Síndrome de Wiskott-Aldrich
Proteína da Síndrome de Wiskott-Aldrich
Proteína Neuronal da Síndrome de Wiskott-Aldrich
Família de Proteínas da Síndrome de Wiskott-Aldrich
Proteína 2 Relacionada a Actina
Proteína 3 Relacionada a Actina
Actinas
Complexo 2-3 de Proteínas Relacionadas à Actina
Proteína cdc42 de Ligação ao GTP
Antígenos CD43
Proteínas
Proteínas do Citoesqueleto
Proteínas dos Microfilamentos
Pseudópodes
Síndromes de Imunodeficiência
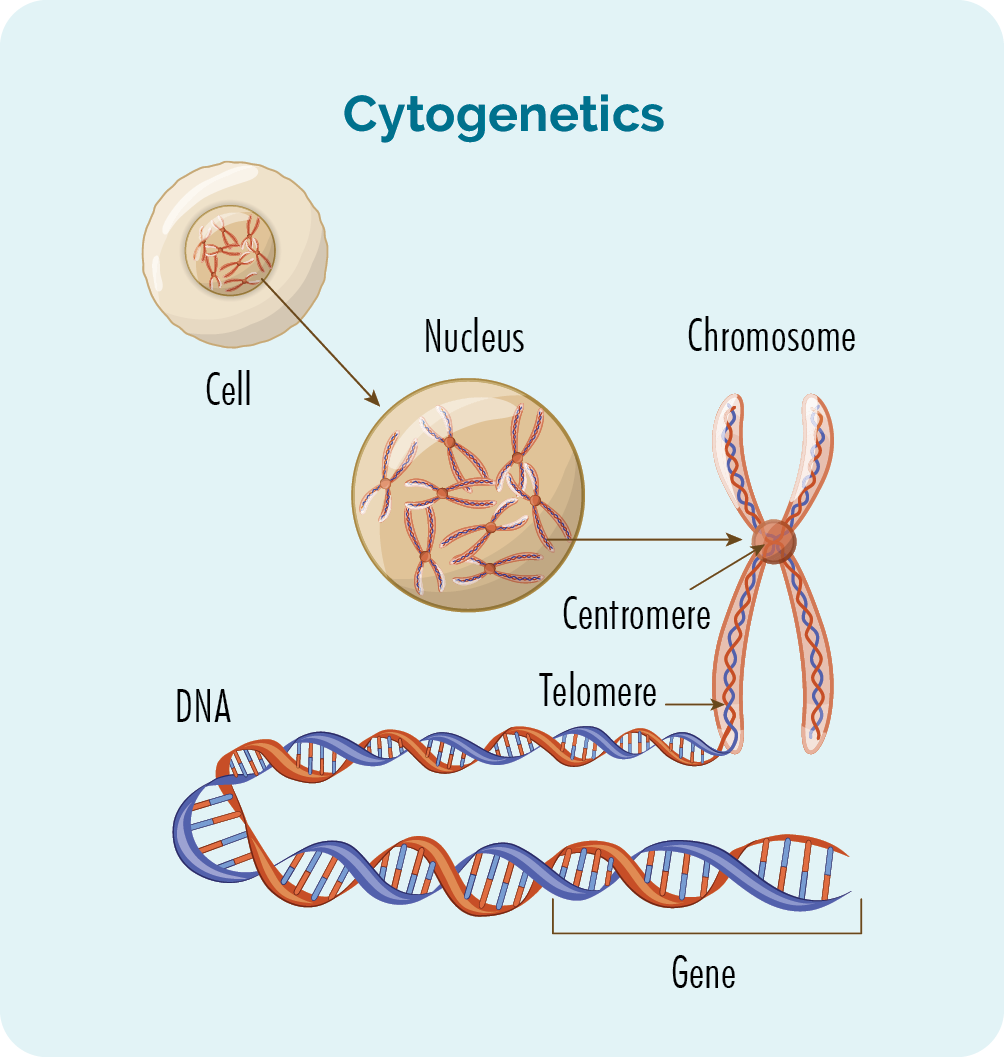
Citoesqueleto
Domínios de Homologia de src
Extensões da Superfície Celular
Transtornos Ovotesticulares do Desenvolvimento Sexual
Cromossomo X
Proteínas de Transporte
Mutação
Sialoglicoproteínas
Citoesqueleto de Actina
Camundongos Knockout
Dados de Sequência Molecular
Nexinas de Classificação
Biopolímeros
Ligação Proteica
Linfócitos
Eczema
Síndrome de Down
Linfócitos T
Proteínas Adaptadoras de Transdução de Sinal
Proteínas do Tecido Nervoso
Síndrome X Metabólica
Doenças Genéticas Ligadas ao Cromossomo X
Sinapses Imunológicas
Sequência de Aminoácidos
Enteropatias Perdedoras de Proteínas
Cortactina
Compensação de Dosagem (Genética)
Ativação Linfocitária
Linfócitos B
Estrutura Terciária de Proteína
Plaquetas
Movimento Celular
Agamaglobulinemia
Camundongos da Linhagem 129
Transdução de Sinal
Proteínas Oncogênicas
A Síndrome de Wiskott-Aldrich é uma doença genética rara que afeta o sistema imunológico e a coagulação sanguínea. É caracterizada por três sintomas principais:
1. Trombocitopenia (contagem baixa de plaquetas), o que leva a facilidade em sangrar e desenvolver hematomas.
2. Eczema, uma condição da pele que causa erupções cutâneas e coceira.
3. Imunodeficiência, que aumenta a suscetibilidade à infecções recorrentes.
Além disso, os indivíduos com a síndrome de Wiskott-Aldrich podem apresentar outros problemas de saúde, como diarreia crônica, infeções respiratórias e do ouvido, alergias, autoimunidade e um aumento no risco de desenvolver câncer, especialmente leucemia.
A síndrome é causada por mutações em um gene chamado WAS (Wiskott-Aldrich Syndrome), localizado no cromossomo X. Isso significa que a doença afeta principalmente os machos, uma vez que eles herdam apenas uma cópia do cromossomo X de suas mães. As mulheres têm duas cópias do cromossomo X e geralmente possuem uma cópia funcional do gene WAS, o que as torna portadoras saudáveis da síndrome.
O tratamento para a síndrome de Wiskott-Aldrich pode incluir transfusões de plaquetas, antibióticos para tratar infecções, imunossupressores para controlar respostas autoimunes e terapia de reposição de células hematopoéticas (TRCH), que envolve o transplante de células-tronco saudáveis de um doador compatível. A pesquisa está em andamento para desenvolver novas opções de tratamento, como terapia gênica, que pode oferecer uma cura duradoura para a síndrome.
A proteína Wiskott-Aldrich (WA Proteína) é um tipo de proteína que está ausente ou funciona de forma anormal em indivíduos com a síndrome de Wiskott-Aldrich, uma doença genética rara. Essa proteína desempenha um papel crucial na regulação da resposta imune e na agregação de plaquetas sanguíneas.
A síndrome de Wiskott-Aldrich é causada por mutações no gene WAS, que fornece instruções para a produção da proteína WA. Quando essa proteína falha em funcionar corretamente, os indivíduos afetados podem apresentar sintomas como uma diminuição na contagem de plaquetas sanguíneas (trombocitopenia), aumentando o risco de sangramento; problemas recorrentes de infecções devido à disfunção do sistema imune; e um aumento no risco de desenvolver certos tipos de câncer, como leucemia.
A proteína WA desempenha um papel importante na organização da estrutura dos glóbulos brancos, chamados leucócitos, e é necessária para a formação de vesículas intracelulares especializadas (chamadas vesículas sinápticas) que desempenham um papel na sinalização celular. Quando essa proteína está ausente ou não funciona corretamente, os leucócitos podem não se mover ou se comunicar adequadamente, o que pode contribuir para a disfunção do sistema imune observada em pessoas com síndrome de Wiskott-Aldrich.
A proteína neuronal da síndrome de Wiskott-Aldrich (WASP, do inglês Wiskott-Aldrich syndrome protein) é uma proteína que desempenha um papel crucial no processo de organização e reorganização dos actos de citoesqueleto de actina nos glóbulos brancos conhecidos como células T. A mutação nesta proteína leva à síndrome de Wiskott-Aldrich, uma doença genética rara que afeta o sistema imunitário e causa problemas de coagulação sanguínea.
A proteína WASP é codificada pelo gene WAS e está presente em células hematopoéticas, incluindo células T, células B, monócitos, macrófagos e plaquetas. A sua função principal é ativar a formação de filamentos de actina, um componente importante do citoesqueleto celular, o que permite a mobilidade e a reorganização da célula.
Na síndrome de Wiskott-Aldrich, as mutações no gene WAS levam a uma proteína WASP anormal ou ausente, resultando em um sistema imunitário deficiente e problemas de coagulação sanguínea. Os indivíduos com esta síndrome apresentam frequentemente infecções recorrentes, hemorragias e uma maior susceptibilidade ao desenvolvimento de câncer.
A família de proteínas da síndrome de Wiskott-Aldrich (WASP, do inglês Wiskott-Aldrich syndrome protein family) é um grupo de proteínas que desempenham papéis importantes na regulação dos processos celulares relacionados à atuação do sistema imune e à organização do citoesqueleto. A proteína WASP original foi primeiramente identificada como a causadora da síndrome de Wiskott-Aldrich, uma doença genética rara que afeta o sistema imunológico e a coagulação sanguínea.
As proteínas WASP estão envolvidas na regulação da atividade dos actináfilos, que são proteínas responsáveis pela formação de filamentos de actina, uma importante componente do citoesqueleto celular. A actina desempenha um papel crucial em diversos processos celulares, como a divisão e movimento celular, o transporte intracelular e a formação de sinapses e aderências entre células.
A família WASP inclui as seguintes proteínas:
1. WASP (Wiskott-Aldrich syndrome protein): É uma proteína expressa predominantemente em células hematopoéticas, como glóbulos brancos e plaquetas. Mutação no gene que codifica a proteína WASP leva à síndrome de Wiskott-Aldrich, caracterizada por uma combinação de problemas imunológicos, como infecções recorrentes, e hemorrágicos, como sangramentos excessivos.
2. N-WASP (Neuronal Wiskott-Aldrich syndrome protein): É uma proteína similar à WASP, mas expressa em células neuronais e outros tipos de células além das hematopoéticas. A proteína N-WASP desempenha um papel importante na formação de sinapses e no transporte vesicular em células neuronais.
3. WAVE (WASP family verprolin homologous protein): É uma família de proteínas relacionadas à WASP, expressa em vários tipos de células. A família WAVE inclui quatro isoformas (WAVE1 a WAVE4) e desempenha um papel importante na regulação da actina nos processos celulares, como o crescimento dos filopódios e lamelipódios, estruturas citoplasmáticas envolvidas no movimento celular.
As proteínas WASP, N-WASP e WAVE são reguladas por sinalização intracelular e atuam como efetores na via de sinalização da Rho GTPase, um tipo de molécula que controla a organização da actina no citoplasma. A activação dessas proteínas leva à formação de complexos multiproteicos denominados "motores de nucleação", que promovem a polimerização da actina e a formação de estruturas celulares especializadas, como filopódios e lamelipódios.
A Proteína 2 Relacionada a Actina, geralmente referida como filamina ou filamento de actina associado, é uma proteína estrutural que desempenha um papel crucial na organização e estabilidade do citoesqueleto de actina. Ela se liga diretamente à actina e promove a formação e manutenção dos feixes de actina, os quais são essenciais para diversos processos celulares, como a motilidade, adesão e divisão celular. A proteína 2 relacionada a actina é expressa em vários tipos de células e está envolvida em uma variedade de funções celulares importantes, incluindo a transdução de sinal, organização da membrana plasmática e regulação do ciclo celular.
A proteína 3 relacionada a actina, também conhecida como Actina-Related Protein 3 (ARP3), é uma proteína que desempenha um papel importante na regulação da organização e dinâmica dos filamentos de actina no citoesqueleto. A ARP3 é um componente fundamental do complexo ARP2/3, que está envolvido na nucleação e extensão dos filamentos de actina. O complexo ARP2/3 é recrutado para locais específicos da célula, onde estimula a formação de novas redes de filamentos de actina, o que é crucial para processos como a motilidade celular, endocitose e organização da membrana plasmática. A proteína ARP3 atua como um regulador negativo do complexo ARP2/3, inibindo sua atividade enquanto não estiver associada ao complexo. Além disso, a ARP3 também pode interagir com outras proteínas e complexos para regular a organização dos filamentos de actina em diferentes contextos celulares.
Em termos médicos, uma "síndrome" refere-se a um conjunto de sinais e sintomas que ocorrem juntos e podem indicar a presença de uma condição de saúde subjacente específica. Esses sinais e sintomas geralmente estão relacionados entre si e podem afetar diferentes sistemas corporais. A síndrome em si não é uma doença, mas sim um conjunto de sintomas que podem ser causados por várias condições médicas diferentes.
Por exemplo, a síndrome metabólica é um termo usado para descrever um grupo de fatores de risco que aumentam a chance de desenvolver doenças cardiovasculares e diabetes. Esses fatores de risco incluem obesidade abdominal, pressão arterial alta, níveis elevados de glicose em jejum e colesterol ruim no sangue. A presença de três ou mais desses fatores de risco pode indicar a presença da síndrome metabólica.
Em resumo, uma síndrome é um padrão característico de sinais e sintomas que podem ajudar os médicos a diagnosticar e tratar condições de saúde subjacentes.
As actinas são proteínas globulares que desempenham um papel fundamental no processo de contrato muscular e também estão envolvidas em outros processos celulares, como a divisão celular, transporte intracelular e mudanças na forma das células. Existem vários tipos diferentes de actinas, mas as duas principais são a actina F (filamentosa) e a actina G (globular). A actina F é responsável pela formação dos feixes de actina que deslizam uns sobre os outros durante a contração muscular, enquanto a actina G está presente em pequenas concentrações em todas as células e pode se associar a outras proteínas para formar estruturas celulares. A actina é uma proteína muito conservada evolutivamente, o que significa que é semelhante em diferentes espécies, desde bactérias até humanos.
O Complexo 2-3 de Proteínas Relacionadas à Actina é um agregado proteico essencial para a regulação do ciclo de vida dos mitocôndrios, as unidades de produção de energia das células. Esse complexo é composto por três principais proteínas: a actina, a proteína associada à actina mitocondrial (MIRO) e a proteína de membrana externa mitocondrial 25 (MER25).
A actina desempenha um papel fundamental nesse complexo, fornecendo uma estrutura dinâmica que pode se alongar e encurtar em resposta a sinais celulares. A MIRO é uma proteína de ligação ao GTP que atua como um sensor de calcios intracelular e controla o transporte mitocondrial ao longo dos microtúbulos da célula. Já a MER25 auxilia na ancoragem do complexo à membrana externa mitocondrial, garantindo sua estabilidade e funcionalidade.
O Complexo 2-3 de Proteínas Relacionadas à Actina desempenha um papel crucial no processo de fissão mitocondrial, que é responsável pela divisão dos mitocôndrios em unidades menores quando necessário. Além disso, esse complexo também participa da resposta celular a estresses oxidativos e do controle da qualidade dos mitocôndrias, auxiliando na remoção de mitocôndrias danificadas ou defeituosas.
Em resumo, o Complexo 2-3 de Proteínas Relacionadas à Actina é um importante componente regulatório do ciclo de vida dos mitocôndrias, envolvido em processos como a fissão mitocondrial, a resposta a estresses oxidativos e o controle da qualidade mitocondrial.
CDC42 é uma proteína de ligação a GTP (guanosina trifosfato) que pertence à família Rho de pequenas proteínas G. É altamente conservada em eucariotos e desempenha um papel fundamental na regulação do citoesqueleto de actina, bem como no processo de organização dos microtúbulos.
A proteína CDC42 alterna entre duas conformações: uma forma ativada quando se liga ao GTP e outra inativa quando se liga ao GDP (guanosina difosfato). A ativação de CDC42 é mediada por guanin nucleotídeo-trocadores específicos, como a Dbl homólogo SOS (Son of Sevenless), que promovem o intercâmbio de GDP por GTP.
Quando ativado, CDC42 desempenha um papel crucial na regulação da formação de filopódios, protrusões celulares ricas em actina que se estendem a partir da membrana plasmática e participam em diversos processos celulares, como adesão celular, migração e polarização. Além disso, CDC42 também regula a ativação de várias cascatas de sinalização, incluindo as vias MAPK (proteín-cinase activada por mitógenos) e PI3K (fosfoinositido 3-quinase), que desempenham papéis importantes em diversos processos celulares, como proliferação, diferenciação e sobrevivência celular.
Devido à sua importância na regulação de vários processos celulares, a proteína CDC42 tem sido implicada em diversas doenças humanas, incluindo câncer, doenças neurodegenerativas e doenças inflamatórias.
CD43, também conhecido como sialoproteína leucocitária (Leu-M5), é uma glicoproteína transmembranar expressa na superfície de vários tipos de células hematopoéticas, incluindo linfócitos T, linfócitos B, monócitos e neutrófilos. Ele desempenha um papel importante em processos celulares como adesão, migração e sinalização.
Como antígeno, CD43 é clinicamente relevante na diferenciação de células hematopoéticas e pode ser usado como marcador para identificar subpopulações específicas de células imunes. Alterações na expressão de CD43 têm sido associadas a várias condições clínicas, incluindo leucemias e linfomas. No entanto, é importante notar que a definição médica geralmente se concentra em sua função como uma proteína e não apenas em seu papel como antígeno.
Proteínas são macromoléculas compostas por cadeias de aminoácidos ligados entre si por ligações peptídicas. Elas desempenham um papel fundamental na estrutura, função e regulação de todos os órgãos e tecidos do corpo humano. As proteínas são necessárias para a crescimento, reparo e manutenção dos tecidos corporais, além de desempenharem funções importantes como enzimas, hormônios, anticorpos e transportadores. Existem diferentes tipos de proteínas, cada uma com sua própria estrutura e função específicas. A síntese de proteínas é regulada geneticamente, ou seja, o tipo e a quantidade de proteínas produzidas em um determinado momento dependem dos genes ativados na célula.
As proteínas do citoesqueleto são um tipo de proteína que desempenham um papel estrutural e funcional crucial no interior das células. Eles formam uma rede dinâmica de filamentos que dão forma, suporte e movimento às células. Existem três tipos principais de proteínas do citoesqueleto: actina, tubulina e intermediate filaments (filamentos intermediários).
A actina é um tipo de proteína que forma filamentos delgados e flexíveis, desempenhando um papel importante em processos como a divisão celular, o movimento citoplasmático e a motilidade das células. A tubulina, por outro lado, forma microtúbulos rígidos e longos que desempenham um papel crucial no transporte intracelular, na divisão celular e na manutenção da forma celular.
Finalmente, os filamentos intermediários são compostos por diferentes tipos de proteínas e formam uma rede resistente que dá suporte à célula e a protege contra tensões mecânicas. Além de seu papel estrutural, as proteínas do citoesqueleto também desempenham funções regulatórias importantes, como o controle da forma celular, da mobilidade e da divisão celular.
As proteínas dos microfilamentos pertencem a um tipo de fibrilas proteicas encontradas no citoplasma das células, desempenhando um papel fundamental na determinação da forma e estrutura celular, além de participarem em diversos processos dinâmicos como o movimento citoplasmático e a divisão celular.
Os microfilamentos são formados principalmente por actina, uma proteína globular que se polimeriza em fibras helicoidais de 6 a 7 nanômetros de diâmetro. A actina é frequentemente encontrada associada com outras proteínas reguladororas e adaptadoras, como a miosina, tropomodina, tropomiosina e a cross-linking protein, que desempenham um papel importante na estabilização e organização dos microfilamentos.
As proteínas dos microfilamentos estão envolvidas em uma variedade de processos celulares, incluindo o movimento citoplasmático, a divisão celular, a adesão celular e a motilidade celular. Além disso, elas também desempenham um papel importante na resposta às forças mecânicas e no estabelecimento de contatos entre células e entre células e a matriz extracelular.
Em resumo, as proteínas dos microfilamentos são uma classe importante de proteínas estruturais que desempenham um papel fundamental na determinação da forma e função celular, participando em uma variedade de processos dinâmicos e mecânicos.
Pseudópodes são estruturas temporárias ou projeções semelhantes a três ou mais a prolongamentos citoplasmáticos que se formam em células vivas por processos de extrusão do citoplasma. Eles são geralmente encontrados em certos tipos de células, como as amebas e outros protistas.
Embora pseudópodes se assemelhem a verdadeiros pés ou extremidades, eles não contêm estruturas esqueléticas ou musculares especializadas. Em vez disso, sua formação é controlada por alterações no citoesqueleto da célula e processos de fluxo citoplasmático.
Os pseudópodes desempenham um papel importante em uma variedade de funções celulares, incluindo a locomoção, fagocitose (ingestão de partículas ou corpos estranhos), e a adesão à superfícies. Eles também podem ser envolvidos no processo de divisão celular em alguns organismos.
Em resumo, pseudópodes são projeções temporárias do citoplasma que se formam em células vivas e desempenham um papel importante em várias funções celulares, especialmente em certos tipos de protistas.
As síndromes de immunodeficiência referen-se a um grupo de condições médicas em que o sistema imunitário está comprometido e incapaz de funcionar adequadamente, tornando o indivíduo susceptível a infeções frequentes e persistentes. Estas síndromes podem ser presentes desde o nascimento (primárias) ou adquiridas posteriormente na vida (secundárias), como resultado de outras condições médicas ou tratamentos, tais como infecção pelo HIV ou uso de medicamentos imunossupressores.
Existem vários tipos de síndromes de immunodeficiência primárias, cada uma delas afetando diferentes partes do sistema imunitário. Algumas das mais conhecidas incluem:
1. Deficiência de Complemento: Ocorre quando o componente proteico do sangue, chamado complemento, está ausente ou não funciona corretamente, resultando em susceptibilidade a infecções bacterianas.
2. Imunodeficiência Combinada Severa (SCID): É uma forma grave de immunodeficiência que afeta os linfócitos T e B, tornando o indivíduo extremamente vulnerável às infecções. A maioria dos casos de SCID é hereditária e pode ser fatal se não for tratada precocemente com um transplante de medula óssea.
3. Neutropenia Congênita: É uma condição em que o número de neutrófilos, um tipo de glóbulo branco importante para combater as infecções bacterianas e fúngicas, está reduzido. Isto resulta em frequentes infecções bacterianas e fúngicas graves.
4. Agammaglobulinemia: É uma condição rara em que o indivíduo é incapaz de produzir anticorpos suficientes, tornando-o susceptível a infecções bacterianas recorrentes, particularmente das vias respiratórias superiores.
5. Deficiência de X-Linked (XLA): É uma forma hereditária de agammaglobulinemia que afeta predominantemente os homens. A deficiência de enzima Bruton's tyrosine kinase resulta em falta de produção de anticorpos e susceptibilidade a infecções bacterianas recorrentes.
Estas são apenas algumas das formas conhecidas de immunodeficiência primária. Existem muitas outras condições raras que podem afetar o sistema imunitário e causar susceptibilidade a infecções recorrentes ou graves. É importante consultar um especialista em doenças do sistema imunitário para obter um diagnóstico preciso e tratamento adequado.
Sim, posso fornecer a definição médica de "citopessoa". O citoesqueleto é uma rede dinâmica e complexa de filamentos proteicos que estão presentes em todas as células vivas. Ele fornece forma, estrutura e suporte à célula, além de desempenhar um papel fundamental em processos celulares importantes, como a divisão celular, o transporte intracelular e a motilidade.
Existem três tipos principais de filamentos no citoesqueleto: actina, microtúbulos e filamentos intermédios. A actina é um tipo de proteína globular que forma filamentos flexíveis e finos, enquanto os microtúbulos são formados por tubulina, uma proteína fibrosa em forma de bastonete. Os filamentos intermédios, por sua vez, são constituídos por proteínas fibrosas mais espessas e rígidas do que a actina.
O citoesqueleto é altamente dinâmico e pode ser remodelado em resposta a estímulos internos ou externos à célula. Essa capacidade de reorganização é fundamental para uma variedade de processos celulares, como o movimento de vesículas intracelulares, a migração celular e a divisão celular. Além disso, o citoesqueleto também desempenha um papel importante na interação das células com o meio ambiente circundante, auxiliando no estabelecimento de contatos entre células e na adesão à matriz extracelular.
Em bioinformática, os domínios de homologia de src referem-se a regiões conservadas em proteínas que desempenham funções similares ou partilham uma origem comum. O termo "src" refere-se especificamente a família de genes de proteínas tirosina quinases, que estão envolvidas em sinalização celular e regulação do crescimento e divisão celular.
A homologia entre esses domínios indica uma relação evolutiva entre as proteínas que os contêm. A identificação de domínios de homologia pode ajudar no processo de classificação e anotação funcional de genes e proteínas desconhecidos, bem como fornecer informações importantes sobre a evolução molecular e a função das proteínas.
Existem diferentes bancos de dados e ferramentas bioinformáticas disponíveis para identificar e analisar domínios de homologia, como Pfam, SMART, InterPro e outros. Essas ferramentas utilizam algoritmos de busca de similaridade e machine learning para identificar padrões recorrentes em sequências de proteínas e predizer a presença de domínios de homologia específicos.
Trombocitopenia é um termo médico que se refere a uma contagem anormalmente baixa de plaquetas (também conhecidas como trombócitos) no sangue. As plaquetas desempenham um papel crucial na coagulação sanguínea, pois ajudam a formar coágulos que impedem o sangramento excessivo quando há uma lesão nos vasos sanguíneos.
Quando os níveis de plaquetas estão abaixo do normal (valores considerados normais variam entre 150.000 e 450.000 plaquetas por microlitro de sangue), o risco de sangramento aumenta. Trombocitopenia leve geralmente não causa sintomas graves, mas casos graves podem resultar em hematomas faciles, sangramentos nas gengivas, nariz ou outras mucosas, sangramento menstrual abundante e, em casos extremos, hemorragias internas.
A trombocitopenia pode ser causada por vários fatores, incluindo doenças do sistema imune que destroem as plaquetas, infecções virais, uso de certos medicamentos, radioterapia ou quimioterapia, deficiências nutricionais (como falta de folato ou vitamina B12), consumo excessivo de álcool e doenças hematológicas como a leucemia. Em alguns casos, a causa da trombocitopenia pode ser desconhecida, o que é chamado de idiopática. O tratamento para a trombocitopenia depende da causa subjacente e pode incluir medicações, transfusões de plaquetas ou, em casos graves, esplenectomia (remoção do baço).
As extensões da superfície celular, também conhecidas como projeções celulares ou apêndices celulares, são estruturas que se projetam além da membrana plasmática de uma célula e aumentam a superfície celular. Elas desempenham um papel importante em várias funções celulares, incluindo a adesão celular, sinalização celular, e endocitose. Algumas das extensões da superfície celular mais comuns são:
1. Microvilos: São pequenas projeções cilíndricas flexíveis que aumentam a superfície de absorção em células como as enterócitos no intestino delgado.
2. Estereocílios: São prolongamentos alongados e rígidos da membrana plasmática encontrados em células especializadas do ouvido interno, responsáveis pela detecção de vibrações sonoras.
3. Flagelos e cílios: São longas projeções celulares que permitem a locomoção em certos tipos de células, como espermatozoides e células epiteliais ciliadas dos pulmões.
4. Filopódios: São finas projeções alongadas da membrana plasmática que participam da adesão celular, migração celular e sinalização celular. Eles são encontrados em vários tipos de células, incluindo neurônios e fibroblastos.
5. Lamelipódios: São grandes projeções planas da membrana plasmática que participam da migração celular e expansão celular durante a divisão celular. Eles são encontrados principalmente em células do tecido conjuntivo, como fibroblastos.
6. Vesículas: São pequenas bolhas revestidas por membrana que se formam na superfície celular e desempenham um papel importante no transporte de substâncias dentro e fora da célula.
Os Transtornos ovotesticulares do desenvolvimento sexual (TODS) são um grupo heterogêneo de condições congênitas em que os órgãos reprodutores apresentam características tanto ovarianas quanto testiculares. Esses transtornos ocorrem devido a distúrbios no desenvolvimento dos genitais internos e/ou externos durante a embriogênese e fetogênese, resultando em uma gama de anormalidades estruturais que podem incluir combinações de gonádas ovotesticulares, útero bicorno (duplo), cérvix duplo, vagina dupla ou ausência parcial ou total do útero e cérvix.
A classificação e a nomenclatura dos TODS foram revisadas recentemente pela Organização Mundial da Saúde (OMS) e agora são chamados de "Disordens do Desenvolvimento Sexual" (DSD). A nova classificação se concentra em três aspectos principais: a condição genética subjacente, a anatomia dos órgãos reprodutores e as características da puberdade e da função sexual.
Os TODS são causados por uma variedade de fatores genéticos e hormonais que interrompem o processo normal de diferenciação sexual durante o desenvolvimento fetal. As mutações em genes específicos, como SRY, WNT4 e SOX9, têm sido associadas a esses transtornos. Além disso, fatores ambientais, como exposição a drogas ou produtos químicos durante a gravidez, também podem desempenhar um papel no desenvolvimento dos TODS.
O tratamento dos TODS geralmente requer uma abordagem multidisciplinar que inclui equipe de especialistas em endocrinologia pediátrica, genética clínica, cirurgia e psicologia. O objetivo do tratamento é normalizar a anatomia reprodutiva, maximizar a função sexual e promover o bem-estar emocional e social da pessoa afetada.
O cromossomo X é um dos dois cromossomos sexuais em humanos (o outro é o cromossomo Y). As pessoas geralmente possuem dois cromossomos idênticos chamados autossomos, e um par de cromossomos sexuais que podem ser either X ou Y. As fêmeas possuem dois cromossomos X (XX), enquanto os machos possuem um X e um Y (XY).
O cromossomo X contém aproximadamente 155 milhões de pares de bases e representa cerca de 5% do DNA total na célula. Carrega entre 800 a 900 genes, muitos dos quais estão relacionados às funções específicas do sexo, como o desenvolvimento das características sexuais secundárias femininas e a produção de hormônios sexuais. No entanto, também contém genes que não estão relacionados ao sexo e são comuns em ambos os sexos.
Algumas condições genéticas estão ligadas ao cromossomo X, como a fibrose quística, distrofia muscular de Duchenne, hemofilia e síndrome de Turner (quando uma pessoa nasce com apenas um cromossomo X). Essas condições geralmente afetam os machos mais frequentemente do que as fêmeas, porque se um homem herdar uma cópia defeituosa do gene em seu único cromossomo X, ele não terá outra cópia para compensar a falha. As mulheres, por outro lado, geralmente têm duas cópias de cada gene no par de cromossomos X, então se uma cópia estiver defeituosa, a outra pode ainda funcionar normalmente.
Proteínas de transporte, também conhecidas como proteínas de transporte transmembranar ou simplesmente transportadores, são tipos específicos de proteínas que ajudam a mover moléculas e ions através das membranas celulares. Eles desempenham um papel crucial no controle do fluxo de substâncias entre o interior e o exterior da célula, bem como entre diferentes compartimentos intracelulares.
Existem vários tipos de proteínas de transporte, incluindo:
1. Canais iónicos: esses canais permitem a passagem rápida e seletiva de íons através da membrana celular. Eles podem ser regulados por voltagem, ligantes químicos ou outras proteínas.
2. Transportadores acionados por diferença de prótons (uniporteres, simportadores e antiporteres): esses transportadores movem moléculas ou íons em resposta a um gradiente de prótons existente através da membrana. Uniporteres transportam uma única espécie molecular em ambos os sentidos, enquanto simportadores e antiporteres simultaneamente transportam duas ou mais espécies moleculares em direções opostas.
3. Transportadores ABC (ATP-binding cassette): esses transportadores usam energia derivada da hidrólise de ATP para mover moléculas contra gradientes de concentração. Eles desempenham um papel importante no transporte de drogas e toxinas para fora das células, bem como no transporte de lípidos e proteínas nas membranas celulares.
4. Transportadores vesiculares: esses transportadores envolvem o empacotamento de moléculas em vesículas revestidas de proteínas, seguido do transporte e fusão das vesículas com outras membranas celulares. Esse processo é essencial para a endocitose e exocitose.
As disfunções nesses transportadores podem levar a várias doenças, incluindo distúrbios metabólicos, neurodegenerativos e câncer. Além disso, os transportadores desempenham um papel crucial no desenvolvimento de resistência à quimioterapia em células tumorais. Portanto, eles são alvos importantes para o desenvolvimento de novas terapias e estratégias de diagnóstico.
Em genética, uma mutação é um cambo hereditário na sequência do DNA (ácido desoxirribonucleico) que pode resultar em um cambio no gene ou região reguladora. Mutações poden ser causadas por erros de replicación ou réparo do DNA, exposição a radiação ionizante ou substancias químicas mutagénicas, ou por virus.
Existem diferentes tipos de mutações, incluindo:
1. Pontuais: afetan un único nucleótido ou pairaxe de nucleótidos no DNA. Pueden ser categorizadas como misturas (cambios na sequencia do DNA que resultan en un aminoácido diferente), nonsense (cambios que introducen un códon de parada prematura e truncan a proteína) ou indels (insercións/eliminacións de nucleótidos que desplazan o marco de lectura).
2. Estruturais: involvan cambios maiores no DNA, como deleciones, duplicacións, inversións ou translocacións cromosómicas. Estas mutações poden afectar a un único gene ou extensos tramos do DNA e pueden resultar en graves cambios fenotípicos.
As mutações poden ser benévolas, neutras ou deletéras, dependendo da localización e tipo de mutación. Algúns tipos de mutações poden estar associados con desordens genéticas ou predisposición a determinadas enfermidades, mentres que outros non teñen efecto sobre a saúde.
Na medicina, o estudo das mutações é importante para o diagnóstico e tratamento de enfermedades genéticas, así como para a investigación da patogénese de diversas enfermidades complexas.
Sialoglicoproteínas são um tipo específico de glicoproteínas que contém altos níveis de ácido siálico, um açúcar derivado da neuraminic acid, ligado à cadeia polissacarídea. Eles estão presentes em grande quantidade na superfície das células e desempenham um papel importante em uma variedade de processos biológicos, incluindo a interação celular, reconhecimento antigênico e regulação da atividade enzimática. Algumas sialoglicoproteínas também servem como marcadores para certas doenças, como o câncer. Eles são particularmente abundantes na membrana plasmática de células nervosas e estão envolvidos em processos neuronais importantes, como a sinapse e a plasticidade sináptica.
O citoesqueleto de actina é um componente fundamental do citoesqueleto, a estrutura que fornece forma e suporte às células. Ele é composto principalmente por filamentos de actina, uma proteína fibrosa altamente conservada em todos os tipos de células e organismos eucarióticos.
Os filamentos de actina são polímeros flexíveis que se organizam em diferentes configurações espaciais, dependendo da função específica da célula. Eles podem formar redes reticuladas, fascículos paralelos ou anéis contínuos ao redor da célula.
O citoesqueleto de actina desempenha várias funções importantes na célula, incluindo a manutenção da integridade estrutural, o transporte intracelular, a divisão celular e a motilidade celular. Ele interage com outros componentes do citoesqueleto, como os microtúbulos e os filamentos intermédios, bem como com proteínas motoras como a miosina, para gerar força mecânica e permitir que a célula se mova e altere sua forma.
Além disso, o citoesqueleto de actina também desempenha um papel importante na interação da célula com o ambiente externo, através do estabelecimento de adesões focais e outras estruturas de adesão à matriz extracelular. Essas interações permitem que a célula receba sinais do ambiente exterior e responda adequadamente a eles, o que é fundamental para processos como a proliferação celular, a diferenciação e a migração.
"Knockout mice" é um termo usado em biologia e genética para se referir a camundongos nos quais um ou mais genes foram desativados, ou "knockout", por meio de técnicas de engenharia genética. Isso permite que os cientistas estudem os efeitos desses genes específicos na função do organismo e no desenvolvimento de doenças. A definição médica de "knockout mice" refere-se a esses camundongos geneticamente modificados usados em pesquisas biomédicas para entender melhor as funções dos genes e seus papéis na doença e no desenvolvimento.
"Dados de sequência molecular" referem-se a informações sobre a ordem ou seqüência dos constituintes moleculares em uma molécula biológica específica, particularmente ácidos nucléicos (como DNA ou RNA) e proteínas. Esses dados são obtidos através de técnicas experimentais, como sequenciamento de DNA ou proteínas, e fornecem informações fundamentais sobre a estrutura, função e evolução das moléculas biológicas. A análise desses dados pode revelar padrões e características importantes, tais como genes, sítios de ligação regulatórios, domínios proteicos e motivos estruturais, que podem ser usados para fins de pesquisa científica, diagnóstico clínico ou desenvolvimento de biotecnologia.
Na medicina, "nexinas de classificação" não são um termo reconhecido ou estabelecido. O termo "nexina" geralmente se refere a proteínas que desempenham um papel na formação de conexões entre células (conhecidas como uniões comunicantes ou gap junctions em inglês), permitindo a comunicação e o fluxo de moléculas entre as células adjacentes.
As nexinas são classificadas em duas categorias principais: conexina e pannexina. As conexinas formam uniões comunicantes clássicas, enquanto as panexinas formam canais hemichannel que podem se encontrar com outros canais hemichannel em células adjacentes para formar canais intercelulares.
Portanto, é importante esclarecer a terminologia exata ou o contexto em que "nexinas de classificação" foi mencionado, pois isso pode se referir a um conceito ou área de pesquisa específica relacionada às nexinas e suas funções.
Biopolímeros são polímeros naturais produzidos por organismos vivos, sejam eles animais, vegetais ou microorganismos. Eles desempenham um papel fundamental em muitas funções biológicas importantes, como a estrutura e suporte mecânico dos tecidos, armazenamento de energia, sinalização celular e proteção contra patógenos.
Existem três principais categorias de biopolímeros:
1. Polissacarídeos: São polímeros de açúcares simples, como a amilose e a celulose, que são encontrados em plantas e animais e desempenham funções estruturais e energéticas.
2. Proteínas: São cadeias longas de aminoácidos que desempenham uma variedade de funções importantes nas células vivas, como a formação de estruturas celulares, enzimas e hormônios.
3. Ácidos nucleicos: São biopolímeros que armazenam e transmitem informações genéticas nas células vivas. O DNA e o RNA são exemplos de ácidos nucleicos.
Biopolímeros sintéticos também podem ser produzidos em laboratório, imitando a estrutura e as propriedades dos biopolímeros naturais. Esses materiais têm atraído muita atenção na pesquisa e desenvolvimento de novas tecnologias, como os bio-materiais para engenharia de tecidos e nanotecnologia.
Em bioquímica, uma ligação proteica refere-se a um tipo específico de interação entre duas moléculas, geralmente entre uma proteína e outa molécula (como outra proteína, peptídeo, carboidrato, lípido, DNA, ou outro ligante orgânico ou inorgânico). Essas interações são essenciais para a estrutura, função e regulação das proteínas. Existem diferentes tipos de ligações proteicas, incluindo:
1. Ligação covalente: É o tipo mais forte de interação entre as moléculas, envolvendo a troca ou compartilhamento de elétrons. Um exemplo é a ligação disulfureto (-S-S-) formada pela oxidação de dois resíduos de cisteínas em proteínas.
2. Ligação iônica: É uma interação eletrostática entre átomos com cargas opostas, como as ligações entre resíduos de aminoácidos carregados positivamente (lisina, arginina) e negativamente (ácido aspártico, ácido glutâmico).
3. Ligação hidrogênio: É uma interação dipolo-dipolo entre um átomo parcialmente positivo e um átomo parcialmente negativo, mantido por um "ponte" de hidrogênio. Em proteínas, os grupos hidroxila (-OH), amida (-CO-NH-) e guanidina (R-NH2) são exemplos comuns de grupos que podem formar ligações de hidrogênio.
4. Interações hidrofóbicas: São as interações entre resíduos apolares, onde os grupos hidrofóbicos tenderão a se afastar da água e agrupar-se juntos para minimizar o contato com o solvente aquoso.
5. Interações de Van der Waals: São as forças intermoleculares fracas resultantes das flutuações quantísticas dos dipolos elétricos em átomos e moléculas. Essas interações são importantes para a estabilização da estrutura terciária e quaternária de proteínas.
Todas essas interações contribuem para a estabilidade da estrutura das proteínas, bem como para sua interação com outras moléculas, como ligantes e substratos.
Os linfócitos são um tipo de glóbulos brancos (leucócitos) que desempenham um papel central no sistema imunológico, especialmente na resposta adaptativa imune. Existem dois tipos principais de linfócitos: linfócitos B e linfócitos T. Os linfócitos B são responsáveis pela produção de anticorpos e desempenham um papel importante na resposta imune humoral, enquanto que os linfócitos T estão envolvidos em células mediadas a respostas imunes, como a ativação de outras células do sistema imunológico e a destruição direta de células infectadas ou tumorais. Os linfócitos são produzidos no medula óssea e amadurecem no timo (para os linfócitos T) ou nos tecidos linfoides (para os linfócitos B).
Eczema é um termo geral utilizado para descrever diversas condições da pele caracterizadas por eritema (cores vermelhas na pele), inchaço, descamação e vesículas que podem se romper e formar crostas ou supurações. A forma mais comum de eczema é a dermatite atópica, que geralmente começa na infância e pode estar associada a alergias e/ou à predisposição genética. Outros tipos de eczema incluem a dermatite de contato, que ocorre em resposta ao contato com substâncias irritantes ou alérgicas, e a neurodermatite, que geralmente afeta braços, pescoço e cabeça e é desencadeada por estresse emocional, entre outros fatores. O tratamento do eczema pode incluir cremes hidratantes, corticosteroides tópicos, antihistamínicos e imunossupressores, dependendo da gravidade e do tipo de eczema.
A Síndrome de Down, também conhecida como Trissomia 21, é um distúrbio genético causado pela presença de todo ou parte de um terceiro cromossomo 21. Normalmente, as pessoas possuem dois cromossomos 21, um herdado de cada pai. No entanto, as pessoas com Síndrome de Down têm três cópias deste cromossomo, o que resulta em alterações físicas distintas e níveis variados de atraso no desenvolvimento intelectual e deficiências na fala e linguagem.
Os sinais e sintomas da Síndrome de Down podem incluir:
1. Características faciais distintas, como olhos arredondados com uma dobra cutânea nas pálpebras internas (puxado para baixo na parte interna do olho), nariz achatado e orelhas pequenas e posicionadas baixas.
2. Baixa estatura e peso ao nascer.
3. Hipotonia muscular, que pode afetar a força e a coordenação motora.
4. Atrasos no desenvolvimento, como sentar, andar, falar e controlar os esfíncteres.
5. Deficiências intelectuais leves a moderadas.
6. Problemas de audição e visão.
7. Problemas cardiovasculares congênitos em aproximadamente 50% dos casos.
8. Alto risco de desenvolver outras condições, como doenças respiratórias, problemas gastrointestinais, hipotiroidismo, leucemia e demência na idade adulta.
A Síndrome de Down é a causa genética mais comum de deficiência intelectual e afeta aproximadamente 1 em cada 700 nascidos vivos nos Estados Unidos. Embora não haja cura para a síndrome, intervenções educativas e terapêuticas precoces podem ajudar a maximizar o potencial de desenvolvimento das pessoas afetadas.
Os linfócitos T são um tipo específico de glóbulos brancos, também conhecidos como leucócitos, que desempenham um papel crucial no sistema imunológico adaptativo dos mamíferos. Eles são produzidos e maduram no tecido linfoide associado ao intestino (TALI) e na medula óssea antes de se moverem para o timo, onde completam a maturação e se diferenciam em diferentes subconjuntos de linfócitos T, como os linfócitos T CD4+ (auxiliares) e os linfócitos T CD8+ (citotóxicos).
Os linfócitos T auxiliares desempenham um papel importante na ativação de outras células do sistema imunológico, como macrófagos e linfócitos B, enquanto os linfócitos T citotóxicos são responsáveis por destruir diretamente as células infectadas ou tumorais.
As membranas dos linfócitos T possuem receptores de superfície específicos, chamados receptores de linfócitos T (TCR), que reconhecem antígenos apresentados em moléculas do complexo principal de histocompatibilidade (MHC) nas células do corpo. Isso permite que os linfócitos T detectem e respondam a células infectadas por vírus, bactérias intracelulares ou outros patógenos.
Além disso, os linfócitos T também possuem moléculas de superfície adicionais, como a CD3, que transmitem sinais intracelulares após o reconhecimento do antígeno e desencadeiam respostas imunes específicas.
Em resumo, os linfócitos T são células importantes do sistema imunológico adaptativo que auxiliam no reconhecimento e destruição de células infectadas ou tumorais, contribuindo assim para a proteção do organismo contra infecções e doenças.
As proteínas adaptadoras de transdução de sinal são moléculas reguladoras importantes em vias de transdução de sinais celulares. Elas não possuem atividade enzimática intrínseca, mas desempenham um papel crucial na organização e coordenação das cascatas de sinalização ao conectar receptores de sinal às proteínas efetoras.
As proteínas adaptadoras geralmente contêm domínios estruturais modulares, como domínios SH2 (Src homology 2), SH3 (Src homology 3) ou PH ( Pleckstrin homology), que permitem sua interação específica com outras proteínas e lipídios. Essas interações facilitam a formação de complexos multiproteicos transitorios, que são necessários para a amplificação, diversificação e integração dos sinais recebidos pelas células.
Algumas proteínas adaptadoras também podem participar na recrutamento de cinases e fosfatases, enzimas que adicionam ou removem grupos fosfato em outras proteínas, respectivamente. Essas modificações químicas desencadeiam alterações conformacionais nas proteínas alvo, levando à sua ativação ou inativação e, consequentemente, ao controle da atividade de vias de sinalização específicas.
Em resumo, as proteínas adaptadoras de transdução de sinal são moléculas cruciais na organização e regulação das cascatas de sinalização celular, permitindo que as células detectem, processem e respondam adequadamente a estímulos externos e internos.
As proteínas do tecido nervoso referem-se a um grande grupo de proteínas específicas que desempenham funções importantes no sistema nervoso central e periférico. Elas estão envolvidas em uma variedade de processos biológicos, incluindo a transmissão sináptica, a manutenção da estrutura das células nervosas (neurônios) e a proteção contra danos celulares.
Algumas proteínas do tecido nervoso bem conhecidas incluem:
1. Neurofilamentos: proteínas estruturais que fornecem suporte e integridade às células nervosas.
2. Tubulina: uma proteína importante na formação de microtúbulos, que desempenham um papel crucial no transporte axonal e no movimento citoplasmático.
3. Canais iônicos: proteínas que regulam o fluxo de íons através da membrana celular, desempenhando um papel fundamental na geração e condução de sinais elétricos nos neurônios.
4. Receptores neurotransmissores: proteínas localizadas nas membranas pré- e pós-sinápticas que permitem a ligação e a ativação dos neurotransmissores, desencadeando respostas celulares específicas.
5. Enzimas: proteínas que catalisam reações químicas importantes no metabolismo e no sinalizamento celular.
6. Proteínas de choque térmico (HSPs): proteínas induzidas por estresse que ajudam a proteger as células nervosas contra danos causados por estressores ambientais, como calor, frio ou hipóxia.
7. Fatores neurotróficos: proteínas que promovem o crescimento, a sobrevivência e a diferenciação dos neurônios, desempenhando um papel crucial no desenvolvimento e na manutenção do sistema nervoso.
As alterações nas expressões e funções dessas proteínas podem contribuir para o desenvolvimento de diversos distúrbios neurológicos e psiquiátricos, como doença de Alzheimer, doença de Parkinson, esclerose múltipla, depressão e transtorno bipolar. Assim, a compreensão dos mecanismos moleculares envolvidos na regulação das proteínas cerebrais pode fornecer informações importantes para o desenvolvimento de novas estratégias terapêuticas para essas condições.
A Síndrome X Metabólica, também conhecida como Síndrome da Resistência à Insulina, é um conjunto de fatores de risco para doenças cardiovasculares e diabetes do tipo 2. Embora não exista um consenso completo sobre os critérios diagnósticos, a síndrome geralmente é definida pela presença de três ou mais dos seguintes fatores:
1. Obesidade abdominal (circunferência da cintura maior que 102 cm em homens e 88 cm em mulheres)
2. Níveis elevados de triglicérides no sangue (> 150 mg/dL)
3. Baixos níveis de HDL colesterol (< 40 mg/dL em homens e < 50 mg/dL em mulheres)
4. Pressão arterial alta (> 130/85 mmHg)
5. Níveis elevados de glicose em jejum (> 100 mg/dL) ou diagnóstico prévio de diabetes do tipo 2
6. Níveis elevados de proteína C-reativa (PCR), um marcador de inflamação (> 2,0 mg/L)
Além disso, a resistência à insulina é considerada um fator central na síndrome X metabólica. Isso significa que as células do corpo tornam-se menos sensíveis à ação da insulina, uma hormona produzida pelo pâncreas que regula o nível de glicose no sangue. A resistência à insulina pode levar ao aumento dos níveis de glicose no sangue e à eventual diabetes do tipo 2.
A síndrome X metabólica é frequentemente associada a fatores genéticos e ambientais, como dieta deficiente em nutrientes e rica em calorias, sedentarismo, obesidade e idade avançada. O tratamento geralmente inclui mudanças no estilo de vida, como exercícios regulares, dieta saudável e perda de peso, bem como medicação para controlar os níveis de glicose, pressão arterial e colesterol.
Doenças genéticas ligadas ao cromossomo X são condições herdadas que resultam de alterações (mutações) em genes localizados no cromossomo X. As mulheres possuem dois cromossomos X (dupla dose), enquanto os homens têm um cromossomo X e um cromossomo Y (dose única). Isso significa que as mulheres geralmente são menos susceptíveis a desenvolver essas doenças, pois podem ter uma cópia funcional do gene no outro cromossomo X. No entanto, os homens não têm esse privilégio e, portanto, apresentam sintomas mais graves ou são mais susceptíveis de desenvolver a doença.
Exemplos de doenças genéticas ligadas ao cromossomo X incluem:
1. Hemofilia - uma condição que afeta a capacidade de coagulação sanguínea, causada por mutações em genes que controlam a produção de fatores de coagulação.
2. Distrofia muscular de Duchenne e Becker - doenças que causam fraqueza e perda de massa muscular, devido a mutações no gene da distrofina.
3. Síndrome de X frágil - uma causa genética comum de atraso mental e transtornos do comportamento, causada por mutações no gene FMR1.
4. Síndrome de Daltonismo - um tipo de deficiência visual que afeta a capacidade de distinguir cores, causada por mutações em genes responsáveis pela produção de proteínas envolvidas na percepção de cores.
5. Doença de von Willebrand - uma condição hemorrágica hereditária causada por mutações no gene que controla a produção do fator de von Willebrand, uma proteína importante para a coagulação sanguínea.
Na medicina e na imunologia, as sinapses imunológicas referem-se às interações específicas e altamente reguladas entre células do sistema imune, que desempenham um papel crucial no reconhecimento, sinalização e resposta a patógenos ou substâncias estranhas. Essas sinapses imunológicas são semelhantes em conceito às sinapses neuronais, onde as moléculas de sinalização são trocadas entre células para permitir a comunicação e coordenação das respostas.
Nas sinapses imunológicas, os linfócitos T e B, que são as principais células do sistema adaptativo imune, interagem com outras células imunes, como macrófagos, células dendríticas e outros linfócitos. Essas interações ocorrem em estruturas especializadas chamadas sinapses imunológicas, nas quais as moléculas de sinalização são apresentadas em uma plataforma organizada que permite a comunicação eficiente entre as células.
A formação das sinapses imunológicas é desencadeada por sinais de reconhecimento, como a interação do receptor de linfócitos T (TCR) com o complexo peptídeo-MHC em células apresentadoras de antígenos, ou a interação do receptor de linfócitos B (BCR) com um antígeno. Essas interações desencadeiam uma cascata de eventos que resultam na formação de uma sinapse imunológica, na qual as moléculas de sinalização, como citocinas e quimiocinas, são secretadas ou apresentadas em vesículas para permitir a comunicação entre as células.
As sinapses imunológicas desempenham um papel fundamental no controle da resposta imune, pois permitem que as células do sistema imune se comuniquem e coordenem suas atividades para eliminar patógenos ou células danificadas. Além disso, as sinapses imunológicas também desempenham um papel importante na regulação da resposta imune, pois permitem que as células do sistema imune se comuniquem com outras células do corpo para garantir que a resposta imune seja adequada e não cause danos colaterais.
Em resumo, as sinapses imunológicas são estruturas especializadas que permitem que as células do sistema imune se comuniquem e coordenem suas atividades para eliminar patógenos ou células danificadas. A formação das sinapses imunológicas é desencadeada por sinais de reconhecimento, como a interação do TCR com o complexo peptídeo-MHC em células apresentadoras de antígenos, e envolve a secreção ou apresentação de moléculas de sinalização para coordenar as atividades das células do sistema imune. As sinapses imunológicas desempenham um papel importante na regulação da resposta imune, pois permitem que as células do sistema imune se comuniquem com outras células do corpo para garantir que a resposta imune seja adequada e não cause danos colaterais.
Uma sequência de aminoácidos refere-se à ordem exata em que aminoácidos específicos estão ligados por ligações peptídicas para formar uma cadeia polipeptídica ou proteína. Existem 20 aminoácidos diferentes que podem ocorrer naturalmente nas sequências de proteínas, cada um com sua própria propriedade química distinta. A sequência exata dos aminoácidos em uma proteína é geneticamente determinada e desempenha um papel crucial na estrutura tridimensional, função e atividade biológica da proteína. Alterações na sequência de aminoácidos podem resultar em proteínas anormais ou não funcionais, o que pode contribuir para doenças humanas.
Protein-losing enteropathies (PLE) referem-se a um grupo de condições clínicas em que ocorre uma perda excessiva de proteínas séricas no trato gastrointestinal, resultando em hipoproteinemia e edema. A perda de proteínas pode ser causada por vários mecanismos, incluindo aumento da permeabilidade intestinal, disfunção linfática ou aumento do fluxo linfático, insuficiência motora intestinal e inflamação crônica.
A condição mais comum associada a PLE é a enteropatia sensível a glúten, seguida por outras causas menos comuns, como doenças inflamatórias intestinais (como doença de Crohn e colite ulcerativa), infecções entéricas, neoplasias gastrointestinais, isquemia intestinal, enteropatias associadas a amiloidose e outras condições sistêmicas.
Os sinais e sintomas clínicos de PLE podem incluir edema periférico, ascites, hipoalbuminemia, anemia, disfunção hepática e diarreia crônica. O diagnóstico geralmente requer uma avaliação clínica cuidadosa, incluindo exames de sangue, radiografias, endoscopia com biópsia e estudos de permeabilidade intestinal. O tratamento depende da causa subjacente e pode incluir dieta especial, medicamentos, cirurgia ou terapia de reposição de proteínas.
Cortactina é uma proteína intracelular que desempenha um papel importante na organização e dinâmica do citoesqueleto de actina, especialmente nas regiões da membrana plasmática. Ela interage com outras proteínas envolvidas no rearranjo dos filamentos de actina, como Arp2/3 e Vasp (Vermiformin, adenilililato/guanilato cinase inibidor, proteína estrutural).
A cortactina contém vários domínios funcionais, incluindo um domínio de ligação à actina, que permite a sua associação com filamentos de actina; e um domínio de repetição rica em prolina, que é responsável pela interação com outras proteínas reguladoras do citoesqueleto.
A cortactina desempenha um papel crucial em diversos processos celulares, como a formação de adesões focais, a manutenção da integridade da membrana plasmática, o transporte vesicular e a migração celular. Alterações na expressão ou atividade da cortactina têm sido associadas a várias doenças, incluindo câncer e distúrbios neurológicos.
A "ativação linfocitária" é um termo usado em medicina e imunologia para descrever o processo em que as células do sistema imune, chamadas linfócitos, são ativadas e se tornam capazes de realizar suas funções específicas, como a produção de anticorpos ou a destruição de células infectadas ou tumorais.
Esse processo é iniciado quando os linfócitos entram em contato com um antígeno, uma substância estrangeira que desencadeia uma resposta imune. A interação entre o antígeno e o receptor de superfície do linfócito leva à ativação da célula, que começa a se dividir e a diferenciar em células especializadas.
A ativação linfocitária é um processo complexo que envolve uma série de sinais e mensageiros químicos, incluindo citocinas e quimiocinas, que auxiliam na comunicação entre as células do sistema imune. Essa comunicação é fundamental para a coordenação da resposta imune e para garantir que as células do sistema imune atuem de forma adequada para combater a infecção ou o tumor.
Em resumo, a "ativação linfocitária" refere-se ao processo em que as células do sistema imune, os linfócitos, são ativadas e se diferenciam em células especializadas capazes de realizar funções específicas de defesa imune.
Os linfócitos B são um tipo de glóbulos brancos (leucócitos) que desempenham um papel central no sistema imunológico adaptativo, especialmente na resposta humoral da imunidade adaptativa. Eles são produzidos e maturam no tufolo dos órgãos linfoides primários, como o baço e a medula óssea vermelha. Após a ativação, os linfócitos B se diferenciam em células plasmáticas que produzem anticorpos (imunoglobulinas) específicos para um antígeno estranho, auxiliando assim na neutralização e eliminação de patógenos como bactérias e vírus. Além disso, os linfócitos B também podem funcionar como células apresentadoras de antígenos, contribuindo para a ativação dos linfócitos T auxiliares.
Em bioquímica e ciência de proteínas, a estrutura terciária de uma proteína refere-se à disposição tridimensional dos seus átomos em uma única cadeia polipeptídica. Ela é o nível de organização das proteínas que resulta da interação entre os resíduos de aminoácidos distantes na sequência de aminoácidos, levando à formação de estruturas secundárias (como hélices alfa e folhas beta) e regiões globulares ou fibrilares mais complexas. A estrutura terciária é mantida por ligações não covalentes, como pontes de hidrogênio, interações ionicamente carregadas, forças de Van der Waals e, em alguns casos, pelos ligantes ou ions metálicos que se ligam à proteína. A estrutura terciária desempenha um papel crucial na função das proteínas, uma vez que determina sua atividade enzimática, reconhecimento de substratos, localização subcelular e interações com outras moléculas.
Na medicina, as plaquetas, também conhecidas como trombócitos, são pequenos fragmentos celulares sem núcleo que desempenham um papel crucial na coagulação sanguínea. Eles são produzidos no tecido ósseo por células chamadas megacariócitos e têm uma vida útil curta de aproximadamente 7 a 10 dias.
A função principal das plaquetas é ajudar a controlar o sangramento em caso de lesão vascular. Quando um vaso sanguíneo é danificado, as plaquetas se agregam no local do dano e formam um coágulo sanguíneo para impedir a perda excessiva de sangue. Além disso, as plaquetas também desempenham um papel na reparação dos tecidos vasculares danificados e na liberação de vários mediadores bioquímicos que participam da resposta inflamatória e do processo de cura.
Uma contagem baixa de plaquetas no sangue, conhecida como trombocitopenia, pode aumentar o risco de hemorragias e sangramentos excessivos. Por outro lado, um número elevado de plaquetas, chamado trombocitose, pode aumentar o risco de formação de coágulos sanguíneos anormais, levando a condições como trombose e embolia. Portanto, é importante manter um equilíbrio adequado no número de plaquetas no sangue para garantir uma coagulação normal e prevenir complicações relacionadas à saúde.
Movimento celular é um termo usado em biologia para descrever o movimento ativo de células, que pode ocorrer em diferentes contextos e por meios variados. Em geral, refere-se à capacidade das células de se deslocarem de um local para outro, processo essencial para diversas funções biológicas, como a embriogênese, a resposta imune, a cicatrização de feridas e o desenvolvimento de tumores.
Existem vários mecanismos responsáveis pelo movimento celular, incluindo:
1. Extensão de pseudópodos: As células podem estender projeções citoplasmáticas chamadas pseudópodos, que lhes permitem se mover em direção a um estímulo específico ou para explorar o ambiente circundante.
2. Contração do citoesqueleto: O citoesqueleto é uma rede de filamentos proteicos presente no citoplasma celular, que pode se contrair e relaxar, gerando forças mecânicas capazes de deslocar a célula.
3. Fluxo de actina: A actina é um tipo de proteína do citoesqueleto que pode se polimerizar e despolimerizar rapidamente, formando estruturas dinâmicas que impulsionam o movimento celular.
4. Movimento amebóide: Algumas células, como as amebas, podem mudar de forma dramaticamente e se mover por fluxos cíclicos de citoplasma em direção a pseudópodos em expansão.
5. Migração dirigida: Em alguns casos, o movimento celular pode ser orientado por sinais químicos ou físicos presentes no ambiente, como gradientes de concentração de moléculas químicas ou a presença de matriz extracelular rica em fibrilas colágenas.
Em resumo, o movimento celular é um processo complexo e altamente regulado que envolve uma variedade de mecanismos e interações entre proteínas e outras moléculas no citoplasma e no ambiente extracelular.
A agamaglobulinemia é uma rara condição genética em que o sistema imunológico está deficiente devido à falta de produção de certos anticorpos, também conhecidos como imunoglobulinas. Isso acontece porque as pessoas com agamaglobulinemia têm um número reduzido ou ausente de células B maduras, que são responsáveis pela produção desses anticorpos.
Existem vários tipos de agamaglobulinemia, mas a forma mais comum é a agamaglobulinemia ligada ao X (XLA), também conhecida como hipogammaglobulinemia do tipo Bruton. Esta condição é herdada de forma recessiva e afeta predominantemente homens, pois o gene defeituoso está localizado no cromossomo X.
As pessoas com agamaglobulinemia são mais susceptíveis a infecções bacterianas recorrentes, especialmente das vias respiratórias superiores e do trato gastrointestinal. Além disso, elas podem desenvolver complicações autoimunes e reações adversas a medicamentos.
O tratamento para a agamaglobulinemia geralmente consiste em administração regular de imunoglobulinas por via intravenosa ou subcutânea, para substituir os anticorpos faltantes e prevenir infecções. Também pode ser necessário tratamento antibiótico profilático para prevenir infecções recorrentes.
Os Camundongos da Linhagem 129 são uma cepa específica de camundongos usados em pesquisas biomédicas. Eles foram desenvolvidos pela primeira vez na década de 1940 e são frequentemente utilizados em estudos genéticos, imunológicos e oncológicos.
A linhagem 129 é geneticamente distinta de outras cepas de camundongos, como a linhagem C57BL/6, e apresenta certas características únicas que a tornam adequada para determinados tipos de pesquisas. Por exemplo, os camundongos da linhagem 129 são suscetíveis a certos tipos de câncer e têm um sistema imunológico que pode ser facilmente manipulado, o que os torna úteis em estudos de imunoterapia e doenças autoimunes.
Além disso, os camundongos da linhagem 129 são geneticamente variáveis, o que significa que podem ser modificados geneticamente para criar diferentes fenótipos e estudar a função de genes específicos em doenças e processos biológicos. No entanto, é importante notar que os resultados obtidos em camundongos podem não se aplicar diretamente a humanos devido às diferenças genéticas e fisiológicas entre as espécies.
A detecção de heterozigoto é um método de análise genética que permite identificar indivíduos que possuem diferentes alelos (versões alternativas de um gene) em cada cópia de um determinado gene. Isso é particularmente útil em doenças genéticas em que a presença de um alelo mutante em uma cópia do gene (mas não necessariamente na outra cópia) pode levar ao desenvolvimento da doença. A detecção de heterozigotos pode ser realizada por meio de várias técnicas, como a electroforese em gel de densidade isográdica (EGDIs), a reação em cadeia da polimerase (PCR) seguida de digestão enzimática ou sequenciamento de DNA. Essa informação é importante em diagnóstico genético, planejamento familiar e pesquisa genética.
Em medicina e biologia, a transdução de sinal é o processo pelo qual uma célula converte um sinal químico ou físico em um sinal bioquímico que pode ser utilizado para desencadear uma resposta celular específica. Isto geralmente envolve a detecção do sinal por um receptor na membrana celular, que desencadeia uma cascata de eventos bioquímicos dentro da célula, levando finalmente a uma resposta adaptativa ou homeostática.
A transdução de sinal é fundamental para a comunicação entre células e entre sistemas corporais, e está envolvida em processos biológicos complexos como a percepção sensorial, o controle do ciclo celular, a resposta imune e a regulação hormonal.
Existem vários tipos de transdução de sinal, dependendo do tipo de sinal que está sendo detectado e da cascata de eventos bioquímicos desencadeada. Alguns exemplos incluem a transdução de sinal mediada por proteínas G, a transdução de sinal mediada por tirosina quinase e a transdução de sinal mediada por canais iónicos.
Esplenectomia é um procedimento cirúrgico em que o baço é removido. O baço é um órgão localizado no quadrante superior esquerdo do abdômen, à direita do estômago e atrás do pâncreas. Ele desempenha um papel importante na defesa do corpo contra infecções, especialmente aquelas causadas por bactérias encapsuladas. Além disso, o baço ajuda no processamento da sangue velho e das células sanguíneas anormais.
Existem várias razões pelas quais alguém pode precisar de uma esplenectomia, incluindo:
1. Doenças hematológicas: Em alguns transtornos sanguíneos, como a talassemia e a anemia falciforme, o baço pode ficar inchado (esplenomegalia) e destruir excessivamente as células vermelhas do sangue saudáveis. Nesses casos, a remoção do baço pode ajudar a controlar os sintomas.
2. Trauma: Se o baço for severamente ferido ou rompido devido a um traumatismo abdominal, uma esplenectomia pode ser necessária para parar a hemorragia interna e salvar a vida do paciente.
3. Infecções: Algumas infecções graves, como a neutropenia febril grave ou a infecção por bactérias encapsuladas (como o pneumococo, meningococo e haemophilus), podem exigir a remoção do baço para prevenir futuras infecções graves.
4. Tumores: Tumores benignos ou malignos no baço também podem ser indicativos de uma esplenectomia.
Após a esplenectomia, o corpo pode ficar mais susceptível às infecções por bactérias encapsuladas. Portanto, é recomendável que os pacientes sejam vacinados contra essas bactérias antes da cirurgia ou imediatamente após a cirurgia, se possível. Além disso, é importante que os pacientes procurem atendimento médico imediato se desenvolverem sinais e sintomas de infecção, como febre, calafrios e fraqueza.
Proteínas oncogénicas referem-se a proteínas que desempenham um papel importante no desenvolvimento do câncer. Elas são geralmente resultado da mutação, amplificação ou sobre-expressão de genes proto-oncogene, os quais normalmente auxiliam no controle do crescimento celular, diferenciação e apoptose (morte celular programada). Quando esses genes sofrem alterações, eles podem se transformar em oncogenes, levando à produção de proteínas oncogénicas que contribuem para a transformação maligna das células e promovem a formação de tumores. Exemplos de proteínas oncogénicas incluem HER2/neu, EGFR, c-MYC e BCR-ABL.
Em medicina e biologia celular, uma linhagem celular refere-se a uma população homogênea de células que descendem de uma única célula ancestral original e, por isso, têm um antepassado comum e um conjunto comum de características genéticas e fenotípicas. Essas células mantêm-se geneticamente idênticas ao longo de várias gerações devido à mitose celular, processo em que uma célula mother se divide em duas células filhas geneticamente idênticas.
Linhagens celulares são amplamente utilizadas em pesquisas científicas, especialmente no campo da biologia molecular e da medicina regenerativa. Elas podem ser derivadas de diferentes fontes, como tecidos animais ou humanos, embriões, tumores ou células-tronco pluripotentes induzidas (iPSCs). Ao isolar e cultivar essas células em laboratório, os cientistas podem estudá-las para entender melhor seus comportamentos, funções e interações com outras células e moléculas.
Algumas linhagens celulares possuem propriedades especiais que as tornam úteis em determinados contextos de pesquisa. Por exemplo, a linhagem celular HeLa é originária de um câncer de colo de útero e é altamente proliferativa, o que a torna popular no estudo da divisão e crescimento celulares, além de ser utilizada em testes de drogas e vacinas. Outras linhagens celulares, como as células-tronco pluripotentes induzidas (iPSCs), podem se diferenciar em vários tipos de células especializadas, o que permite aos pesquisadores estudar doenças e desenvolver terapias para uma ampla gama de condições médicas.
Em resumo, linhagem celular é um termo usado em biologia e medicina para descrever um grupo homogêneo de células que descendem de uma única célula ancestral e possuem propriedades e comportamentos similares. Estas células são amplamente utilizadas em pesquisas científicas, desenvolvimento de medicamentos e terapias celulares, fornecendo informações valiosas sobre a biologia das células e doenças humanas.
 Síndrome de Wiskott-Aldrich - Wikipedia
Síndrome de Wiskott-Aldrich - Wikipedia
 Síndrome de Wiskott-Aldrich - Imunologia; distúrbios alérgicos - Manuais MSD edição para profissionais
Síndrome de Wiskott-Aldrich - Imunologia; distúrbios alérgicos - Manuais MSD edição para profissionais
DeCS 2011 - versão 22 de dezembro de 2011
DeCS 2014 - versão 16 de dezembro de 2014
 DeCS 2014 - versão 16 de dezembro de 2014
DeCS 2014 - versão 16 de dezembro de 2014
DeCS 2011 - versão 22 de dezembro de 2011
DeCS 2010 - versão 12 de fevereiro de 2010
DeCS 2016 - versão 12 de junho de 2016
DeCS 2013 - versão 15 de julho de 2013
DeCS 2012 - versão 22 de fevereiro de 2012
DeCS 2013 - versão 17 de dezembro de 2013
DeCS 2012 - versão 22 de fevereiro de 2012
DeCS 2017 - versão 04 de julho de 2017
DeCS 2011 - versão 06 de janeiro de 2011
DeCS 2009 - versão 20 de fevereiro de 2009
 Imunodeficiência Combinada Severa | Concise Medical Knowledge
Imunodeficiência Combinada Severa | Concise Medical Knowledge
DeCS
 Instrução de uso: Intratect - DR. DOPING
Instrução de uso: Intratect - DR. DOPING
 Linfócitos baixos: principais causas e sintomas | QBemQFaz
Linfócitos baixos: principais causas e sintomas | QBemQFaz
 Conquista - Instituto TMO
Conquista - Instituto TMO
 DII - Epidemiologia - Coggle Diagram
DII - Epidemiologia - Coggle Diagram
Servi o de Hematologia Cl nica - Centro Hospitalar e Universit rio de Coimbra
 Imunologia - Maurílio de Almeida - Precisão Absoluta
Imunologia - Maurílio de Almeida - Precisão Absoluta
 Perguntas frequentes | Diseasemaps
Perguntas frequentes | Diseasemaps
Sintomas1
- Dr. Wiskott descobriu os sintomas em 3 meninos da mesma família e o Dr. Aldrich a sua relação com o cromossomo X. A criança apresenta infecções constantes e de repetição (otite, pneumonia…), plaquetopenia (baixa contagem de plaquetas), eczemas na pele (grosseirões, manchas de tom púrpura…) e sangramentos espontâneos (principalmente nasais e gengivares). (wikipedia.org)
DiGeorge1
- Certos distúrbios de imunodeficiência hereditária, como a síndrome de DiGeorge, a síndrome de Wiskott-Aldrich, uma imunodeficiência grave combinada e a ataxia-telangiectasia. (qbemqfaz.com.br)
Imunoglobulina1
- A imunoglobulina subcutânea é indicada para as seguintes síndromes de imunodeficiência primária: agamaglobulinemia e hipogamaglobulinemia congênitas, imunodeficiência comum variável, imunodeficiência combinada grave e síndrome de Wiskott-Aldrich. (fleury.com.br)
Ligada1
- A Síndrome de Wiskott-Aldrich caracteriza-se por desencadear uma imunodeficiência infantil ligada ao cromossomo X, sendo manifestada exclusivamente em meninos. (wikipedia.org)
Transplante1
- O Trabalho sobre transplantes de medula em pacientes portadores de Síndrome de Wiskott Aldrich, realizado pela médica hematologista e encologista do Programa de Transplante Pediátrico do Hospital de Clínicas, Carmem Bonfim, conquistou o primeiro lugar no Prêmio Julio Voltarelli, concedido pela Sociedade Brasileira de Transplante. (institutotmo.org.br)
Combinado1
- A síndrome de Wiskott-Aldrich é uma imunodeficiência que resulta de um defeito combinado de células B e T e se caracteriza por infecção recorrente, eczema e trombocitopenia. (msdmanuals.com)
Grave1
- Eventos adversos raros: cefaleia grave, meningite asséptica, azotemia, síndrome de leucoencefalopatia posterior reversível (PRES) e fenômenos tromboembólicos (infarto agudo do miocárdio, infarto cerebral e tromboembolismo pulmonar). (fleury.com.br)
Syndrome1
- Pathological events in platelets of Wiskott-Aldrich syndrome patients. (msdmanuals.com)