Displasia Ectodérmica
Displasia Ectodermal Anhidrótica Tipo 1
Hipohidrosis
Ectodisplasinas
Anodoncia
Displasia Ectodérmica Hipohidrótica Autosómica Recesiva
Uñas Malformadas
Receptor Edar
Receptor Xedar
Receptores de la Ectodisplasina
Dentadura Parcial Removible
Sindactilia
Displasia Ectodérmica Anhidrótica Tipo 3
Dentadura Completa Inferior
Displasia Fibrosa Ósea
Enfermedades de la Uña
Proteína de Dominio de Muerte Asociada a Edar
Labio Leporino
Síndromes de Inmunodeficiencia
Glándulas Sudoríparas
Dentición
Enfermedades Genéticas Ligadas al Cromosoma X
Linaje
Deformidades Congénitas de las Extremidades
Displasia Broncopulmonar
Anomalías Múltiples
Cromosoma X
Diseño de Dentadura
Enfermedades del Desarrollo Óseo
Queratinas Tipo II
Cabello
Enfermedades del Cabello
Incontinencia Pigmentaria
Queratodermia Palmoplantar
Hiperpigmentación
Deformidades Congénitas de la Mano
Ectromelia
Quinasa I-kappa B
Fotofobia
Síndrome de Ellis-Van Creveld
Dentadura Completa
Displasia del Cuello del Útero
Mutación
Ligamiento Genético
Displasia Fibromuscular
Dentadura Parcial
Párpados
Displasia Fibrosa Poliostótica
Deformidades Congénitas del Pie
Displasia Pélvica Canina
Displasia Cleidocraneal
Fenotipo
Displasia Retiniana
Mutación Missense
Sudoración
Luxación Congénita de la Cadera
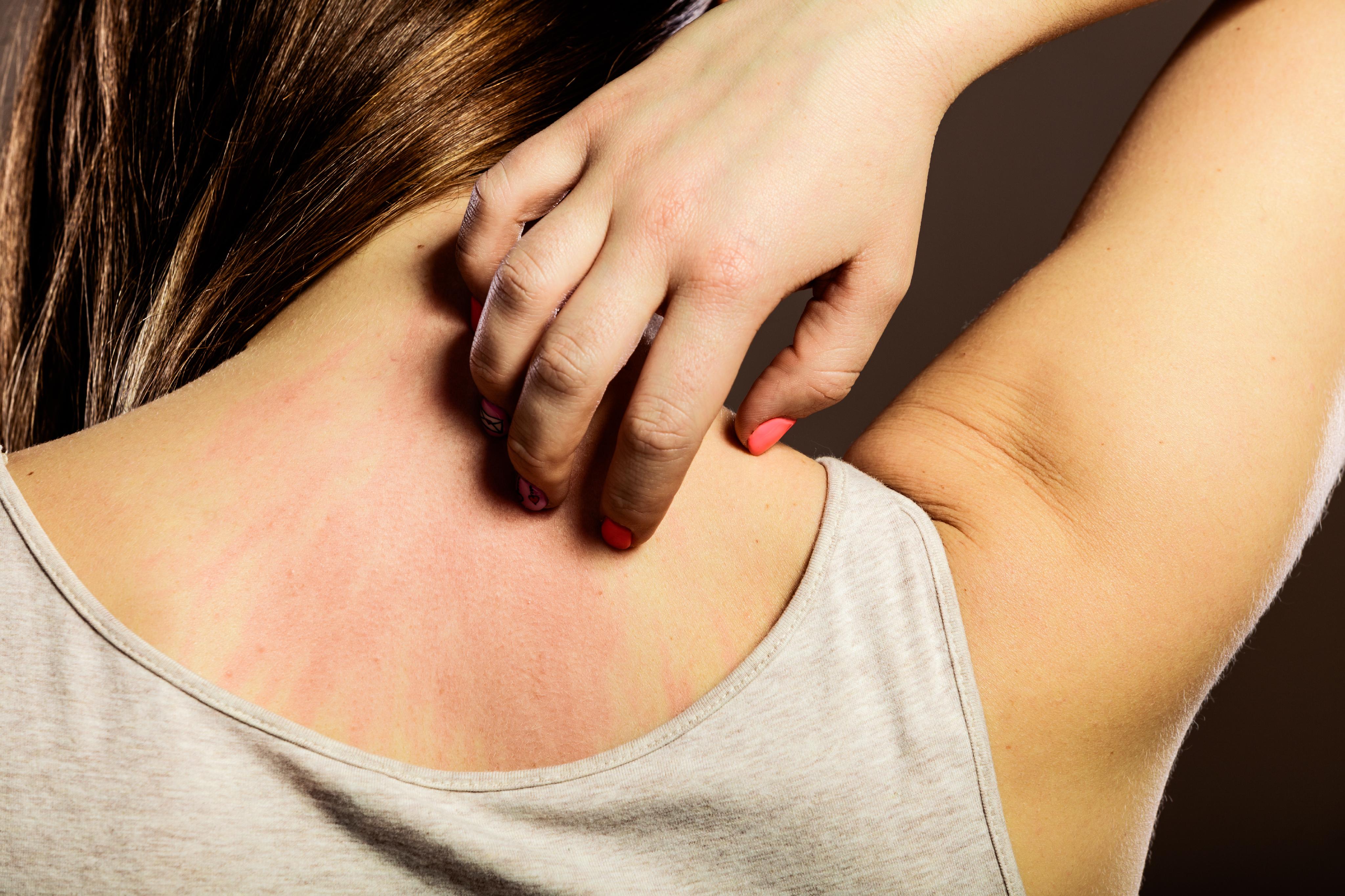
La displasia ectodérmica es un término general que se utiliza para describir un grupo heterogéneo de trastornos congénitos hereditarios que afectan el desarrollo y la diferenciación de las estructuras derivadas de la cresta neural y la ectodermis. Estos tejidos incluyen la piel, el pelo, las uñas, los dientes, las glándulas sudoríparas y algunas partes del sistema nervioso periférico.
Existen varios tipos de displasia ectodérmica, cada uno con diferentes conjuntos de síntomas y grados de gravedad. Algunos de los signos y síntomas comunes asociados con estas condiciones incluyen: piel seca o escamosa, falta de sudoración, uñas débiles o incurvadas, dientes anormales o ausentes, cabello fino, rizado o ausente, y anomalías del sistema nervioso periférico.
La displasia ectodérmica puede ser causada por mutaciones en varios genes diferentes, y hereda a menudo de forma autosómica dominante o recesiva. El tratamiento suele ser sintomático y depende de los síntomas específicos presentes en cada individuo.
La Displasia Ectodermal Anhidrótica Tipo 1 (DEA-Type 1), también conocida como Síndrome de Christ-Siemens-Touraine, es una enfermedad genética hereditaria que afecta el desarrollo y la función de las glándulas ectodérmicas. Las glándulas ectodérmicas incluyen glándulas sudoríparas, glándulas sebáceas, pelo, uñas, dientes y parte del sistema nervioso periférico.
La DEA-Type 1 es causada por mutaciones en el gen EDA (Ectodysplasin A) o en los genes relacionados EDAR y EDARADD. Estos genes desempeñan un papel importante en la señalización durante el desarrollo embrionario, especialmente en la formación de estructuras derivadas de la ectodermis.
Los síntomas más comunes de la DEA-Type 1 incluyen:
1. Anhidrosis o hipohidrosis: Incapacidad para sudar normalmente, lo que puede llevar a episodios de hipertermia (aumento de la temperatura corporal).
2. Hipodoncia: Ausencia congénita o retraso en la erupción de algunos dientes permanentes.
3. Onicodistrofia: Uñas anómalas, delgadas y frágiles.
4. Piel seca y escamosa.
5. Ausencia o crecimiento lento del cabello.
6. Alteraciones en la forma y estructura de las orejas.
7. Paladar hendido o labio leporino en algunos casos.
El diagnóstico se realiza mediante la observación clínica, la historia familiar y los estudios genéticos. El tratamiento es sintomático y puede incluir medidas para mantener la hidratación, el uso de protectores solares, la estimulación artificial del crecimiento de los dientes y la corrección quirúrgica de las malformaciones craneofaciales. La asesoría genética es importante para la planificación familiar y el consejo sobre el riesgo de recurrencia en futuras generaciones.
La hipohidrosis es un término médico que se refiere a una condición en la cual una persona tiene una reducción significativa en la capacidad de sudar. La sudoración es un proceso normal y vital para el cuerpo humano, ya que ayuda a regular la temperatura corporal. Cuando una persona suda, los líquidos se liberan a través de las glándulas sudoríparas en la piel, lo que ayuda a refrescar el cuerpo y a disipar el calor.
La hipohidrosis puede afectar a todo el cuerpo o solo a ciertas áreas de la piel. Puede ser causada por varios factores, incluyendo enfermedades genéticas, lesiones en la piel, quemaduras graves, infecciones, trastornos neurológicos y el uso de algunos medicamentos.
Los síntomas de la hipohidrosis pueden variar dependiendo de su gravedad. En casos leves, una persona puede experimentar sudoración escasa o ausente en respuesta al calor o al ejercicio físico. Sin embargo, en casos más graves, la falta de sudoración puede conducir a un aumento de la temperatura corporal, lo que puede provocar síntomas como debilidad, mareos, náuseas, vómitos y confusión. En los casos más extremos, la hipohidrosis puede incluso causar insuficiencia orgánica y ser potencialmente mortal.
El tratamiento de la hipohidrosis depende de la causa subyacente de la afección. Si se identifica y se trata la causa subyacente, la sudoración normal puede volver a la normalidad. En algunos casos, el médico puede recomendar terapias para estimular la sudoración, como la iontoforesis o la estimulación nerviosa eléctrica transcutánea. En los casos más graves, se pueden considerar opciones quirúrgicas, como la transferencia de glándulas sudoríparas o la implantación de un dispositivo para ayudar a regular la temperatura corporal.
Las ectodisplasias se refieren a un grupo de trastornos genéticos poco frecuentes que afectan el desarrollo de tejidos derivados del ectodermo, que es una de las tres capas germinales en el embrión temprano. El ectodermo da origen a la piel, el sistema nervioso, los dientes y varias glándulas, incluyendo las glándulas sudoríparas y las glándulas mamarias.
Las ectodisplasias pueden causar una variedad de síntomas, dependiendo del tipo específico de la afección. Estos síntomas pueden incluir anormalidades en el crecimiento del cabello, uñas y dientes; anomalías en la estructura y función de las glándulas sudoríparas y mamarias; y defectos en el desarrollo de los huesos, músculos y nervios.
Existen más de 150 tipos diferentes de ectodisplasias, cada uno con su propio patrón específico de síntomas y causas genéticas subyacentes. Algunos de los tipos más comunes de ectodisplasias incluyen la anquilosis de las articulaciones y el cráneo, la síndrome de EEC (ectrodactilia, ectodermal dysplasia y displasia oral/facial), y la displasia ectodérmica hidropisciforme.
El tratamiento de las ectodisplasias depende del tipo específico de afección y puede incluir una variedad de enfoques, desde el cuidado de apoyo y la terapia de reemplazo hormonal hasta la cirugía reconstructiva. El pronóstico para las personas con ectodisplasias varía ampliamente, dependiendo de la gravedad de los síntomas y la disponibilidad de tratamientos efectivos.
Anodoncia es un término médico que se utiliza para describir la ausencia congénita o la falta congénita de uno o más dientes permanentes en las arcadas dentarias. Esta condición está presente desde el nacimiento y puede afectar a cualquier diente permanente, aunque es más común en los incisivos laterales superiores.
La anodoncia se produce cuando los folículos dentarios, que son las estructuras especializadas de tejido conectivo que dan lugar al desarrollo y crecimiento de los dientes, no se forman o no se desarrollan correctamente durante la embriogénesis. La causa exacta de esta afección es desconocida, pero se cree que puede estar relacionada con factores genéticos y ambientales.
La anodoncia puede presentarse en forma aislada o asociada con otras anomalías dentales o síndromes genéticos. En algunos casos, la ausencia de dientes permanentes puede causar problemas estéticos y funcionales, como dificultad para masticar y hablar, así como alteraciones en el desarrollo y alineación de los dientes restantes.
El tratamiento de la anodoncia depende de la gravedad y extensión de la afección. Puede incluir la colocación de implantes dentales, puentes fijos o prótesis removibles para reemplazar los dientes ausentes y restaurar la función y estética dental. En algunos casos, también puede ser necesario realizar un tratamiento ortodóncico para corregir la alineación de los dientes restantes.
La displasia ectodérmica hipohidrótica autosómica recesiva (EDH, por sus siglas en inglés) es un trastorno genético extremadamente raro que afecta el desarrollo de varias partes del cuerpo relacionadas con la ectodermis embrionaria, que es uno de los tres tipos básicos de tejido corporal durante las primeras etapas del desarrollo fetal.
Las características clínicas principales de EDH incluyen:
1. Hipohidrosis o anhidrosis (incapacidad para sudar normalmente) lo que lleva a una alta sensibilidad al calor y problemas de temperatura reguladora.
2. Anomalías del cabello y las uñas, como el pelo escaso, fino, lanoso o ondulado, cejas y pestañas escasas, y uñas frágiles o de crecimiento anormal.
3. Problemas dentales graves, incluyendo retrasos en la erupción dental, dientes con malformaciones, ausencia congénita de dientes y una mayor susceptibilidad a las caries.
4. Anomalías de la piel, como sequedad excesiva, palidez, falta de sudoración, aumento de la sensibilidad al frío y al calor, y enrojecimiento fácil.
5. Problemas oculares, como el ojo seco, la incapacidad para producir lágrimas y una mayor susceptibilidad a las infecciones oculares.
EDH es heredado de manera autosómica recesiva, lo que significa que un individuo debe heredar dos copias del gen anormal (una de cada padre) para desarrollar el trastorno. Los padres de un niño afectado generalmente no muestran signos o síntomas de la enfermedad, pero son portadores y tienen un 25% de probabilidades de transmitir el gen anormal a cada uno de sus hijos.
El diagnóstico de EDH se basa en los hallazgos clínicos y puede confirmarse mediante pruebas genéticas. El tratamiento es sintomático y multidisciplinario, involucrando a especialistas en dermatología, odontología, oftalmología, otorrinolaringología y fisioterapia. La terapia de reemplazo de lágrimas artificiales, la hidratación regular de la piel y los cuidados dentales preventivos son esenciales para el manejo adecuado del trastorno.
Las uñas malformadas, también conocidas como uñas dismórficas o anómalas, se refieren a un grupo de afecciones en las que las uñas presentan alteraciones en su forma, tamaño, textura o coloración, debido a anomalías congénitas, infecciosas, traumáticas o asociadas a diversas patologías sistémicas. Estas malformaciones pueden afectar a una o varias uñas y pueden presentarse de forma aislada o como parte de un síndrome más complejo.
Existen diferentes tipos de uñas malformadas, entre los que se incluyen:
1. Uña ondulada o en embudo (inglés: "Koilonychia" o "Platyonychia"): Se caracteriza por una uña con forma de cucharón o acanalada, aplanada o engrosada. Puede ser el resultado de deficiencias nutricionales, como la falta de hierro o proteínas, o estar asociada a enfermedades reumatológicas y dermatológicas.
2. Uña engrosada (inglés: "Hypertrophic Nail" o "Onychauxis"): Se presenta cuando la uña se vuelve más gruesa y abultada de lo normal, a menudo con estrías y surcos. Puede ser el resultado de traumatismos repetidos, infecciones fúngicas o enfermedades sistémicas como psoriasis y liquen plano.
3. Uña quebradiza (inglés: "Brittle Nails" o "Onychoschizia"): Se refiere a uñas frágiles, con tendencia a agrietarse, romperse o desprenderse fácilmente. Esto puede deberse al envejecimiento, exposición a productos químicos agresivos, deficiencias nutricionales o trastornos tiroideos.
4. Uña con manchas blancas (inglés: "Leukonychia"): Se caracteriza por la presencia de pequeñas manchas blancas en la uña. Esto puede ser el resultado de traumatismos menores, deficiencias nutricionales o enfermedades sistémicas como la diabetes y problemas hepáticos.
5. Uña con líneas rojas (inglés: "Splinter Hemorrhage"): Se presentan pequeñas líneas rojas debajo de la uña, que pueden ser el resultado de traumatismos menores o estar asociadas a enfermedades sistémicas como endocarditis infecciosa y lupus eritematoso sistémico.
6. Uña encarnada (inglés: "Onychocryptosis"): Ocurre cuando la uña crece hacia dentro, penetrando en la piel y causando dolor e inflamación. Esto puede ser el resultado de malos cortes de uñas, calzado inadecuado o predisposición genética.
7. Uña con hongos (inglés: "Onychomycosis"): Se caracteriza por la presencia de manchas amarillentas y engrosamiento de la uña, que puede causar dolor e incomodidad. Esto es causado por hongos que se desarrollan en ambientes cálidos y húmedos.
8. Uña con psoriasis (inglés: "Psoriatic Nail Disease"): Se presenta engrosamiento, decoloración y descamación de la uña, que puede estar asociada a psoriasis en otras partes del cuerpo.
9. Uña con paroniquia (inglés: "Paronychia"): Es una infección bacteriana o fúngica alrededor de la uña, que puede causar hinchazón, enrojecimiento y dolor. Esto puede ser el resultado de traumatismos menores o malos cortes de uñas.
10. Uña con melanoma (inglés: "Subungual Melanoma"): Se presenta una mancha oscura debajo de la uña, que puede ser un signo de melanoma. Es importante buscar atención médica inmediata si se observa este síntoma.
Las anomalías dentarias se refieren a cualquier tipo de condición o trastorno que afecta el desarrollo, la estructura, el número o la posición de los dientes. Estas anomalías pueden presentarse en forma de dientes adicionales (supernumerarios), ausencia de dientes (agénesis), dientes con forma anormal, tamaño anormal o coloración anormal, y malposiciones dentarias (como mordidas abiertas, cruzadas o profundas).
Las anomalías dentarias pueden ser causadas por una variedad de factores, incluyendo la genética, la exposición a ciertos medicamentos durante el desarrollo dental y los traumatismos. Algunas de estas anomalías pueden requerir tratamiento dental o ortodóncico para corregirlas y prevenir posibles problemas funcionales o estéticos.
Es importante tener en cuenta que algunas anomalías dentarias pueden estar asociadas con otras condiciones médicas o síndromes, por lo que es recomendable buscar atención dental especializada si se presentan este tipo de situaciones.
El receptor Edar, o receptor del factor de crecimiento épidermis EDA, es un tipo de proteína que se encuentra en la superficie celular y desempeña un papel crucial en el desarrollo y mantenimiento de varios tejidos y órganos. Es parte del sistema de señalización EDA (factor de crecimiento épidermis) y está involucrado en una variedad de procesos biológicos, como la morfogénesis, la homeostasis tisular y la respuesta inmunitaria.
El receptor Edar consta de tres dominios principales: un dominio extracelular que se une al ligando EDA, un dominio transmembrana y un dominio intracelular que participa en la transducción de señales. Cuando el ligando EDA se une al receptor Edar, induce la formación de un complejo de señalización que incluye a la proteína adaptadora Edaradd y la cinasa Nedd4l. Esta interacción desencadena una cascada de eventos que conducen a la activación de diversas vías de transducción de señales, lo que finalmente conduce a la regulación de la expresión génica y los procesos celulares subsiguientes.
Las mutaciones en el gen del receptor Edar se han asociado con varias anomalías congénitas, como el síndrome anhidrótico-ectodermal (AEC), que se caracteriza por la ausencia de glándulas sudoríparas y dientes, así como por anomalías en el cabello, las uñas y la piel. Además, estudios recientes han sugerido que el receptor Edar puede desempeñar un papel en el desarrollo y progressión del cáncer, particularmente en los tumores de mama y ovario.
La hipotricosis es un término médico que se refiere a una condición capilar en la cual hay una disminución anormal o pérdida notable del crecimiento del cabello. Puede afectar el cuero cabelludo o todo el cuerpo. La gravedad de la pérdida del cabello puede variar desde leve hasta severa, e incluso puede estar asociada con otras anomalías, como defectos en las uñas o problemas en el desarrollo. Es importante destacar que existen diferentes tipos y causas de hipotricosis, incluyendo factores genéticos, enfermedades autoinmunes, infecciosas, deficiencias nutricionales o exposición a ciertos fármacos o tratamientos. El diagnóstico y manejo de la hipotricosis requieren una evaluación médica completa para determinar la causa subyacente y así poder ofrecer el tratamiento más apropiado.
Después de realizar una extensa investigación, puedo informarte que no existe una definición médica establecida o reconocida para "Receptor Xedar". Es posible que te refieras a un término específico o concepto relacionado con la medicina o la biología molecular que no es ampliamente utilizado o aceptado en la comunidad científica. En tal caso, te agradecería que proporcionaras más detalles o contexto adicionales para poder brindarte una respuesta más precisa y útil.
Sin embargo, si nos referimos a los receptores en general dentro del campo de la biología molecular y la farmacología, se trata de proteínas específicas presentes en las membranas celulares o dentro del citoplasma que se unen a determinadas moléculas (ligandos) y desencadenan una respuesta bioquímica o fisiológica dentro de la célula. Algunos ejemplos comunes de receptores incluyen los receptores de neurotransmisores, hormonas, factores de crecimiento y fármacos.
Si tienes más información sobre el "Receptor Xedar" o si deseas obtener información sobre un tema relacionado, no dudes en preguntar nuevamente. Estoy aquí para ayudarte.
Los receptores de la ectodisplasia (EDA-R) son un tipo de receptor localizado en la superficie celular, que pertenecen a la familia de las proteínas TNF (Tumor Necrosis Factor). Estos receptores desempeñan un papel crucial en la señalización celular y están implicados en varios procesos biológicos importantes, como el desarrollo embrionario, la homeostasis tisular y la respuesta inmune.
La ectodisplasia es una enfermedad genética rara que afecta al desarrollo de las estructuras derivadas del ectodermo, como el pelo, los dientes, las glándulas sudoríparas y algunas partes del sistema nervioso. La proteína EDA (Ectodisplasia-A) es una ligando que se une a estos receptores y desempeña un papel fundamental en el desarrollo de los tejidos afectados por esta enfermedad.
Los receptores de la ectodisplasia están formados por una cadena extracelular, una transmembrana y una intracelular. La cadena extracelular contiene varios dominios que le permiten unirse a su ligando, la proteína EDA. Cuando la proteína EDA se une al receptor, desencadena una cascada de eventos que conducen a la activación de diversas vías de señalización celular.
La activación de los receptores de la ectodisplasia está asociada con varios procesos biológicos importantes, como la proliferación y diferenciación celular, la apoptosis (muerte celular programada), la inflamación y la respuesta inmune. Por lo tanto, los receptores de la ectodisplasia desempeñan un papel crucial en el desarrollo y mantenimiento de varios tejidos y órganos del cuerpo humano.
Una dentadura parcial removible, también conocida como prótesis parcial removible, es un dispositivo protésico utilizado en odontología para reemplazar los dientes perdidos. Está diseñada para adaptarse a las encías y mordidas específicas de cada paciente. Se fabrica generalmente con una base de acrílico o nylon termoendurecido que contiene los dientes falsos, los cuales pueden ser de resina o porcelana.
Este tipo de prótesis se mantiene en su lugar gracias a retenedores metálicos o plásticos que se acoplan a los dientes naturales adyacentes a través de ganchos, ataches o attaching. La dentadura parcial removible es una opción versátil y económica en comparación con otras alternativas como las prótesis fijas o implantes dentales.
Es importante mencionar que el éxito del tratamiento con este tipo de prótesis depende en gran medida del cumplimiento del paciente en términos de higiene oral, uso adecuado y controles periódicos con su profesional dental.
La sindactilia es un término médico que se refiere a la fusión congénita o anormal de dos o más dedos en las manos o los pies. Puede involucrar tejido óseo, tejido conectivo o ambos. La gravedad de esta condición puede variar desde una leve unión de la piel entre los dedos hasta dedos completamente fusionados en sus longitudes.
La sindactilia puede ocurrir aisladamente como un defecto congénito aislado, pero también se asocia con varias anomalías cromosómicas y síndromes genéticos, como el síndrome de Apert, el síndrome de Poland, el síndrome de Pfeiffer y el síndrome de Down.
El tratamiento suele implicar cirugía reconstructiva para separar los dedos y mejorar la función y apariencia. La intervención quirúrgica generalmente se realiza en la infancia, ya que es cuando los tejidos son más flexibles y el potencial de recuperación es mayor.
La displasia ectodérmica anhidrótica tipo 3 (DEA III), también conocida como Síndrome de Clouston, es una enfermedad hereditaria autosómica dominante que afecta el desarrollo y la función de las estructuras derivadas de la ectodermis embrionaria. La DEA III se caracteriza principalmente por anomalías en la piel, el cabello, las uñas y los dientes.
Las personas con DEA III presentan una falta total o parcial de glándulas sudoríparas ecrinas, lo que resulta en hipohidrosis o anhidrosis, es decir, disminución o ausencia de sudoración, especialmente en las palmas de las manos y plantas de los pies. Esto puede llevar a episodios de hipertermia y problemas para regular la temperatura corporal, especialmente durante el ejercicio o en climas cálidos.
Otros signos clínicos de DEA III incluyen:
1. Alopecia: Pérdida del pelo en las cejas, pestañas y cabello, que puede ser parcial o total. El cabello restante es fino, despigmentado y quebradizo.
2. Anomalías ungueales: Las uñas pueden ser gruesas, engrosadas, de crecimiento lento, frágiles y con estrías transversales. En algunos casos, las uñas pueden estar ausentes en los dedos de manos y pies.
3. Anomalías dentales: Los dientes suelen ser pequeños, con esmalte dental anormal, hipoplásico o ausente, lo que puede causar sensibilidad dental e incrementar el riesgo de caries dental.
4. Piel: La piel puede ser seca, escamosa y vulnerable a infecciones. También pueden presentarse hiperqueratosis palmoplantar (callosidades en las palmas y plantas de los pies).
5. Otras manifestaciones menos frecuentes incluyen anormalidades del esmalte de las uñas, onicodistrofia (deformación de las uñas), hiperhidrosis (exceso de sudoración) y displasia ectodérmica (anomalías en la piel, pelo, uñas, dientes y glándulas sudoríparas).
El diagnóstico de DEA III se basa en los hallazgos clínicos y puede ser confirmado mediante estudios genéticos. La mayoría de los casos de DEA III están asociados con mutaciones en el gen KRT14, que codifica la queratina 14, una proteína importante para la integridad estructural de la piel y las uñas.
El tratamiento de DEA III se centra en aliviar los síntomas y mejorar la calidad de vida del paciente. Las recomendaciones incluyen:
1. Higiene dental adecuada para prevenir caries y enfermedades periodontales.
2. Uso de protectores bucales durante la noche para prevenir el bruxismo (apretar o rechinar los dientes).
3. Protección de las uñas con esmaltes protectores o láminas de gel para evitar su rotura y desprendimiento.
4. Uso de cremas hidratantes y emolientes para mantener la piel hidratada y prevenir la sequedad y descamación.
5. Evitar el contacto con sustancias irritantes o alérgenos que puedan empeorar los síntomas.
6. En caso de hiperhidrosis, se pueden utilizar antitranspirantes tópicos o inyecciones de toxina botulínica para controlar el exceso de sudoración.
7. La fisioterapia y la terapia ocupacional pueden ayudar a mejorar la movilidad y la función de las manos y los pies.
8. En algunos casos, se puede considerar la cirugía para corregir malformaciones o deformidades graves.
9. El apoyo psicológico y emocional es fundamental para ayudar a las personas con esta enfermedad a enfrentar los desafíos diarios y mejorar su calidad de vida.
La dentadura completa inferior, también conocida como prótesis completa mandibular o dentadura postiza inferior, es un dispositivo protésico removible que se utiliza para reemplazar todos los dientes ausentes en la mandíbula inferior. Está compuesta generalmente por una base de acrílico que imita el color de las encías y dientes de resina o porcelana que reproducen la apariencia y función de los dientes naturales. Se adhiere a la encía mediante un sellante adhesivo y se retira fácilmente para su limpieza y mantenimiento. La dentadura completa inferior ayuda a restaurar la capacidad masticatoria, fonación y estética de la persona que la usa.
La displasia fibrosa ósea es un trastorno poco frecuente del tejido conectivo que afecta el crecimiento y desarrollo normal de los huesos. Esta afección se caracteriza por la formación excesiva de tejido fibroso en lugar del tejido óseo normal durante el proceso de crecimiento y reparación de los huesos. Esto puede llevar a una amplia gama de signos y síntomas, dependiendo de dónde y cuán extensamente se presente la displasia fibrosa ósea en el esqueleto.
Los síntomas más comunes incluyen dolor óseo, hinchazón o sensibilidad en las áreas afectadas, cojera, fracturas espontáneas y deformidades esqueléticas progresivas. En algunos casos, la displasia fibrosa ósea puede afectar la columna vertebral, resultando en una postura encorvada o joroba. También se han informado complicaciones más graves, como insuficiencia cardíaca y problemas respiratorios, cuando la displasia fibrosa ósea afecta los huesos que forman el tórax y restringen la capacidad pulmonar.
Aunque la causa exacta de la displasia fibrosa ósea sigue siendo desconocida, se sabe que está asociada con mutaciones en genes específicos, como GNAS1 y ACVR1. Estas mutaciones pueden heredarse o pueden ocurrir espontáneamente durante el desarrollo embrionario. El diagnóstico de displasia fibrosa ósea generalmente se realiza mediante radiografías, tomografías computarizadas o resonancias magnéticas que muestran las características típicas de la enfermedad en los huesos afectados. El tratamiento suele ser multidisciplinario e incluye medicamentos para aliviar el dolor, fisioterapia y, en algunos casos, cirugía para corregir las deformidades óseas o extirpar tumores benignos asociados con la enfermedad.
Las enfermedades de la uña, también conocidas como patologías ungueales, se refieren a un grupo diverso de condiciones que afectan la integridad estructural y función de las uñas. Estas afecciones pueden afectar tanto a las uñas de las manos como a las de los pies. Las enfermedades de la uña pueden ser el resultado de infecciones, trastornos sistémicos, traumatismos o procesos tumorales.
Algunas de las enfermedades de la uña más comunes incluyen:
1. Onicomicosis: Es una infección fúngica de la uña que puede causar decoloración, engrosamiento, debilitamiento y distorsión de la placa ungueal. Puede ser difícil de tratar y a menudo requiere terapia antifúngica prolongada.
2. Oniquia lunula negra: Se caracteriza por una mancha negra en la lúnula (la media luna blanca en la base de la uña). Por lo general, es el resultado de un hematoma subungueal causado por un traumatismo menor o microtrauma repetitivo.
3. Psoriasis ungueal: La psoriasis puede afectar las uñas, causando decoloración amarillenta, engrosamiento, desprendimiento y picados en la placa ungueal. A menudo se asocia con lesiones psoriásicas en la piel.
4. Paroniquia: Es una infección bacteriana del tejido blando alrededor de la uña. Puede causar enrojecimiento, hinchazón, dolor y pus alrededor de la uña.
5. Onicodistrofia: Se refiere a una alteración en la forma, textura o crecimiento de las uñas. Puede ser el resultado de una variedad de factores, como infecciones, lesiones, trastornos sistémicos y uso de medicamentos.
6. Onicomicosis: Es una infección fúngica de la uña que puede causar decoloración amarillenta, engrosamiento, fragilidad y desprendimiento de la placa ungueal.
7. Uñas encarnadas: Ocurre cuando el borde lateral o la punta de la uña se clava en la piel al crecer, lo que puede causar dolor, enrojecimiento e hinchazón.
8. Linfedema ungueal: Se caracteriza por una hinchazón y engrosamiento de la uña y el tejido blando circundante, a menudo asociado con linfedema crónico.
9. Síndrome del túnel carpiano: Puede causar entumecimiento, hormigueo y debilidad en las manos, que a veces se extienden a las uñas.
10. Enfermedad de Haglund: También conocida como espolón calcáneo, puede causar dolor e hinchazón en el talón y cambios en la forma y el crecimiento de la uña del dedo gordo del pie.
La Proteína de Dominio de Muerte Asociada a Edar (Edward Death Domain Associated protein, EDDA o EDARDD) es un término médico que se refiere a una proteína involucrada en la apoptosis o muerte celular programada. EDARDD es un adaptador citoplásmico que contiene un dominio de muerte (DD) y media la señalización intracelular del receptor de muerte Edar (Edward Death Receptor).
La proteína EDARDD se une al dominio de muerte del receptor Edar y a la proteína FADD (Fas-associated protein with death domain), lo que lleva a la activación de la cascada de señalización que desencadena la apoptosis. Las mutaciones en el gen que codifica para EDARDD se han asociado con enfermedades genéticas, como el síndrome de ectodermal divergente (EDS) y el síndrome de hiperplasia adrenal con defectos del desarrollo ectodérmico (HADES). Estas condiciones se caracterizan por anormalidades en la formación y desarrollo de los tejidos derivados del ectodermo, como la piel, el cabello, las uñas y los dientes.
El labio leporino, también conocido como fisura labial, es una malformación congénita que afecta la línea media de la cara. Se caracteriza por una hendidura o grieta en el labio, que puede variar en longitud desde un simple surco hasta una división completa que se extiende hasta la nariz. La gravedad de la afección puede variar desde formas leves que solo involucran el borde del labio hasta formas más severas que también afectan el paladar (techo de la boca), lo que se conoce como labio leporino y paladar hendido.
Esta condición ocurre durante el desarrollo fetal, alrededor de las cuatro a siete semanas de gestación, cuando los tejidos que forman el rostro del feto no se fusionan correctamente. Aunque la causa exacta es desconocida, se cree que está relacionada con una combinación de factores genéticos y ambientales.
El tratamiento generalmente involucra cirugía para cerrar la hendidura y restaurar la apariencia y función normales del labio y la nariz. La cirugía suele realizarse cuando el bebé tiene entre tres y seis meses de edad. En algunos casos, pueden ser necesarias múltiples intervenciones quirúrgicas para lograr los mejores resultados posibles. Además de la cirugía, también pueden ser necesarios otros tratamientos, como terapia del habla y audición, ortodoncia y counseling psicológico.
Los síndromes de inmunodeficiencia se refieren a un grupo de trastornos en los que el sistema inmunitario está deprimido o ausente, lo que hace que una persona sea más susceptible a las infecciones. Estas enfermedades pueden ser presentes desde el nacimiento (congenitales) o adquiridas más tarde en la vida.
Los síndromes de inmunodeficiencia congénita incluyen, entre otros, el Síndrome de Inmunodeficiencia Adquirida (SIDA), causado por el virus de inmunodeficiencia humana (VIH), y el déficit de adenosina desaminasa (ADA). El SIDA se caracteriza por una destrucción gradual del sistema inmunitario, haciendo que la persona sea vulnerable a diversas infecciones oportunistas y ciertos tipos de cáncer. El déficit de ADA es una enfermedad hereditaria que afecta la capacidad del cuerpo para producir glóbulos blancos normales, aumentando el riesgo de infección.
Los síndromes de inmunodeficiencia adquiridos pueden ser el resultado de diversas causas, como enfermedades, medicamentos o infecciones. Por ejemplo, algunos tratamientos contra el cáncer, como la quimioterapia y la radioterapia, pueden dañar las células del sistema inmunitario y debilitar su función. Algunas enfermedades, como la diabetes y la fibrosis quística, también pueden aumentar el riesgo de infección al afectar negativamente al sistema inmunológico.
En general, los síndromes de inmunodeficiencia se manifiestan con una mayor susceptibilidad a las infecciones, que pueden ser recurrentes o más graves de lo normal. El tratamiento depende de la causa subyacente y puede incluir antibióticos, terapia de reemplazo de células sanguíneas o medicamentos para estimular el sistema inmunitario.
Las anomalías de la boca, también conocidas como defectos de labio y paladar, son afecciones congénitas que ocurren durante el desarrollo fetal. Estas anormalidades pueden variar desde pequeñas hendiduras en el labio o el paladar hasta fusiones completas de los lados de la cara.
Existen tres tipos principales de anomalías de la boca:
1. Fisura labial: Esta es una abertura en el labio superior, que puede extenderse desde la base del nariz hasta la comisura de los labios. Puede afectar un lado (unilateral) o ambos lados (bilateral) del labio.
2. Fisura palatina: Esta es una abertura en el techo de la boca (paladar), que puede extenderse desde la parte posterior de la boca hasta el borde frontal del paladar duro. Puede ocurrir con o sin fisura labial.
3. Síndrome de Pierre Robin: Esta es una combinación de anomalías, incluyendo un paladar hendido, una mandíbula inferior pequeña (micrognatia) y una lengua grande que puede bloquear las vías respiratorias (glosoptosis).
Las causas de estas anomalías no se conocen completamente, pero se cree que son el resultado de una combinación de factores genéticos y ambientales. El consumo de alcohol, el uso de ciertos medicamentos y la exposición a sustancias químicas durante el embarazo pueden aumentar el riesgo de anomalías de la boca.
El tratamiento de las anomalías de la boca generalmente implica cirugía para cerrar la hendidura y restaurar la apariencia y la función normal de la boca. La cirugía suele realizarse durante los primeros meses de vida, aunque el momento exacto depende del tipo y la gravedad de la anomalía. Después de la cirugía, se pueden necesitar otros tratamientos, como terapia del habla y audiología, para ayudar al niño a desarrollarse normalmente.
Las glándulas sudoríparas son glándulas exocrinas que producen sudor, un líquido compuesto principalmente de agua con sales inorgánicas disueltas, productos de desecho y pequeñas cantidades de proteínas y lípidos. Estas glándulas desempeñan un papel vital en la termorregulación del cuerpo, ayudando a mantener una temperatura corporal constante al liberar sudor en la superficie de la piel, el cual se evapora y enfría el cuerpo cuando la temperatura ambiente es superior a la temperatura corporal.
Existen dos tipos principales de glándulas sudoríparas:
1. Glándulas sudoríparas eccrinas: Son las más numerosas y se encuentran distribuidas por toda la superficie de la piel, excepto en las orejas y los labios. Producen un sudor agua y ligeramente alcalino que no contiene proteínas ni lípidos. Las glándulas eccrinas desempeñan un papel importante en la termorregulación y también pueden responder a estímulos emocionales, como el estrés o la ansiedad, lo que provoca sudoración excesiva en las manos, los pies y las axilas.
2. Glándulas sudoríparas apocrinas: Son mucho menos numerosas y se localizan principalmente en las axilas, alrededor de los pezones y en los genitales. Las glándulas apocrinas secretan un sudor espeso y oloroso que contiene proteínas y lípidos. Este tipo de sudor no desempeña un papel significativo en la termorregulación, pero puede interactuar con las bacterias cutáneas para producir compuestos volátiles que causan el característico olor corporal.
Las glándulas sudoríparas están controladas por el sistema nervioso simpático y responden a diversos estímulos, como el calor, el ejercicio, las emociones y las hormonas. La disfunción de las glándulas sudoríparas puede dar lugar a trastornos como la hiperhidrosis (sudoración excesiva) o la anhidrosis (incapacidad para sudar).
La dentición, en términos médicos, se refiere al proceso y el patrón de desarrollo y erupción de los dientes en humanos y otros animales. Es un evento normal del crecimiento que ocurre en etapas específicas, comenzando generalmente desde la infancia hasta la edad adulta.
En los seres humanos, la dentición temporal (conocida como "dientes de leche") comienza alrededor de los 6 meses de edad, donde por lo general erupcionan los incisivos inferiores. Todos los dientes temporales suelen haber erupcionado a la edad de aproximadamente 2-3 años. Posteriormente, entre los 6 y 12 años, se caen gradualmente estos dientes para dar paso a la dentición permanente o definitiva.
La dentición permanente consta de 32 dientes (incluyendo los terceros molares o "muelas del juicio"), aunque no siempre todas éstas últimas logran erupcionar. El patrón y tiempo de erupción pueden variar entre individuos.
La correcta dentición es importante para la función oral, como masticar y hablar, así como también para el desarrollo facial y la salud general. Anomalías en el proceso de dentición, como la agenesia (falta congénita de uno o más dientes) o la displasia (desarrollo anormal de los dientes), pueden requerir tratamientos odontológicos especiales.
En términos médicos, un síndrome se refiere a un conjunto de signos y síntomas que ocurren juntos y pueden indicar una condición particular o enfermedad. Los síndromes no son enfermedades específicas por sí mismos, sino más bien una descripción de un grupo de características clínicas.
Un síndrome puede involucrar a varios órganos y sistemas corporales, y generalmente es el resultado de una combinación de factores genéticos, ambientales o adquiridos. Algunos ejemplos comunes de síndromes incluyen el síndrome de Down, que se caracteriza por retraso mental, rasgos faciales distintivos y problemas de salud congénitos; y el síndrome metabólico, que implica una serie de factores de riesgo cardiovascular como obesidad, diabetes, presión arterial alta e hiperlipidemia.
La identificación de un síndrome a menudo ayuda a los médicos a hacer un diagnóstico más preciso y a desarrollar un plan de tratamiento apropiado para el paciente.
Las Enfermedades Genéticas Ligadas al Cromosoma X (X-Linked Diseases) se refieren a un grupo de trastornos genéticos causados por genes mutados en el cromosoma X. Los hombres suelen ser más afectados que las mujeres, ya que los hombres tienen una sola copia del cromosoma X y una copia del cromosoma Y, mientras que las mujeres tienen dos copias del cromosoma X.
Si un gen en el cromosoma X de un hombre tiene una mutación que causa una enfermedad, él desarrollará la enfermedad porque no tiene otra copia funcional del gen para compensar. Las mujeres que hereden la misma mutación tienen otra copia normal del gen en su otro cromosoma X, por lo que generalmente son menos afectadas o no presentan síntomas en absoluto.
Ejemplos de enfermedades genéticas ligadas al cromosoma X incluyen la hemofilia, la distrofia muscular de Duchenne, el daltonismo y la enfermedad de Fragile X. Estas condiciones pueden afectar gravemente la vida y tienen diferentes grados de severidad. La herencia de estas enfermedades se produce siguiendo un patrón recesivo ligado al cromosoma X, lo que significa que para que un hombre desarrolle la enfermedad, solo necesita heredar el gen anormal de su madre. Las mujeres, por otro lado, necesitan heredar el gen anormal de ambos padres para desarrollar la enfermedad.
En el contexto de la medicina y la biología, un linaje se refiere a una sucesión o serie de organismos relacionados genéticamente que descienden de un antepasado común más reciente. Puede hacer referencia a una secuencia particular de genes que se heredan a través de generaciones y que ayudan a determinar las características y rasgos de un organismo.
En la genética, el linaje mitocondrial se refiere a la línea de descendencia materna, ya que las mitocondrias, que contienen su propio ADN, se transmiten generalmente de madre a hijo. Por otro lado, el linaje del cromosoma Y sigue la línea paterna, ya que los cromosomas Y se heredan del padre y se mantienen intactos durante la meiosis, lo que permite rastrear la ascendencia masculina.
Estos linajes pueden ser útiles en la investigación genética y antropológica para estudiar la evolución y la migración de poblaciones humanas y otras especies.
Las deformidades congénitas de las extremidades se refieren a anomalías estructurales o funcionales presentes en los miembros superiores e inferiores al momento del nacimiento. Estas condiciones pueden variar desde malformaciones menores hasta problemas graves que afectan la capacidad de una persona para usar sus extremidades de manera normal.
Las causas de estas deformidades pueden ser genéticas, ambientales o resultado de una combinación de factores. Algunos ejemplos comunes de deformidades congénitas de las extremidades incluyen:
1. Displasia esquelética: Es una condición que afecta el desarrollo normal del hueso y puede resultar en extremidades cortas, anormales o deformadas. Un ejemplo es la acondroplasia, que es una forma común de enanismo.
2. Artrogriposis múltiple congénita: Esta es una condición que causa rigidez articular y contracturas musculares en varias articulaciones del cuerpo, incluyendo las extremidades.
3. Síndrome de Poland: Es una anomalía congénita caracterizada por la ausencia parcial o total del músculo pectoral mayor en el pecho, junto con afectación variable de los huesos, músculos, nervios y vasos sanguíneos de la extremidad superior.
4. Hemimelia: Es una malformación congénita donde falta todo o parte de un hueso en una extremidad. Por ejemplo, la hemimelia fibular es una afección en la que falta parte o todo el hueso fibular de la pierna.
5. Luxación congénita: Se trata de una luxación (desalineación) permanente de una articulación presente al nacer, como la luxación congénita de cadera.
El tratamiento para las deformidades congénitas de las extremidades depende del tipo y gravedad de la afección. Puede incluir terapia física, dispositivos ortopédicos, cirugía o una combinación de estos. El objetivo es mejorar la función y apariencia de la extremidad afectada y prevenir complicaciones futuras.
La displasia broncopulmonar (DBP) es una afección pulmonar crónica que afecta principalmente a los bebés prematuros. Se caracteriza por una inflamación y un crecimiento anormal de los conductos bronquiales (vías respiratorias más grandes en los pulmones) y los alvéolos (sacos de aire pequeños en los pulmones donde ocurre el intercambio de oxígeno y dióxido de carbono).
La DBP puede variar en gravedad desde formas leves que solo requieren oxígeno suplementario hasta formas más graves que pueden causar insuficiencia respiratoria crónica. Los síntomas más comunes incluyen dificultad para respirar, sibilancias, tos, infecciones pulmonares recurrentes y retraso del crecimiento.
La DBP es una enfermedad multifactorial que puede ser causada por una combinación de factores, como la prematuridad, la infección pulmonar, la exposición a oxígeno suplementario durante un largo período de tiempo y los daños causados por los ventiladores mecánicos. El tratamiento puede incluir oxígeno suplementario, broncodilatadores, corticosteroides y, en algunos casos, cirugía. La fisioterapia respiratoria y la nutrición adecuada también son importantes para el manejo de esta enfermedad.
La fisura del paladar, también conocida como fisura palatina o labio leporino con fisura palatina, es un defecto congénito en la boca y el rostro. Se produce durante el desarrollo fetal, alrededor de la séptima a la novena semana de embarazo, cuando los tejidos que forman el labio y el paladar no se fusionan completamente.
En una fisura del paladar, hay una abertura entre el lado izquierdo y derecho del paladar (el techo blando de la boca), lo que puede extenderse a través del labio superior e incluso afectar la nariz. Esto puede dar lugar a problemas en la alimentación, el habla, el crecimiento dental y los oídos.
La gravedad de la fisura del paladar varía de una persona a otra. Algunas personas pueden tener solo una pequeña abertura, mientras que otras pueden tener una fisura extensa que involucra tanto el labio como el paladar. El tratamiento generalmente implica cirugía para cerrar la fisura y restaurar la apariencia y la función normal de la boca y el rostro.
Las anomalías múltles son una condición médica en la que un individuo presenta más de una anomalía congénita o malformación. Estas anomalías pueden afectar diferentes partes del cuerpo y pueden variar en gravedad desde leves hasta graves.
Las causas de las anomalías múltiples pueden ser genéticas, ambientales o una combinación de ambas. Algunos ejemplos de síndromes que involucran anomalías múltiples incluyen el síndrome de Down, el síndrome de Turner y el síndrome de Noonan.
El tratamiento para las anomalías múltiples depende del tipo y la gravedad de las malformaciones. Puede incluir cirugía, terapia física o ocupacional, y management médico a largo plazo. En algunos casos, el pronóstico puede ser favorable con un tratamiento y manejo adecuados, mientras que en otros casos las anomalías múltiples pueden ser letales.
Es importante que los individuos con anomalías múltiples reciban atención médica especializada y seguimiento regular para garantizar la mejor calidad de vida posible.
El cromosoma X es uno de los dos cromosomas sexuales en humanos (el otro es el cromosoma Y), que juegan un papel fundamental en la determinación del sexo. Las mujeres tienen dos cromosomas X (llamadas genotipo XX) y los hombres tienen un cromosoma X y un cromosoma Y (genotipo XY).
Los cromosomas X contienen alrededor de 155 millones de pares de bases y representan aproximadamente el 5% del ADN total en las células somáticas. Contiene entre un 1,5 y un 2 por ciento más de genes que el cromosoma Y y codifica para alrededor de 1.500 proteínas diferentes.
El cromosoma X también contiene una gran cantidad de ADN repetitivo y pseudogenes, así como regiones no codificantes reguladoras importantes que controlan la expresión génica. Además, el cromosoma X presenta un fenómeno llamado inactivación del cromosoma X, en el que uno de los dos cromosomas X se comprime y silencia en cada célula somática femenina, lo que garantiza que las mujeres expresen cantidades aproximadamente iguales de genes del cromosoma X que los hombres.
Las mutaciones en los genes del cromosoma X pueden causar una variedad de trastornos genéticos, como la hemofilia, el daltonismo y la distrofia muscular de Duchenne. Estos trastornos se denominan a menudo enfermedades ligadas al cromosoma X porque los hombres, que solo tienen un cromosoma X, tienen más probabilidades de desarrollarlas que las mujeres, quienes tienen dos copias del cromosoma X y por lo tanto una copia de respaldo en caso de que haya una mutación.
El término "diseño de dentadura" no es un término médico específico, pero supongo que te refieres a algo similar al "diseño de prótesis dental" o "plan de tratamiento protésico".
Un diseño de prótesis dental o plan de tratamiento protésico es un proceso en el que se crea una solución artificial para reemplazar dientes perdidos o dañados. Esto puede incluir la fabricación y colocación de dentaduras postizas completas o parciales, puentes fijos o removibles, implantes dentales y otros dispositivos protésicos.
El proceso generalmente comienza con una evaluación exhaustiva de la boca del paciente, incluyendo radiografías y modelos de estudio para determinar las mejores opciones de tratamiento. Luego, se crea un plan de tratamiento detallado que describe los procedimientos necesarios, el tipo de prótesis dental recomendada y los costos asociados.
La prótesis dental se diseña y fabrica utilizando materiales como resina acrílica, metal o porcelana. El objetivo es crear una prótesis que sea cómoda, funcional y estéticamente agradable para el paciente. Después de la colocación, se realizan ajustes adicionales si es necesario para garantizar un ajuste perfecto y una buena función.
Las Enfermedades del Desarrollo Óseo (EDO) son un grupo de trastornos que afectan el crecimiento y desarrollo normal de los huesos. Estas condiciones pueden ser presentes desde el nacimiento o adquirirse más tarde en la vida, y pueden ser causadas por una variedad de factores, incluyendo genéticos, hormonales, nutricionales y ambientales.
Las EDO se caracterizan por anomalías en la forma, tamaño, densidad y fuerza de los huesos. Pueden afectar a uno o más huesos y pueden causar una variedad de síntomas, dependiendo de la gravedad y la ubicación de la afección. Algunos de los síntomas más comunes incluyen dolor óseo, deformidades esqueléticas, crecimiento anormal, fracturas espontáneas o repetidas, y discapacidad física.
Algunos ejemplos de EDO incluyen la enfermedad de Paget del hueso, la osteogénesis imperfecta, la displasia fibrosa poliestóptica, el síndrome de Marfan, y el síndrome de Hurler. El tratamiento de estas condiciones depende del tipo y la gravedad de la afección, pero puede incluir medicamentos, terapia física, cirugía ortopédica y cambios en el estilo de vida.
Las queratinas tipo II son un subgrupo de proteínas estructurales fibrosas llamadas queratinas que se encuentran en los epitelios escamosos estratificados y las células de la capa externa de la piel humana, conocida como epidermis. Estas queratinas desempeñan un papel crucial en el proceso de keratinización, proporcionando resistencia y rigidez a la superficie exterior de la piel.
Las queratinas tipo II se clasifican como queratinas intermedias y forman filamentos intermedios junto con las queratinas tipo I. Los dos tipos de queratinas se unen entre sí para formar heterodímeros, que luego se agrupan en grupos más grandes llamados filamentos intermedios. Estos filamentos intermedios son responsables de proporcionar integridad estructural a las células epiteliales y desempeñan un papel importante en la resistencia mecánica de la piel.
Las queratinas tipo II se expresan en una variedad de tejidos, incluidos los folículos pilosos, las uñas y las glándulas sudoríparas eccrinas. La expresión de diferentes tipos de queratina tipo II varía durante el desarrollo y la diferenciación celular, lo que sugiere que desempeñan funciones específicas en diferentes etapas del crecimiento y desarrollo celular.
Las mutaciones en los genes que codifican las queratinas tipo II se han relacionado con varias afecciones cutáneas, como la epidermólisis ampollosa adquirida y hereditaria, el síndrome de Debré-acantosis-pseudocélulas y la displasia epidérmica adquirida. Estas enfermedades se caracterizan por fragilidad cutánea, ampollas y lesiones en la piel.
El cabello es una fibra proteica muerta y queratinizada que se encuentra en la piel de los mamíferos. En humanos, el crecimiento del cabello ocurre en folículos pilosos específicos en la dermis (capa interna de la piel). El cabello está compuesto principalmente por una proteína llamada queratina y está formado por tres partes: el tallo, la cutícula y la raíz.
El tallo del cabello es la parte visible que sobresale de la superficie de la piel. La cutícula es la capa externa protectora del tallo del cabello, compuesta por células escamosas aplanadas que se superponen unas a otras como las tejas de un tejado. La raíz del cabello está ubicada en el folículo piloso y es la parte responsable del crecimiento del cabello.
El ciclo de vida del cabello consta de tres fases: crecimiento (anágena), transición (catágena) y caída (telógena). El crecimiento del cabello ocurre durante la fase anágena, que dura aproximadamente entre 2 a 6 años y el cabello puede crecer hasta una longitud de entre 18 a 30 pulgadas. Durante la fase catágena, que dura alrededor de 2-3 semanas, el crecimiento del cabello se detiene y la raíz se desprende del suministro de sangre. Finalmente, en la fase telógena, que dura aproximadamente de 100 a 150 días, el folículo piloso permanece inactivo y el cabello cae, comenzando un nuevo ciclo de crecimiento.
El crecimiento y la apariencia del cabello pueden verse afectados por diversos factores, como la genética, la edad, las hormonas, los medicamentos, el estrés y las enfermedades. Las condiciones médicas que pueden causar pérdida de cabello incluyen la alopecia areata, la alopecia androgenética, la deficiencia nutricional y el hipertiroidismo, entre otras. El tratamiento de la pérdida de cabello depende de la causa subyacente y puede incluir medicamentos, terapias hormonales, cambios en el estilo de vida y, en algunos casos, procedimientos quirúrgicos como el trasplante de cabello.
Las enfermedades del cabello se refieren a diversas condiciones que pueden afectar la apariencia, el crecimiento y la salud general del cabello. Estas afecciones pueden ser el resultado de factores genéticos, infecciosos, nutricionales o ambientales. Algunos ejemplos comunes de enfermedades del cabello incluyen:
1. Alopecia Areata: Una enfermedad autoinmune que causa la pérdida de pelo en parches redondos en el cuero cabelludo o cualquier otra parte del cuerpo donde haya pelo.
2. Caída del Cabello (Efluvio Telógeno): La caída excesiva del cabello que puede ser causada por factores estresantes físicos o emocionales, cambios hormonales, medicamentos o enfermedades graves.
3. Folliculitis: Inflamación e infección de los folículos pilosos, generalmente causados por bacterias u hongos. Puede presentarse como pequeños granos, pústulas o protuberancias en el cuero cabelludo.
4. Pediculosis Capitis (Piojos de la Cabeza): Infestación parasitaria del cuero cabelludo por piojos, que se alimentan de la sangre del scalp y causan picazón intensa.
5. Psoriasis Capitis: Una afección cutánea crónica que causa enrojecimiento, descamación e inflamación del cuero cabelludo.
6. Seborrea: Un trastorno de la piel que afecta el cuero cabelludo y provoca caspa excesiva, picazón y enrojecimiento. En casos graves, puede causar calvicie temporal.
7. Tricoptilosis (Puntas Partidas): Un trastorno del cabello que causa las puntas abiertas o partidas, lo que hace que el cabello sea quebradizo y propenso a romperse fácilmente.
8. Tricofilia: Un trastorno poco común en el que una persona se arranca deliberadamente el cabello, causando calvicie y daño al folículo piloso.
9. Tricosquisis (Cabello Enredado): Un trastorno del cabello que causa nudos apretados y difíciles de deshacer, especialmente en personas con cabello rizado o ondulado.
10. Triquiasis: Condición en la que las pestañas crecen hacia adentro, irritando el ojo y provocando lagrimeo excesivo, enrojecimiento e infecciones oculares.
La incontinencia pigmentaria es una afección rara y progresiva de la piel y el sistema nervioso. Se caracteriza por manchas irregulares de color marrón o negro en la piel (lentigos), hiperpigmentación en las palmas de las manos y las plantas de los pies, y despigmentación en otras áreas de la piel. También puede afectar al sistema nervioso, causando problemas neurológicos como ataxia, parkinsonismo o demencia.
La enfermedad se hereda de forma autosómica recesiva, lo que significa que una persona debe heredar dos copias del gen defectuoso (una de cada padre) para desarrollar la afección. La incontinencia pigmentaria está causada por mutaciones en varios genes diferentes, incluyendo el PIGA, PIGT y PCBP2.
El tratamiento de la incontinencia pigmentaria se centra generalmente en aliviar los síntomas y mejorar la calidad de vida del paciente. Puede incluir terapia física para ayudar con los problemas de movilidad, medicamentos para controlar los espasmos musculares o la rigidez, y terapias para ayudar con los problemas cognitivos. En algunos casos, se puede considerar un trasplante de células madre hematopoyéticas como tratamiento.
La queratodermia palmoplantar es un término médico que se refiere a un grupo de trastornos dermatológicos caracterizados por el engrosamiento y endurecimiento (hiperqueratosis) de la piel en las palmas de las manos y/o plantas de los pies. Esta condición puede ser congénita o adquirida, y su gravedad varía desde formas leves y asintomáticas hasta formas severas que pueden causar discapacidad funcional.
La queratodermia palmoplantar puede presentarse como una enfermedad aislada o como parte de un síndrome más complejo que involucra otros órganos y sistemas corporales. Algunas de las causas conocidas incluyen trastornos genéticos, exposición a ciertos químicos y fármacos, infecciones virales y enfermedades sistémicas como el cáncer y la diabetes.
El diagnóstico de queratodermia palmoplantar generalmente se realiza mediante una evaluación clínica y, a veces, con el apoyo de pruebas adicionales, como biopsias cutáneas o análisis genéticos. El tratamiento puede incluir medidas generales, como el uso de humectantes y la eliminación regular del exceso de piel engrosada, así como terapias específicas, como el uso de retinoides orales o tópicos, fototerapia y cirugía. El pronóstico varía según la causa subyacente y la gravedad de la enfermedad.
La hiperpigmentación es un término médico que se refiere al oscurecimiento de la piel. Este proceso ocurre cuando hay un exceso de producción de melanina, el pigmento que da color a la piel, los cabellos y los ojos. La melanina es producida por células llamadas melanocitos. Cualquier condición que cause a estas células a producir más melanina puede resultar en hiperpigmentación.
La hiperpigmentación puede ser el resultado de diversos factores, como la exposición al sol, las lesiones cutáneas, los trastornos hormonales, ciertos medicamentos y enfermedades de la piel. Algunas condiciones específicas que pueden causar hiperpigmentación incluyen el melasma, el lentigo solar (manchas solares), la dermatitis de contacto y la pitiriasis versicolor.
La hiperpigmentación es generalmente inofensiva, aunque puede ser una fuente de preocupación estética para algunas personas. Existen tratamientos disponibles para reducir o eliminar la hiperpigmentación, como cremas despigmentantes, peelings químicos y láseres dérmicos. Sin embargo, es importante tratar la causa subyacente de la hiperpigmentación para prevenir su recurrencia.
Las deformidades congénitas de la mano se refieren a un grupo diverso de anomalías estructurales y funcionales presentes en el nacimiento que afectan la morfología y función de la mano. Estas condiciones pueden variar desde leves a graves, y pueden afectar a uno o ambos lados.
Las causas de las deformidades congénitas de la mano pueden ser genéticas, adquiridas o debido a factores ambientales durante el desarrollo fetal. Algunas de estas condiciones pueden estar asociadas con síndromes o trastornos genéticos más amplios.
Los ejemplos comunes de deformidades congénitas de la mano incluyen: polidactilia (dedos adicionales), sindactilia (dedos fusionados), braquidactilia (dedos cortos), clinodactilia (curvatura anormal de los dedos), hipoplasia (bajo desarrollo) o aplasia (ausencia) de parte o todo el esqueleto de la mano, y anomalías en la posición o alineación de los huesos y articulaciones de la mano.
El tratamiento de las deformidades congénitas de la mano depende del tipo y gravedad de la afección. Puede incluir observación, terapia ocupacional, fisioterapia, ortesis o dispositivos de asistencia, y en algunos casos, cirugía reconstructiva. El objetivo del tratamiento es mejorar la función y apariencia estética de la mano, así como prevenir complicaciones futuras.
Ectromelia, también conocida como Rattfinger o enfermedad del dedo de ratón, es una enfermedad viral que afecta principalmente a los roedores. Es causada por el virus de la ectromelia (ECTV), un miembro del género Orthopoxvirus, que también incluye el virus de la viruela humana y el virus de la viruela del mono.
La enfermedad se caracteriza por lesiones cutáneas, ulceraciones y necrosis en los tejidos. Puede causar malformaciones en las extremidades de los roedores nacidos de madres infectadas, como falta total o parcial de una o más extremidades (ectromelia literalmente significa "extremidad cortada" en griego).
Aunque es principalmente una enfermedad de los roedores, se ha demostrado que el virus de la ectromelia puede infectar a otros animales y, en raras ocasiones, a humanos. Sin embargo, no representa una amenaza importante para la salud humana.
En un contexto médico, el término 'ectromelia' se utiliza para describir una malformación congénita en la que una extremidad está parcial o totalmente ausente.
En genética, los genes recesivos son aquellos que para expresar su fenotipo (característica visible) necesitan que las dos copias del gen (una heredada de cada padre) sean idénticas y exhiben este gen. Si un individuo tiene una sola copia de un gen recesivo, no mostrará el rasgo asociado con ese gen, ya que el gen dominante cubre o encubre la expresión del gen recesivo. Los genes recesivos solo se manifiestan en la ausencia de un gen dominante. Esto significa que ambos padres pueden no mostrar el rasgo fenotípico, pero aún pueden llevar y pasar el gen recesivo a su descendencia. Un ejemplo común de genes recesivos son los asociados con la enfermedad de la fibrosis quística o la anemia falciforme.
La quinasa I-kappa B (IKK) es una enzima que desempeña un papel crucial en la activación del factor nuclear kappa B (NF-kB), un importante regulador de la respuesta inflamatoria y del sistema inmunitario. La IKK fosforila específicamente los residuos de serina en las proteínas inhibidoras I-kappa B (IkB), lo que provoca su posterior degradación por parte del proteasoma y la liberación y activación subsiguiente del NF-kB. Este proceso es fundamental para la translocación nuclear del NF-kB y la transcripción de genes diana involucrados en respuestas inmunitarias, inflamatorias y celulares al estrés. La IKK está compuesta por tres subunidades: IKKα (IKK1), IKKβ (IKK2) y NEMO (IKKγ), y su activación puede ocurrir a través de diversos estímulos, como citocinas proinflamatorias, radicales libres, radiación UV y agentes infecciosos. La regulación adecuada de la IKK es esencial para mantener el equilibrio homeostático y prevenir enfermedades inflamatorias y autoinmunes.
La fotofobia es una condición médica en la cual un individuo experimenta una sensibilidad excesiva a la luz, especialmente a la luz brillante. La persona puede sentir incomodidad o dolor en los ojos cuando están expuestos a la luz, ya sea natural del sol o artificial de luces eléctricas. Esta reacción exagerada se debe generalmente a una inflamación o irritación en la superficie o interior del ojo, o puede ser un síntoma de ciertas afecciones oculares subyacentes.
La fotofobia no es en sí misma una enfermedad, sino más bien un signo asociado con diversas condiciones oftalmológicas y neurológicas. Algunos de los trastornos que pueden causar fotofobia incluyen conjuntivitis, queratitis, uveítis, meningitis, migrañas y ciertos tipos de lesiones cerebrales. El tratamiento de la fotofobia implica abordar la afección subyacente que está causando la sensibilidad a la luz.
El síndrome de Ellis-Van Creveld es un trastorno genético extremadamente raro que afecta el crecimiento y el desarrollo. Se caracteriza por enanismo (bajo crecimiento), anomalías esqueléticas, problemas cardíacos congénitos, uñas de las manos y los pies anormales y paladar hendido.
Las personas afectadas suelen tener extremidades cortas con dedos cortos y anchos, costillas cortas y un tórax pequeño. También pueden tener una lengua bífida (lengua dividida) y dientes anormales. Los problemas cardíacos más comunes asociados con este síndrome son defectos del septo atrial y ventricular, que son aberturas anómalas en las paredes entre las cámaras superiores e inferiores del corazón, respectivamente.
Este síndrome es causado por mutaciones en genes específicos (EVC y EVC2) y se hereda con un patrón autosómico recesivo, lo que significa que una persona debe heredar dos copias del gen alterado (una de cada padre) para tener la afección. El síndrome de Ellis-Van Creveld afecta aproximadamente a 1 de cada 60.000 recién nacidos vivos en poblaciones generalmente no aisladas.
En terminología dental, una "dentadura completa" se refiere a un conjunto completo de prótesis dentales que están diseñadas para reemplazar la totalidad de los dientes en un maxilar superior o inferior. Una dentadura completa upper (maxilar) consta de 14 a 16 dientes, mientras que una dentadura completa lower (mandíbula) tiene generalmente 12 a 14 dientes. Estos dientes de reemplazo están hechos típicamente de resina acrílica y se colocan sobre una base de plástico que se ajusta a la encía del paciente. Las dentaduras completas pueden ser removibles, lo que significa que el usuario puede quitarlas fácilmente para su limpieza y mantenimiento, o pueden ser support-ed por implantes dentales para una mejor sujeción y funcionalidad. La pérdida total de dientes es comúnmente causada por caries dental no tratadas, enfermedad de las encías avanzada, traumatismos o edentulismo congénito. Las dentaduras completas ayudan a restaurar la capacidad de masticación, mejorar la apariencia estética y aumentar la confianza en uno mismo del paciente.
La alopecia es una condición médica que se caracteriza por la pérdida parcial o completa del cabello en la cabeza, cejas, barba u otras partes del cuerpo. Existen diferentes tipos y causas de alopecia, y su diagnóstico y tratamiento pueden variar dependiendo del tipo específico.
El tipo más común de alopecia es la alopecia androgenética, también conocida como calvicie común, que afecta principalmente a los hombres pero también puede ocurrir en las mujeres. Esta forma de alopecia está relacionada con factores genéticos y hormonales y se caracteriza por un patrón específico de pérdida de cabello en la parte superior e inferior de la cabeza.
Otro tipo común de alopecia es la alopecia areata, que se produce cuando el sistema inmunológico ataca los folículos pilosos, causando la caída del cabello en parches redondos y bien definidos. La alopecia areata puede ocurrir en cualquier parte del cuerpo y afecta tanto a hombres como a mujeres de todas las edades.
La alopecia universal es una forma más grave de alopecia areata que causa la pérdida completa del cabello en todo el cuerpo, incluyendo cejas, pestañas y vello corporal. La alopecia totalis es una forma similar que solo afecta el cuero cabelludo.
El tratamiento de la alopecia depende del tipo y gravedad de la afección. Algunos casos pueden resolverse por sí solos, mientras que otros pueden requerir tratamientos médicos, como corticosteroides tópicos o inyectables, terapia de luz o incluso trasplantes de cabello. En algunos casos, el uso de pelucas, postizos o maquillaje permanente puede ayudar a mejorar la apariencia estética.
En resumen, la alopecia es una afección que causa la pérdida del cabello en diversas áreas del cuerpo y puede tener diferentes causas y presentaciones. El tratamiento depende del tipo y gravedad de la afección y puede incluir opciones médicas o cosméticas. Si experimenta pérdida de cabello inexplicable, es recomendable consultar con un especialista en el tema para obtener un diagnóstico y tratamiento adecuados.
La displasia del cuello del útero, también conocida como displasia cervical, es un trastorno precanceroso que afecta el revestimiento del cuello del útero. Se caracteriza por el crecimiento anormal y desorganizado de células en este área, el cual suele ser detectado mediante pruebas de detección como el Papanicolaou (Pap).
Existen diferentes grados de displasia cervical, clasificados según la gravedad del crecimiento celular anormal:
1. Displasia leve (CIN I): Las células anormales se encuentran únicamente en la capa más superficial del cuello del útero.
2. Displasia moderada (CIN II): El crecimiento celular anormal afecta a las capas intermedias del cuello del útero.
3. Displasia severa (CIN III) o carcinoma in situ: Las células anormales se extienden hasta la base del cuello del útero, pero no han invadido los tejidos circundantes.
La displasia del cuello del útero es generalmente causada por una infección persistente con el virus del papiloma humano (VPH), que se transmite sexualmente. Existen más de 100 tipos diferentes de VPH, y aproximadamente 13 de ellos están relacionados con el desarrollo de displasia cervical y cáncer cervical.
El tratamiento de la displasia del cuello del útero depende del grado de afección, la edad de la paciente, y otros factores. Las opciones de tratamiento pueden incluir:
- Seguimiento cuidadoso con pruebas de Papanicolaou regulares
- Escisión o destrucción de las células anormales mediante procedimientos como la conización o la crioterapia
- Extirpación quirúrgica del útero (histerectomía) en casos más graves o recurrentes.
La prevención es fundamental para reducir el riesgo de desarrollar displasia cervical y cáncer cervical. Las medidas preventivas incluyen:
- Vacunación contra el VPH: Se recomienda la vacunación contra el VPH a las niñas y los niños entre los 11 y los 12 años de edad, aunque también se puede administrar hasta los 45 años. La vacuna protege contra los tipos más comunes del virus que causan el cáncer cervical y otras enfermedades relacionadas con el VPH.
- Uso de preservativos: El uso correcto y consistente de preservativos puede ayudar a reducir el riesgo de transmisión del VPH durante las relaciones sexuales, aunque no elimina por completo el riesgo.
- Pruebas de detección regulares: Las pruebas de Papanicolao pueden detectar cambios precancerosos en el cuello del útero, lo que permite un tratamiento temprano y evita la progresión a cáncer cervical. Se recomienda iniciar las pruebas de detección a los 21 años o dentro de los tres años posteriores al inicio de la actividad sexual, lo que ocurra primero. Las mujeres entre 25 y 65 años deben realizarse una prueba de Papanicolao cada tres a cinco años, dependiendo de su edad y resultados previos.
En términos médicos, una mutación se refiere a un cambio permanente y hereditable en la secuencia de nucleótidos del ADN (ácido desoxirribonucleico) que puede ocurrir de forma natural o inducida. Esta alteración puede afectar a uno o más pares de bases, segmentos de DNA o incluso intercambios cromosómicos completos.
Las mutaciones pueden tener diversos efectos sobre la función y expresión de los genes, dependiendo de dónde se localicen y cómo afecten a las secuencias reguladoras o codificantes. Algunas mutaciones no producen ningún cambio fenotípico visible (silenciosas), mientras que otras pueden conducir a alteraciones en el desarrollo, enfermedades genéticas o incluso cancer.
Es importante destacar que existen diferentes tipos de mutaciones, como por ejemplo: puntuales (sustituciones de una base por otra), deletérreas (pérdida de parte del DNA), insercionales (adición de nuevas bases al DNA) o estructurales (reordenamientos más complejos del DNA). Todas ellas desempeñan un papel fundamental en la evolución y diversidad biológica.
El ligamiento genético, en términos médicos, se refiere al fenómeno en el que dos o más loci (regiones específicas del ADN) en un cromosoma tienden a heredarse juntos durante la reproducción porque están demasiado próximos entre sí para ser separados por el proceso de recombinación genética. La medida de cuán a menudo se heredan juntos se expresa como una unidad llamada "unidades de mapa centimorgan" (cM), que refleja la probabilidad de recombinación entre ellos. Cuanto más cerca estén los loci uno del otro en un cromosoma, mayor será su ligamiento y menor será la probabilidad de recombinación entre ellos. Por lo tanto, el ligamiento genético proporciona información importante sobre la ubicación relativa y la organización de los genes en un cromosoma.
Las enfermedades de las glándulas sudoríparas se refieren a un grupo de trastornos que afectan el funcionamiento normal de estas glándulas, las cuales son responsables de producir y secretar el sudor. Existen dos tipos principales de glándulas sudoríparas en el cuerpo humano: eccrinas y apocrinas.
Las glándulas sudoríparas eccrinas se encuentran distribuidas por toda la superficie corporal y secretan un sudor agua, sal e inodoro que ayuda a regular la temperatura corporal. Las enfermedades que afectan a estas glándulas incluyen:
1. Hiperhidrosis: es una afección en la cual una persona suda más de lo normal, incluso sin razón aparente o en respuesta al calor o el ejercicio físico. Puede afectar a diferentes partes del cuerpo, como las manos, los pies, las axilas y la cara.
2. Anhidrosis: es una enfermedad en la que una persona no puede sudar normalmente o no suda en absoluto. Puede ser causada por lesiones en la piel, quemaduras graves, enfermedades neurológicas o el uso de ciertos medicamentos.
Las glándulas sudoríparas apocrinas se encuentran principalmente en las axilas y la ingle y secretan un sudor más espeso y oloroso que se mezcla con las bacterias de la piel para producir el olor corporal. Las enfermedades que afectan a estas glándulas incluyen:
1. Bromhidrosis: es una afección en la cual una persona produce un sudor excesivamente oloroso, incluso sin haber realizado ejercicio físico o estar bajo estrés.
2. Moluscum contagiosum: es una infección viral de la piel que puede causar lesiones elevadas y llenas de líquido en las axilas y otras partes del cuerpo donde hay glándulas sudoríparas apocrinas.
El tratamiento de estas enfermedades depende de su causa y gravedad. Puede incluir medicamentos, terapias hormonales, cirugía o cambios en el estilo de vida. Es importante consultar a un médico si se experimentan síntomas relacionados con las glándulas sudoríparas para recibir un diagnóstico y tratamiento adecuados.
La displasia fibromuscular (DFM) es un trastorno que afecta principalmente a los vasos sanguíneos pequeños y medianos, especialmente las arterias renales y carótidas. Se caracteriza por el crecimiento anormal de células musculares y fibrosas en la pared de los vasos sanguíneos, lo que puede causar estrechamiento (estenosis), dilatación (aneurisma) o both de las arterias afectadas.
La DFM se clasifica en tres tipos principales según el patrón de afectación:
1. Tipo I (también conocido como tipo medial): Afecta predominantemente a la capa media de la pared arterial, con múltiples anillos de engrosamiento y estrechamiento alternando con áreas normales o dilatadas.
2. Tipo II (también conocido como tipo intimomedial): Afecta a las capas media e íntima de la pared arterial, causando un engrosamiento difuso y continuo de estas capas.
3. Tipo III (también conocido como tipo periférico): Se limita a la capa más externa de la pared arterial (la adventicia) y se caracteriza por engrosamientos irregulares y estrechamientos focales.
La displasia fibromuscular puede causar una variedad de síntomas, dependiendo de la gravedad y la ubicación de las lesiones arteriales. Algunos de los síntomas más comunes incluyen:
- Hipertensión arterial resistente al tratamiento
- Dolor de cabeza (especialmente dolores de cabeza en racimo o migrañas)
- Síncope (desmayos)
- Pulsos débiles o ausentes en las extremidades
- Claudicación intermitente (dolor en las piernas al caminar)
- Dolor torácico
- Insuficiencia renal crónica
El diagnóstico de la displasia fibromuscular generalmente se realiza mediante una combinación de estudios de imagen, como angiografía por resonancia magnética (ARM), tomografía computarizada (TC) o angiografía digital subtráctiva (DSA). El tratamiento puede incluir medicamentos para controlar la presión arterial y aliviar los síntomas, así como procedimientos quirúrgicos o endovasculares para reparar las lesiones arteriales.
Una dentadura parcial, también conocida como prótesis parcial removible, es un dispositivo protésico utilizado en odontología para reemplazar uno o varios dientes naturales que faltan. Está diseñada para adaptarse cómodamente y firmemente a la encía y los dientes restantes del paciente, proporcionando así soporte y estabilidad.
Las dentaduras parciales suelen estar hechas de una combinación de materiales, como acrílicos, metal o nylon reforzado con fibras, que brindan resistencia y durabilidad. Presentan reemplazos de dientes artificiales colocados estratégicamente en la base de la prótesis, que se asemeja a las encías del paciente.
Este tipo de prótesis permite al paciente masticar, hablar y sonreír con confianza, además de prevenir el desplazamiento o cambio de posición de los dientes naturales restantes, evitando problemas como la maloclusión o pérdida ósea adicional. Las dentaduras parciales se pueden quitar fácilmente para su limpieza y mantenimiento regular.
La consanguinidad es un término utilizado en genética y medicina que se refiere a la relación de parentesco entre dos personas que descienden de un antepasado común. Cuanto más reciente sea el ancestro común, mayor será el grado de consanguinidad.
La consanguinidad aumenta la probabilidad de que dos personas compartan genes recesivos para ciertas características o enfermedades, ya que tienen una mayor probabilidad de heredar los mismos alelos (variantes de un gen) de su antepasado común.
Cuando dos personas consanguíneas se aparean y tienen hijos, existe un riesgo aumentado de que sus hijos nazcan con trastornos genéticos recesivos, especialmente si los padres están emparentados en grado cercano, como primos o más cercanos.
Es importante destacar que la consanguinidad no es sinónimo de infertilidad o de malformaciones congénitas, sino que simplemente aumenta el riesgo de presentarlas. Además, existen programas de consejo genético y pruebas diagnósticas prenatales que pueden ayudar a identificar y gestionar los riesgos asociados con la consanguinidad.
Las osteocondrodisplasias son un grupo heterogéneo de trastornos esqueléticos congénitos que afectan el crecimiento y desarrollo del hueso y el cartílago. Estos trastornos están asociados con anomalías en la diferenciación y función de los condrocitos, las células responsables del crecimiento y mantenimiento del tejido cartilaginoso. La mayoría de las osteocondrodisplasias se heredan de forma autosómica dominante o recesiva.
Las manifestaciones clínicas de estas enfermedades varían desde leves a graves y pueden incluir displasia esquelética, disostosis (malformación ósea), enanismo, trastornos de la columna vertebral, luxaciones congénitas de caderas, deformidades articulares, y anomalías faciales y craneales. Algunos tipos de osteocondrodisplasias también pueden presentar problemas respiratorios, cardíacos o neurológicos.
Existen más de 450 tipos diferentes de osteocondrodisplasias, cada uno con características específicas y patrones hereditarios distintivos. Algunos ejemplos comunes incluyen la acondroplasia, el síndrome de Down, el síndrome de Turner, la displasia espondiloepifisaria congénita y la enfermedad de Morquio. El diagnóstico se realiza mediante una combinación de examen físico, radiografías, pruebas genéticas y, en algunos casos, biopsia del tejido óseo o cartilaginoso.
El tratamiento de las osteocondrodisplasias depende del tipo específico y la gravedad de los síntomas. Puede incluir fisioterapia, ortopedia, cirugía correctiva y, en casos graves, cuidados paliativos. El pronóstico varía ampliamente según el tipo de osteocondrodisplasia; algunos tipos tienen un curso benigno con una esperanza de vida normal, mientras que otros pueden causar discapacidades graves y reducir la esperanza de vida.
La displasia fibrosa monostótica es una condición benigna y poco común que afecta el crecimiento y desarrollo del hueso. Se caracteriza por la formación de tejido fibroso en lugar del tejido óseo normal durante el proceso de crecimiento del hueso. Esta afección generalmente afecta a un solo hueso (de ahí el término "monostótica"), aunque en algunos casos puede involucrar múltiples huesos ("poliostótica").
La displasia fibrosa monostótica puede ocurrir en cualquier hueso del cuerpo, pero se observa con mayor frecuencia en los huesos largos como el fémur, el húmero y las costillas. La afección suele presentarse en la infancia o adolescencia, aunque también puede manifestarse en adultos jóvenes.
Los síntomas de la displasia fibrosa monostótica varían dependiendo del tamaño y la ubicación del crecimiento anormal del tejido fibroso dentro del hueso. Algunas personas con esta afección pueden no presentar ningún síntoma, mientras que otras pueden experimentar dolor, debilidad o fragilidad ósea, deformidades óseas y fracturas espontáneas.
El diagnóstico de displasia fibrosa monostótica generalmente se realiza mediante radiografías, tomografías computarizadas (TC) o resonancias magnéticas (RM) que muestran la presencia de tejido fibroso anormal dentro del hueso. La confirmación del diagnóstico puede requerir una biopsia ósea para examinar el tejido afectado bajo un microscopio.
El tratamiento de displasia fibrosa monostótica depende de la gravedad y los síntomas de la afección. En algunos casos, no se requiere ningún tratamiento específico más allá del seguimiento regular con radiografías u otras pruebas de imagen para controlar el crecimiento del tejido fibroso anormal.
Sin embargo, si la afección causa dolor o fracturas recurrentes, se pueden considerar opciones de tratamiento adicionales, como el uso de medicamentos para aliviar el dolor y fortalecer los huesos, la cirugía para corregir las deformidades óseas o la radioterapia para reducir el crecimiento del tejido fibroso anormal.
En general, la displasia fibrosa monostótica es una afección benigna que rara vez pone en peligro la vida. Sin embargo, puede causar problemas significativos con la función y la forma de los huesos afectados, especialmente si no se diagnostica y trata adecuadamente.
Los párpados son estructuras móviles que cubren y protegen el ojo. Están compuestos por varias capas, incluyendo la piel, músculo (el músculo orbicular), tejido conectivo y conjuntiva (membrana mucosa). Los párpados ayudan a mantener la humedad en el ojo al distribuir las lágrimas durante los parpadeos. También desempeñan un papel importante en la comunicación no verbal y la percepción visual, ya que cierran los ojos para evitar la entrada de luz excesiva o peligros potenciales. La incapacidad de mover adecuadamente los párpados se conoce como blefarospasmo o parálisis del párpado.
La displasia fibrosa poliostótica (DFP) es un trastorno poco frecuente del tejido conectivo que afecta el crecimiento y desarrollo de los huesos. Es una enfermedad benigna, aunque puede causar diversas complicaciones médicas. La DFP se caracteriza por la formación excesiva de tejido fibroso en lugar del tejido óseo normal durante el proceso de crecimiento y remodelación ósea. Esto puede llevar a una amplia gama de signos y síntomas, dependiendo de dónde y cuán extensamente se presente la displasia fibrosa.
La palabra "poliostótica" se refiere al patrón común de afectación en múltiples sitios óseos, aunque también existen formas monoostóticas (que afectan a un solo hueso). Los huesos largos, como los fémures, las tibias y los húmeros, son los más comúnmente afectados, pero cualquier hueso en el cuerpo puede verse involucrado. La displasia fibrosa puede causar lesiones óseas expansivas y líticas (disminución de la densidad mineral ósea), lo que lleva a una variedad de complicaciones, como fracturas espontáneas, deformidades óseas y dolor óseo.
La displasia fibrosa poliostótica puede presentarse en cualquier edad, pero generalmente se diagnostica durante la infancia o la adolescencia. Aproximadamente el 70% de los casos se diagnostican antes de los 30 años. La gravedad y el pronóstico de la DFP varían ampliamente, desde formas asintomáticas con lesiones óseas incidentales hasta formas más graves con afectación multisistémica y complicaciones significativas que pueden afectar la calidad de vida. El tratamiento suele ser multidisciplinario e implica el control de los síntomas, la prevención de las complicaciones y, en algunos casos, la intervención quirúrgica para corregir las deformidades óseas o estabilizar las fracturas.
Las deformidades congénitas del pie se refieren a una serie de anomalías estructurales y funcionales presentes en el nacimiento que afectan la forma, la alineación o la función normal del pie. Estas condiciones pueden variar desde formas leves que no necesitan tratamiento hasta deformidades graves que requieren intervención quirúrgica. Algunos ejemplos comunes de deformidades congénitas del pie incluyen el pie zambo o equinovaro, en el cual el talón está elevado y la punta del pie se dobla hacia dentro, y el pie plano congénito, en el que el arco del pie está ausente o deprimido. Otras deformidades pueden incluir un pie varo o valgo, donde el pie se desvía hacia afuera o hacia adentro, respectivamente. El tratamiento depende de la gravedad y el tipo de deformidad congénita del pie y puede incluir medidas no quirúrgicas como el uso de dispositivos ortopédicos, fisioterapia o terapia ocupacional, o intervenciones quirúrgicas en casos más graves.
La masticación es un proceso fisiológico que involucra el movimiento coordinado de los músculos maxilares y la acción de los dientes para triturar, desgarrar o moler los alimentos sólidos en partículas más pequeñas y manejables. Este proceso mecánico facilita la deglución (swallowing) y la digestión posterior de los nutrientes. La masticación es una función importante de los sistemas musculoesquelético y gastrointestinal, y su correcto funcionamiento contribuye a una buena salud oral y general. Los trastornos en la masticación pueden derivar en problemas como la disfagia (dificultad para deglutir), la malnutrición o el dolor articular y muscular.
La displasia pélvica canina (DPCC) es una enfermedad ortopédica degenerativa que afecta a los perros, particularmente a las razas grandes. Se caracteriza por un desarrollo anormal o una laxitud de la articulación de la cadera, lo que provoca dolor, cojera y, en etapas avanzadas, artritis degenerativa. La displasia pélvica canina es el resultado de una combinación de factores genéticos y ambientales, como la dieta y el ejercicio excesivo en etapas tempranas de crecimiento. El diagnóstico se realiza mediante radiografías y la evaluación de la morfología y la gravedad del trastorno. El tratamiento puede incluir cambios en el estilo de vida, medicamentos para aliviar el dolor y, en casos severos, cirugía reconstructiva o reemplazo total de cadera.
La palabra "verrugas" se refiere a un crecimiento benigno (no canceroso) de la piel. Se producen cuando el virus del papiloma humano (VPH) infecta la capa superior de la piel, causando una sobreproliferación de las células de la piel.
Las verrugas suelen tener un aspecto áspero y abultado y pueden ser de diferentes formas y tamaños. Pueden aparecer solas o en grupos. A menudo se encuentran en las manos, los pies, las áreas genitales y alrededor de la boca.
Las verrugas no suelen causar dolor a menos que estén en una zona donde son propensas a ser lastimadas, como el borde de una uña o la planta del pie. Algunas verrugas pueden desaparecer por sí solas, pero otras pueden persistir durante meses o incluso años si no se tratan.
Existen diferentes tipos de verrugas, incluyendo:
1. Verrugas comunes: Suelen aparecer en las manos y los brazos. Tienen un aspecto áspero y abultado y pueden tener pequeños puntos negros en su superficie.
2. Verrugas plantares: Se encuentran en la planta de los pies y pueden ser planas o con forma de coliflor. Pueden causar dolor al caminar.
3. Verrugas genitales: Aparecen en los genitales y alrededor de la boca. Pueden tener un aspecto liso y plano o abultado y áspero. A diferencia de otras verrugas, las verrugas genitales pueden estar asociadas con cáncer de cuello uterino si no se tratan.
4. Verrugas filiformes: Son delgadas y alargadas y suelen aparecer en la cara, especialmente alrededor de la boca y la nariz.
El tratamiento de las verrugas puede incluir el uso de medicamentos tópicos o químicos, crioterapia (congelación), electrocauterización (quemado) o cirugía. En algunos casos, las verrugas pueden desaparecer por sí solas sin tratamiento. Sin embargo, si una verruga es dolorosa, infectada o no desaparece después de varios meses de tratamiento, se recomienda consultar a un médico.
La displasia cleidocraneal es un trastorno genético que afecta el desarrollo normal del esqueleto. Se caracteriza por anomalías en la formación y fusión de los huesos, particularmente en el cráneo y el esternón (hueso en forma de corazón en el centro del pecho).
Las principales manifestaciones clínicas incluyen:
1. Cráneo: El crecimiento y desarrollo normal del cráneo se ven afectados, resultando en una fontanela anterior abierta (el "punto blando" en la parte superior de la cabeza) que puede permanecer presente hasta la edad adulta. También hay un retraso en la soldadura de los huesos craneales, lo que lleva a una cabeza más grande de lo normal (macrocefalia).
2. Cara: Los ojos pueden estar más separados de lo usual (hipertelorismo), y hay una frente prominente y protuberante.
3. Dientes: Hay anomalías dentales significativas, como retraso en la erupción de los dientes temporales y permanentes, dientes adicionales (supernumerarios) o ausencia congénita de algunos dientes.
4. Hombros: El esternón está hipoplásico (subdesarrollado), y los omóplatos (huesos de los hombros) no están unidos al esternón, lo que resulta en una movilidad excesiva de los hombros.
5. Extremidades: A menudo hay dedos cortos o fusionados (sindactilia), caderas anchas y pies planos.
6. Columna vertebral: Puede haber escoliosis o cifosis, que son curvaturas anormales de la columna vertebral.
Este trastorno es hereditario y se transmite con un patrón autosómico dominante, lo que significa que una copia del gen anormal heredado de uno de los padres es suficiente para causar la enfermedad. Sin embargo, aproximadamente el 20-30% de los casos son debidos a nuevas mutaciones y no tienen antecedentes familiares conocidos. El tratamiento incluye fisioterapia, ortopedia y cirugía dental, así como la corrección quirúrgica de las deformidades esqueléticas graves.
El término 'fenotipo' se utiliza en genética y medicina para describir el conjunto de características observables y expresadas de un individuo, resultantes de la interacción entre sus genes (genotipo) y los factores ambientales. Estas características pueden incluir rasgos físicos, biológicos y comportamentales, como el color de ojos, estatura, resistencia a enfermedades, metabolismo, inteligencia e inclinaciones hacia ciertos comportamientos, entre otros. El fenotipo es la expresión tangible de los genes, y su manifestación puede variar según las influencias ambientales y las interacciones genéticas complejas.
La displasia retiniana es una afección congénita del ojo que afecta el desarrollo y la función normal de la retina. Se trata de un grupo de enfermedades hereditarias raras que pueden causar ceguera parcial o completa.
La retina es una capa delgada y sensible a la luz en la parte posterior del ojo que contiene células fotorreceptoras llamadas bastones y conos, las cuales convierten la luz en señales eléctricas que se envían al cerebro a través del nervio óptico. En la displasia retiniana, estas células fotorreceptoras no se desarrollan o funcionan correctamente, lo que resulta en una visión disminuida o distorsionada.
Existen diferentes tipos de displasia retiniana, cada uno con diferentes grados de severidad y síntomas. Algunos afectan solo a pequeñas áreas de la retina, mientras que otros pueden involucrar toda la retina. Los síntomas más comunes incluyen visión borrosa o distorsionada, dificultad para ver en condiciones de poca luz, movimientos anormales de los ojos (nistagmo) y sensibilidad a la luz (fotofobia).
La displasia retiniana puede ser causada por mutaciones en varios genes diferentes, muchos de los cuales están involucrados en el desarrollo y mantenimiento de la retina. La mayoría de los casos se heredan de forma autosómica recesiva, lo que significa que una persona debe heredar dos copias del gen anormal (una de cada padre) para desarrollar la afección. Sin embargo, algunos tipos se heredan de forma dominante, lo que significa que solo es necesario heredar una copia del gen anormal para desarrollar la enfermedad.
No existe cura conocida para la displasia retiniana, y el tratamiento generalmente se centra en mejorar la visión y reducir los síntomas. Las opciones de tratamiento pueden incluir anteojos o lentes de contacto, cirugía para corregir problemas estructurales en el ojo, y terapia visual para ayudar a mejorar la visión. En algunos casos, se puede considerar un trasplante de córnea o retina, aunque estos procedimientos son experimentales y tienen riesgos significativos asociados.
Una mutación missense es un tipo específico de mutación en el ADN que causa la sustitución de un solo nucleótido (la unidad básica de los genes), lo que resulta en la producción de un aminoácido diferente en la proteína codificada. Esta alteración puede tener diversos efectos en la función de la proteína, dependiendo de dónde ocurra y cuán crucial sea el aminoácido reemplazado.
En algunos casos, una mutación missense podría no afectar significativamente la función de la proteína, especialmente si el aminoácido original y el nuevo son químicamente similares. Sin embargo, cuando el cambio ocurre en un dominio crucial de la proteína o involucra aminoácidos con propiedades químicas muy diferentes, esto puede conducir a una pérdida total o parcial de la función de la proteína.
Las mutaciones missense pueden asociarse con diversas enfermedades genéticas, dependiendo del gen y la proteína afectados. Por ejemplo, algunas mutaciones missense en el gen BRCA1 aumentan el riesgo de cáncer de mama y ovario hereditario.
La sudoración, también conocida como transpiración, es un proceso fisiológico normal en el que las glándulas sudoríparas secretan un líquido llamado sudor. Este líquido ayuda a regular la temperatura corporal al evaporarse sobre la piel y disipar el calor.
Existen dos tipos principales de glándulas sudoríparas: las ecrinas, que se encuentran por todo el cuerpo y secretan un sudor agua principalmente con sales y pequeñas cantidades de otras sustancias; y las apocrinas, que están localizadas en áreas específicas como las axilas y la ingle, y secretan un sudor más espeso y oloroso ya que contiene proteínas y lípidos que pueden ser descompuestos por bacterias presentes en la piel.
La sudoración excesiva o anormal se denomina hiperhidrosis y puede ser causada por diversas condiciones médicas, como trastornos endocrinos o neurológicos, infecciones, reacciones a ciertos medicamentos o factores emocionales. La hipohidrosis, o sudoración insuficiente, también puede ocurrir y puede ser el resultado de lesiones en las glándulas sudoríparas, quemaduras graves u otras afecciones médicas.
La detección de heterocigotos es un proceso de identificación de individuos que tienen diferentes alelos (variantes genéticas) de un gen en cada uno de sus cromosomas homólogos. Este término se utiliza a menudo en el contexto de la genética médica y la counseleía genética.
En condiciones genéticas recesivas, los individuos que heredan una copia normal y una copia alterada/dañada del gen aún no mostrarán síntomas de la enfermedad, ya que solo se manifiesta cuando ambas copias del gen están alteradas. Sin embargo, estos individuos son portadores de la condición y pueden transmitirla a su descendencia.
La detección de heterocigotos permite identificar a estos portadores asintomáticos, lo que puede ser útil en la planificación familiar y el asesoramiento genético. Por ejemplo, si ambos miembros de una pareja son portadores de una mutación recesiva particular, hay un 25% de probabilidad de que su hijo herede las dos copias alteradas del gen y desarrolle la enfermedad correspondiente.
Es importante tener en cuenta que algunas pruebas genéticas solo detectan mutaciones en uno o ambos alelos, pero no siempre pueden determinar con certeza si un individuo es heterocigoto o no. Por lo tanto, los resultados deben interpretarse con precaución y bajo la guía de profesionales médicos y genéticos calificados.
La luxación congénita de la cadera, también conocida como displasia del desarrollo de la hancha o enfermedad de Legg-Calvé-Perthes, es una afección en la cual el extremo superior del fémur (hueso del muslo) no encaja correctamente en la cavidad de la pelvis (socket). En condiciones normales, el extremo del fémur se conecta perfectamente con la cavidad de la pelvis formando una articulación esférica. Sin embargo, en la luxación congénita de la cadera, esta articulación no se forma correctamente desde el nacimiento o durante los primeros meses de vida.
Esta afección puede causar que el niño camine con una cojera o mantenga una postura anormal. Si no se trata, la luxación congénita de la cadera puede provocar dolor y problemas de movilidad en la edad adulta. El tratamiento temprano es fundamental para prevenir complicaciones a largo plazo y garantizar un desarrollo normal del niño. Los métodos de tratamiento pueden incluir el uso de férulas, dispositivos ortopédicos o, en casos graves, cirugía.
La displasia tanatofórica es un término médico que se utiliza para describir una condición genética rara y letalmente grave que afecta el desarrollo normal de los huesos y otros tejidos en el cuerpo. Esta enfermedad se caracteriza por la presencia de esqueletos anormalmente grandes o excesivamente pequeños, así como por una variedad de anomalías estructurales en los huesos y otras partes del cuerpo.
La displasia tanatofórica se puede presentar en dos formas principales: tipo I y tipo II. El tipo I se caracteriza por un esqueleto grande con extremidades cortas, una cabeza grande y prominente, y una columna vertebral anormalmente curvada. Por otro lado, el tipo II se distingue por un esqueleto pequeño con extremidades largas y delgadas, una caja torácica estrecha y deformada, y una cabeza grande y abultada.
Ambos tipos de displasia tanatofórica suelen ir acompañados de otras anomalías, como problemas cardíacos, renales y pulmonares, así como retraso mental y convulsiones. La enfermedad es letal y la mayoría de los niños con esta afección mueren antes de nacer o durante el primer año de vida.
La displasia tanatofórica se produce por mutaciones en genes específicos, como el gen FGFR3 (factor de crecimiento fibroblástico receptor 3) y el gen FGFR2 (factor de crecimiento fibroblástico receptor 2). Estas mutaciones causan una sobreactivación del receptor, lo que lleva a un crecimiento anormal de los huesos y otros tejidos.
El diagnóstico de displasia tanatofórica se realiza mediante estudios de imagenología, como radiografías y resonancias magnéticas, así como pruebas genéticas. No existe un tratamiento curativo para la enfermedad, aunque se pueden administrar medicamentos para aliviar los síntomas y mejorar la calidad de vida del niño.
 Displasia Ectodérmica - Dra. Candela Mellado - - "Investigadores por el Mundo" y " EERR " - Podcast en iVoox
Displasia Ectodérmica - Dra. Candela Mellado - - "Investigadores por el Mundo" y " EERR " - Podcast en iVoox VII Conferencia Internacional de Displasia Ectodérmica, en abril en Murcia | Somos Pacientes
VII Conferencia Internacional de Displasia Ectodérmica, en abril en Murcia | Somos Pacientes Alopecia - Wikipedia
Alopecia - Wikipedia Congenital Cutis Aplasia of the Lower Limbs. A Case Report and Literature Review
Congenital Cutis Aplasia of the Lower Limbs. A Case Report and Literature Review Listado de proyectos - Fundación Ramón Areces
Listado de proyectos - Fundación Ramón Areces Los cuidados orales para personas con discapacidades | Colgate®
Los cuidados orales para personas con discapacidades | Colgate® Primera Dentición: Información de ausencia o retraso en crecimiento de dientes.
Primera Dentición: Información de ausencia o retraso en crecimiento de dientes. Hipohidrosis - Trastornos dermatológicos - Manual MSD versión para profesionales
Hipohidrosis - Trastornos dermatológicos - Manual MSD versión para profesionales S10045 archivos - PRICAI
S10045 archivos - PRICAI Anodoncia parcial relacionada con quiste dentígero y quiste traumático. Revisión de la literatura y presentación de un caso
Anodoncia parcial relacionada con quiste dentígero y quiste traumático. Revisión de la literatura y presentación de un caso síndromes neurocutáneos; Síndromes de Displasia Neuroectodérmica; Facomatosis
síndromes neurocutáneos; Síndromes de Displasia Neuroectodérmica; Facomatosis BVS Paraguay
BVS Paraguay Búsqueda | BVS CLAP/SMR-OPS/OMS
Búsqueda | BVS CLAP/SMR-OPS/OMS Anomalías Congénitas. Vocabulario de Ciencias de la Salud para Argentina
Anomalías Congénitas. Vocabulario de Ciencias de la Salud para Argentina DeCS
DeCS Melanie Gaydos. Una lección sobre ¿Belleza o Tolerancia? - OCHO Marketing
Melanie Gaydos. Una lección sobre ¿Belleza o Tolerancia? - OCHO Marketing cap15
cap15 Síndromes Asociados - Fundación Gantz
Síndromes Asociados - Fundación Gantz Revista Más Dermatología
Revista Más Dermatología Acumulos y pigmentos
Acumulos y pigmentos PATOLOGÍA : Alteración de Tamaño en las piezas dentales - Directorio Odontológico
PATOLOGÍA : Alteración de Tamaño en las piezas dentales - Directorio Odontológico Anodoncia - My Dentist
Anodoncia - My Dentist