of AT-rich DNA sequences, while HMGB proteins have two "HMG-box" domains that enable them to bend and unwind DNA.
HMG proteins play important roles in many physiological and pathological processes, such as embryonic development, inflammation, and cancer. Dysregulation of HMG protein function has been implicated in various diseases, including neurodegenerative disorders, diabetes, and cancer. Therefore, understanding the structure, function, and regulation of HMG proteins is crucial for developing new therapeutic strategies for these diseases.
Human chromosome pair 2 consists of two rod-shaped structures present in the nucleus of each cell of the human body. Each member of the pair contains thousands of genes and other genetic material, encoded in the form of DNA molecules. Chromosomes are the physical carriers of inheritance, and human cells typically contain 23 pairs of chromosomes for a total of 46 chromosomes.
Chromosome pair 2 is one of the autosomal pairs, meaning that it is not a sex chromosome (X or Y). Each member of chromosome pair 2 is approximately 247 million base pairs in length and contains an estimated 1,000-1,300 genes. These genes play crucial roles in various biological processes, including development, metabolism, and response to environmental stimuli.
Abnormalities in chromosome pair 2 can lead to genetic disorders, such as cat-eye syndrome (CES), which is characterized by iris abnormalities, anal atresia, hearing loss, and intellectual disability. This disorder arises from the presence of an extra copy of a small region on chromosome 2, resulting in partial trisomy of this region. Other genetic conditions associated with chromosome pair 2 include proximal 2q13.3 microdeletion syndrome and Potocki-Lupski syndrome (PTLS).
'Abnormalities, Multiple' is a broad term that refers to the presence of two or more structural or functional anomalies in an individual. These abnormalities can be present at birth (congenital) or can develop later in life (acquired). They can affect various organs and systems of the body and can vary greatly in severity and impact on a person's health and well-being.
Multiple abnormalities can occur due to genetic factors, environmental influences, or a combination of both. Chromosomal abnormalities, gene mutations, exposure to teratogens (substances that cause birth defects), and maternal infections during pregnancy are some of the common causes of multiple congenital abnormalities.
Examples of multiple congenital abnormalities include Down syndrome, Turner syndrome, and VATER/VACTERL association. Acquired multiple abnormalities can result from conditions such as trauma, infection, degenerative diseases, or cancer.
The medical evaluation and management of individuals with multiple abnormalities depend on the specific abnormalities present and their impact on the individual's health and functioning. A multidisciplinary team of healthcare professionals is often involved in the care of these individuals to address their complex needs.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
The neural crest is a transient, multipotent embryonic cell population that originates from the ectoderm (outermost layer) of the developing neural tube (precursor to the central nervous system). These cells undergo an epithelial-to-mesenchymal transition and migrate throughout the embryo, giving rise to a diverse array of cell types and structures.
Neural crest cells differentiate into various tissues, including:
1. Peripheral nervous system (PNS) components: sensory neurons, sympathetic and parasympathetic ganglia, and glial cells (e.g., Schwann cells).
2. Facial bones and cartilage, as well as connective tissue of the skull.
3. Melanocytes, which are pigment-producing cells in the skin.
4. Smooth muscle cells in major blood vessels, heart, gastrointestinal tract, and other organs.
5. Secretory cells in endocrine glands (e.g., chromaffin cells of the adrenal medulla).
6. Parts of the eye, such as the cornea and iris stroma.
7. Dental tissues, including dentin, cementum, and dental pulp.
Due to their wide-ranging contributions to various tissues and organs, neural crest cells play a crucial role in embryonic development and organogenesis. Abnormalities in neural crest cell migration or differentiation can lead to several congenital disorders, such as neurocristopathies.
Dominant genes refer to the alleles (versions of a gene) that are fully expressed in an individual's phenotype, even if only one copy of the gene is present. In dominant inheritance patterns, an individual needs only to receive one dominant allele from either parent to express the associated trait. This is in contrast to recessive genes, where both copies of the gene must be the recessive allele for the trait to be expressed. Dominant genes are represented by uppercase letters (e.g., 'A') and recessive genes by lowercase letters (e.g., 'a'). If an individual inherits one dominant allele (A) from either parent, they will express the dominant trait (A).
Melanocytes are specialized cells that produce, store, and transport melanin, the pigment responsible for coloring of the skin, hair, and eyes. They are located in the bottom layer of the epidermis (the outermost layer of the skin) and can also be found in the inner ear and the eye's retina. Melanocytes contain organelles called melanosomes, which produce and store melanin.
Melanin comes in two types: eumelanin (black or brown) and pheomelanin (red or yellow). The amount and type of melanin produced by melanocytes determine the color of a person's skin, hair, and eyes. Exposure to UV radiation from sunlight increases melanin production as a protective response, leading to skin tanning.
Melanocyte dysfunction or abnormalities can lead to various medical conditions, such as albinism (lack of melanin production), melasma (excessive pigmentation), and melanoma (cancerous growth of melanocytes).
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Transcription factors are proteins that play a crucial role in regulating gene expression by controlling the transcription of DNA to messenger RNA (mRNA). They function by binding to specific DNA sequences, known as response elements, located in the promoter region or enhancer regions of target genes. This binding can either activate or repress the initiation of transcription, depending on the properties and interactions of the particular transcription factor. Transcription factors often act as part of a complex network of regulatory proteins that determine the precise spatiotemporal patterns of gene expression during development, differentiation, and homeostasis in an organism.
DNA-binding proteins are a type of protein that have the ability to bind to DNA (deoxyribonucleic acid), the genetic material of organisms. These proteins play crucial roles in various biological processes, such as regulation of gene expression, DNA replication, repair and recombination.
The binding of DNA-binding proteins to specific DNA sequences is mediated by non-covalent interactions, including electrostatic, hydrogen bonding, and van der Waals forces. The specificity of binding is determined by the recognition of particular nucleotide sequences or structural features of the DNA molecule.
DNA-binding proteins can be classified into several categories based on their structure and function, such as transcription factors, histones, and restriction enzymes. Transcription factors are a major class of DNA-binding proteins that regulate gene expression by binding to specific DNA sequences in the promoter region of genes and recruiting other proteins to modulate transcription. Histones are DNA-binding proteins that package DNA into nucleosomes, the basic unit of chromatin structure. Restriction enzymes are DNA-binding proteins that recognize and cleave specific DNA sequences, and are widely used in molecular biology research and biotechnology applications.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
DNA Mutational Analysis is a laboratory test used to identify genetic variations or changes (mutations) in the DNA sequence of a gene. This type of analysis can be used to diagnose genetic disorders, predict the risk of developing certain diseases, determine the most effective treatment for cancer, or assess the likelihood of passing on an inherited condition to offspring.
The test involves extracting DNA from a patient's sample (such as blood, saliva, or tissue), amplifying specific regions of interest using polymerase chain reaction (PCR), and then sequencing those regions to determine the precise order of nucleotide bases in the DNA molecule. The resulting sequence is then compared to reference sequences to identify any variations or mutations that may be present.
DNA Mutational Analysis can detect a wide range of genetic changes, including single-nucleotide polymorphisms (SNPs), insertions, deletions, duplications, and rearrangements. The test is often used in conjunction with other diagnostic tests and clinical evaluations to provide a comprehensive assessment of a patient's genetic profile.
It is important to note that not all mutations are pathogenic or associated with disease, and the interpretation of DNA Mutational Analysis results requires careful consideration of the patient's medical history, family history, and other relevant factors.
Anophthalmos is a medical condition where an individual is born without one or both eyes. It is a congenital disorder, which means it is present at birth. In cases where only one eye is affected, it is called unilateral anophthalmos, and when both eyes are missing, it is referred to as bilateral anophthalmos.
Anophthalmos is different from microphthalmia, another congenital condition where the eye is present but abnormally small. In some cases, anophthalmos may be accompanied by other developmental anomalies or syndromes. The exact cause of anophthalmos is not always known, but it can be associated with genetic mutations or environmental factors that affect fetal development.
Individuals with anophthalmos require specialized medical care and management to ensure proper eye socket development, visual rehabilitation, and overall well-being. This may include the use of prosthetic eyes, orthoptic therapy, and other supportive measures.
Genetic linkage is the phenomenon where two or more genetic loci (locations on a chromosome) tend to be inherited together because they are close to each other on the same chromosome. This occurs during the process of sexual reproduction, where homologous chromosomes pair up and exchange genetic material through a process called crossing over.
The closer two loci are to each other on a chromosome, the lower the probability that they will be separated by a crossover event. As a result, they are more likely to be inherited together and are said to be linked. The degree of linkage between two loci can be measured by their recombination frequency, which is the percentage of meiotic events in which a crossover occurs between them.
Linkage analysis is an important tool in genetic research, as it allows researchers to identify and map genes that are associated with specific traits or diseases. By analyzing patterns of linkage between markers (identifiable DNA sequences) and phenotypes (observable traits), researchers can infer the location of genes that contribute to those traits or diseases on chromosomes.