Epilepsies, Myoclonic
Myoclonic Epilepsy, Juvenile
Epilepsy
Epilepsy, Generalized
Myoclonie
Myoclonic Epilepsies, Progressive
Epilepsy, Temporal Lobe
Epilepsy, Reflex
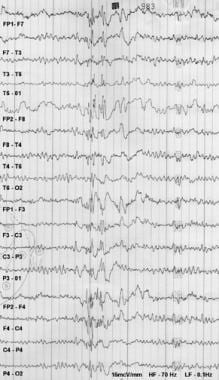
Electroencephalography
Grand Mal
Syndrome Merrf
Dyssynergie Cérébelleuse Myoclonique
Crises
Petit Mal
NAV1.1 Voltage-Gated Sodium Channel
Spasmes Infantiles
Valproate
Epilepsy, Complex Partial
Crises Convulsives Fébriles
Pentétrazole
Epilepsy, Frontal Lobe
Epilepsy, Rolandic
Status Epilepticus
Epilepsy, Post-Traumatic
Clonazépam
Ataxie
Convulsivants
Remnographie
Maladie De Lafora
Syndrome Melas
Généalogie
Arn Transfert Lysine
Enregistrement Vidéo
Photodermatoses
Facteurs Déclenchants
Encéphale
Triazines
Age of Onset
Sclérose
Carbamazépine
Encéphalomyopathies Mitochondriales
Cyclopentolate
Hyperglycinémie Non Cétosique
Pilocarpine
Lobectomie Temporale Antérieure
Thalamus
Intellectual Disability
Tremblement
Mutation
Chromosomes Humains De La Paire 16
Arn Transfert Leucine
Troubles Dystoniques
Canaux Sodiques
Récepteur Gaba-Benzodiazépine
Phénotype
Mort Subite
Troubles De La Motricité
Lobe Occipital
Neuroimaging
Epilepsies, Partial
Malformations of Cortical Development
Hippocampe
Lobe Temporal
Psychochirurgie
Retrospective Studies
Epilepsy, Benign Neonatal
Leucoencéphalite Sclérosante Subaiguë
Ketogenic Diet
Mutation Faux Sens
Family Health
Embrasement
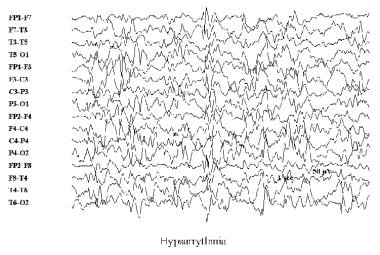
L'épilepsie myoclonique est un type d'épilepsie caractérisé par des spasmes musculaires brusques et involontaires appelés myoclonies. Ces spasmes peuvent affecter une seule partie du corps ou tout le corps et peuvent varier en fréquence de quelques fois par jour à plusieurs fois par semaine.
Les épilepsies myocloniques peuvent être causées par divers facteurs, y compris des anomalies génétiques, des lésions cérébrales, certaines infections ou intoxications. Dans certains cas, la cause peut rester inconnue.
Ce type d'épilepsie peut se manifester à tout âge, mais il est plus fréquent chez les enfants et les adolescents. Les épilepsies myocloniques peuvent être associées à d'autres symptômes, tels que des absences, des convulsions tonico-cloniques ou une déficience intellectuelle.
Le diagnostic d'épilepsie myoclonique repose sur l'observation des crises, la réalisation d'un électroencéphalogramme (EEG) et l'exclusion de toute autre cause possible de myoclonies. Le traitement dépend de la gravité et de la fréquence des crises, mais peut inclure des médicaments antiépileptiques, une alimentation spéciale ou, dans certains cas, une intervention chirurgicale.
Il est important de noter que les épilepsies myocloniques peuvent être bien contrôlées avec un traitement approprié, mais sans traitement, elles peuvent avoir des répercussions importantes sur la qualité de vie et le fonctionnement quotidien des personnes atteintes.
La myoclonie épilepsie juvénile (JME) est un type d'épilepsie caractérisée par des secousses musculaires brusques et involontaires appelées myoclonies. Ces secousses peuvent affecter une partie du corps ou tout le corps et peuvent se produire de manière isolée ou en série. Les crises de JME sont généralement déclenchées par des stimuli sensoriels tels que le bruit, la lumière ou le toucher, et ont tendance à s'aggraver avec l'excitation émotionnelle ou le stress.
Les symptômes de JME commencent généralement à l'adolescence ou au début de l'âge adulte, bien que des cas précoces aient été décrits. Les personnes atteintes de JME peuvent également présenter d'autres types de crises épileptiques, telles que des absences et des convulsions tonico-cloniques généralisées.
JME est souvent héréditaire et est causée par des mutations dans certains gènes qui affectent la façon dont les neurones communiquent dans le cerveau. Le diagnostic de JME repose sur l'histoire clinique, les résultats de l'examen neurologique et les résultats de l'électroencéphalogramme (EEG), qui peuvent montrer des modèles caractéristiques d'activité cérébrale.
Le traitement de JME implique généralement la prise de médicaments anticonvulsivants pour contrôler les crises. Les options de traitement comprennent la lamotrigine, le valproate et le topiramate. Dans certains cas, une intervention chirurgicale peut être envisagée pour les personnes qui ne répondent pas au traitement médicamenteux.
Les personnes atteintes de JME peuvent également bénéficier d'un soutien psychologique et éducatif pour faire face aux défis associés à la gestion des crises et à la vie avec une maladie chronique.
L'épilepsie est une condition médicale caractérisée par des crises récurrentes et imprévisibles, qui résultent d'une activité électrique anormale et excessive dans le cerveau. Ces crises peuvent se manifester de différentes manières, allant de secousses musculaires brèves et localisées à des périodes de confusion ou de perte de conscience. Pour poser un diagnostic d'épilepsie, une personne doit avoir eu au moins deux crises non provoquées, séparées par au moins 24 heures, à moins que la cause sous-jacente et le risque de futures crises ne justifient un traitement.
L'épilepsie peut être causée par divers facteurs, tels que des lésions cérébrales traumatiques, des infections cérébrales, des troubles congénitaux du cerveau, des tumeurs cérébrales ou un accident vasculaire cérébral. Dans certains cas, la cause peut être inconnue.
Le traitement de l'épilepsie dépend de la gravité et du type de crises, ainsi que des facteurs sous-jacents qui peuvent contribuer à la condition. Les options de traitement comprennent souvent des médicaments anticonvulsivants, une intervention chirurgicale, un régime alimentaire spécial ou d'autres thérapies complémentaires. Dans certains cas, les personnes atteintes d'épilepsie peuvent être en mesure de contrôler leurs crises avec un traitement approprié, tandis que d'autres peuvent continuer à avoir des crises malgré les efforts de traitement.
La définition médicale de l'épilepsie généralisée est la suivante :
L'épilepsie généralisée est un type d'épilepsie caractérisé par des crises qui affectent les deux hémisphères cérébraux simultanément. Les personnes atteintes de ce type d'épilepsie peuvent présenter différents types de crises, notamment des convulsions tonico-cloniques (grand mal), des absences (petit mal) ou des myoclonies.
Les crises tonico-cloniques se manifestent par une perte de conscience soudaine, suivie d'une rigidité du corps (phase tonique) et de secousses musculaires rythmiques (phase clonique). Les crises d'absence sont caractérisées par une brève interruption de la conscience, sans perte de posture ni chute. Les myoclonies se manifestent par des secousses musculaires soudaines et brèves qui peuvent affecter n'importe quelle partie du corps.
L'épilepsie généralisée peut être causée par des facteurs génétiques ou acquis, tels que des lésions cérébrales traumatiques, des infections cérébrales, des tumeurs cérébrales ou un manque d'oxygène à la naissance. Le diagnostic est généralement posé sur la base de l'histoire clinique du patient, de l'examen neurologique et de l'enregistrement des crises par électroencéphalogramme (EEG).
Le traitement de l'épilepsie généralisée repose sur l'utilisation de médicaments anticonvulsivants, qui peuvent aider à prévenir ou à réduire la fréquence des crises. Dans certains cas, une intervention chirurgicale peut être recommandée pour les personnes qui ne répondent pas au traitement médicamenteux.
La myoclonie est un type de spasme musculaire involontaire caractérisé par des contractions soudaines et brèves des muscles, souvent comparées à des secousses ou des mouvements ressemblant à des battements. Ces mouvements peuvent affecter un seul muscle ou un groupe de muscles et peuvent survenir au repos ou pendant des activités volontaires. Les myoclonies peuvent être causées par divers facteurs, tels que des lésions cérébrales, des maladies neurodégénératives, des infections, des troubles métaboliques ou des réactions médicamenteuses. Elles peuvent également se produire sans cause apparente et sont alors appelées myoclonies essentielles. Dans certains cas, les myoclonies peuvent être traitées avec des médicaments ou d'autres thérapies, mais dans d'autres situations, elles peuvent être difficiles à gérer et avoir un impact significatif sur la qualité de vie d'une personne.
La définition médicale des "Myoclonies Progessives" ou "Epilepsies Myocloniques Progressives" (EMP) est un groupe de troubles neurologiques caractérisés par la présence de myoclonies, qui sont des secousses musculaires brusques et involontaires, ainsi que d'autres types de crises épileptiques. Les myoclonies peuvent affecter n'importe quel muscle du corps, mais elles sont souvent observées dans les membres supérieurs et inférieurs.
Les EMP sont considérées comme progressives car les symptômes ont tendance à s'aggraver avec le temps. Les personnes atteintes de ces troubles peuvent présenter des difficultés croissantes pour effectuer des tâches quotidiennes, telles que marcher, parler ou manger, en raison de l'aggravation des myoclonies et d'autres symptômes.
Les EMP peuvent être causées par diverses affections sous-jacentes, notamment des maladies génétiques, des infections cérébrales, des traumatismes crâniens ou des tumeurs cérébrales. Dans certains cas, la cause peut rester inconnue même après une évaluation approfondie.
Le traitement des EMP dépend de la cause sous-jacente et peut inclure des médicaments anticonvulsivants pour contrôler les crises épileptiques, ainsi que des thérapies de réadaptation pour aider à gérer les difficultés fonctionnelles. Dans certains cas, une intervention chirurgicale peut être envisagée pour traiter les foyers épileptogènes spécifiques dans le cerveau.
Les anticonvulsivants sont une classe de médicaments utilisés pour prévenir et contrôler les convulsions ou les crises épileptiques. Ils fonctionnent en régulant l'activité électrique dans le cerveau, ce qui peut aider à prévenir les saisies.
Les anticonvulsivants sont souvent prescrits pour traiter divers types d'épilepsie, mais ils peuvent également être utilisés hors indication pour traiter d'autres conditions telles que la douleur neuropathique, les troubles bipolaires et certains types de migraines.
Il existe plusieurs anticonvulsivants disponibles sur le marché, chacun ayant des mécanismes d'action et des profils d'effets secondaires uniques. Certains exemples courants incluent la phénytoïne, la carbamazépine, le valproate, le lamotrigine, le topiramate et le gabapentin.
Comme avec tout médicament, les anticonvulsivants peuvent entraîner des effets secondaires indésirables. Les effets secondaires courants comprennent la somnolence, les étourdissements, la nausée, les vomissements et les éruptions cutanées. Dans de rares cas, certains anticonvulsivants peuvent également entraîner des effets secondaires graves tels que des dommages hépatiques, des pensées suicidaires ou une diminution de la production de globules blancs.
Il est important de travailler en étroite collaboration avec un professionnel de la santé pour déterminer le plan de traitement anticonvulsivant le plus approprié et pour surveiller tout effet secondaire potentiel.
L'épilepsie du lobe temporal est un type spécifique d'épilepsie, qui est une condition caractérisée par des crises récurrentes. Dans cette forme, les crises commencent dans le lobe temporal du cerveau. Les lobes temporaux sont situés sur les côtés de votre tête, juste derrière vos yeux.
Il existe deux types principaux de crises dans l'épilepsie du lobe temporal : les crises complexes et les crises simples. Les crises complexes, également appelées crises avec perte de conscience ou crises focales compliquées, impliquent souvent une altération de la conscience ou du comportement. La personne peut regarder vide, se comporter bizarrement, ou effectuer des mouvements automatiques comme se frotter les mains ou se mordre la langue. Les crises simples, ou crises focales simples, ne causent pas de perte de conscience et la personne peut continuer à interagir avec son environnement pendant la crise.
Les causes de l'épilepsie du lobe temporal peuvent inclure des lésions cérébrales antérieures, telles qu'un accident vasculaire cérébral ou une infection, ou des anomalies congénitales. Dans certains cas, la cause est inconnue. Le diagnostic est généralement posé sur la base d'un examen clinique, d'une évaluation des antécédents médicaux et de résultats d'examens complémentaires tels qu'un électroencéphalogramme (EEG) ou une imagerie cérébrale.
Le traitement de l'épilepsie du lobe temporal implique généralement des médicaments anticonvulsivants. Dans certains cas, la chirurgie peut être recommandée si les médicaments ne contrôlent pas adéquatement les crises.
La définition médicale de « Epilepsy, Reflex » est la suivante :
Le reflex epilepsy est un type d'épilepsie qui est déclenché par des stimuli spécifiques et répétitifs. Ces stimuli peuvent être sensoriels, cognitifs ou psychogènes. Les exemples courants de reflex epilepsy comprennent l'epilepsie photosensible, où les crises sont déclenchées par des flashs lumineux ou des motifs à haute fréquence ; l'epilepsie tactile, où les crises sont provoquées par des stimuli tactiles spécifiques ; et l'epilepsie auditive, où les crises sont déclenchées par des sons spécifiques.
Dans certains cas, les personnes atteintes de reflex epilepsy peuvent développer des crises sans la présence du stimulus déclencheur après une période de temps. Cette condition est appelée épilepsie réflexe symptomatique. Le traitement de l'épilepsie réflexe dépend de la cause sous-jacente et peut inclure des médicaments antiépileptiques, la modification du mode de vie pour éviter les déclencheurs et, dans certains cas, la chirurgie.
L'électroencéphalographie (EEG) est une procédure diagnostique non invasive utilisée pour enregistrer l'activité électrique du cerveau. Elle est réalisée en attachant de petits capteurs, appelés électrodes, à la surface du cuir chevelu avec un gel conducteur ou une pâte. Ces électrodes détectent les impulsions électriques minuscules et imperceptibles produites par l'activité neuronale dans le cerveau et transmettent ces informations à un amplificateur, où elles sont affichées sous forme d'ondes sur un moniteur.
L'EEG est utilisée pour aider au diagnostic et à la gestion de divers troubles neurologiques, tels que l'épilepsie, les convulsions, les tumeurs cérébrales, les accidents vasculaires cérébraux, l'encéphalite, la méningite, la sclérose en plaques et d'autres conditions qui affectent le fonctionnement du cerveau. Elle peut également être utilisée pour évaluer l'efficacité de certains médicaments, surveiller l'activité cérébrale pendant le sommeil et aider à la planification des interventions chirurgicales cérébrales.
L'EEG est considérée comme sûre et indolore, sans aucun risque connu associé à sa réalisation. Cependant, il peut être difficile pour certaines personnes de rester assises ou allongées immobiles pendant la durée de l'enregistrement, ce qui peut entraîner des artefacts dans les enregistrements EEG. Dans ces cas, des médicaments relaxants peuvent être administrés pour aider à réduire l'agitation et améliorer la qualité de l'enregistrement.
Le syndrome de MERRF (Myoclonic Epilepsy with Ragged Red Fibers) est un trouble mitochondrial héréditaire rare, lié à l'X, caractérisé par une épilepsie myoclonique, des troubles du mouvement, une démence et une faiblesse musculaire progressive. Le nom vient de ses principales caractéristiques : myoclonies (secousses musculaires soudaines et incontrôlables), épilepsie et fibres rouges déchiquetées (observées dans les biopsies musculaires). Cette maladie est généralement causée par une mutation du gène mitochondrial MT-TK (MT-TK, qui code pour la tRNA lysine). Les symptômes peuvent varier considérablement d'une personne à l'autre et apparaissent généralement pendant l'enfance ou l'adolescence. Le traitement est principalement axé sur les symptômes et peut inclure des médicaments contre l'épilepsie, la physiothérapie et d'autres thérapies de soutien.
La dyssynergie cérébelleuse myoclonique est un trouble du mouvement caractérisé par des contractions musculaires brusques et incontrôlables (myoclonies) associées à une mauvaise coordination musculaire (dyssynergie) et à une instabilité posturale. Ces symptômes sont dus à une dysfonction du cervelet, une structure du cerveau responsable de la coordination des mouvements volontaires.
Les myoclonies peuvent affecter un muscle ou un groupe de muscles et peuvent être déclenchées par des mouvements volontaires, des réflexes ou des stimulations sensorielles telles que des bruits soudains ou des touches légères. Les personnes atteintes de dyssynergie cérébelleuse myoclonique peuvent également présenter une variété d'autres symptômes tels qu'une démarche instable, des mouvements oculaires anormaux, une dysarthrie (parole difficile), une dysphagie (difficulté à avaler) et une ataxie (perte de coordination des mouvements volontaires).
La dyssynergie cérébelleuse myoclonique peut être causée par divers facteurs, notamment des lésions cérébrales, des maladies dégénératives du système nerveux central, des infections, des intoxications et des troubles métaboliques. Dans certains cas, la cause de la dyssynergie cérébelleuse myoclonique est inconnue, ce qui est alors qualifié d'idiopathique.
Le traitement de la dyssynergie cérébelleuse myoclonique dépend de la cause sous-jacente et peut inclure des médicaments anticonvulsivants, des agents antiépileptiques, des benzodiazépines ou d'autres thérapies. La physiothérapie et l'ergothérapie peuvent également être bénéfiques pour améliorer la fonction musculaire et la coordination.
La chaîne de navires voltage-gated NaV1.1, également connue sous le nom de SCN1A, est un type spécifique de canal sodium qui joue un rôle crucial dans la génération et la propagation des potentiels d'action dans les neurones. Il s'agit d'un canal sodium dépendant du voltage qui est sensible aux changements du potentiel membranaire et qui permet le flux d'ions sodium dans la cellule lorsqu'il est actif.
Le canal NaV1.1 est largement exprimé dans le système nerveux central, en particulier dans les corps corticaux et les fibres myélinisées des neurones. Il joue un rôle important dans l'initiation et la propagation des potentiels d'action le long des axones, en particulier dans les régions non myélinisées de l'arbre dendritique.
Les mutations du gène SCN1A peuvent entraîner diverses maladies neurologiques, notamment l'épilepsie généralisée avec pointes-ondes polyspiques et absence (GEFS+), le syndrome de Dravet et d'autres formes d'épilepsie. Ces mutations peuvent entraîner une altération de la fonction du canal NaV1.1, ce qui peut perturber l'équilibre des ions sodium dans les neurones et entraîner une hyperexcitabilité neuronale et une épilepsie.
Le Piracétam est un médicament cognitivement stimulant, appartenant à la classe des nootropiques. Il est largement utilisé dans le traitement de divers troubles neurologiques et cognitifs, tels que la démence, les troubles de l'apprentissage et de la mémoire, ainsi que pour aider à prévenir les accidents vasculaires cérébraux et améliorer les fonctions cérébrales globales.
Le Piracétam agit en modulant l'activité des neurotransmetteurs dans le cerveau, tels que l'acétylcholine et le glutamate, ce qui peut entraîner une amélioration de la mémoire, de l'humeur, de la concentration et de la capacité d'apprentissage. Il est également connu pour améliorer la circulation sanguine dans le cerveau, ce qui peut aider à protéger les cellules cérébrales contre les dommages et à favoriser leur survie.
Le Piracétam est disponible sous forme de comprimés, de gélules ou de solution liquide et est généralement bien toléré, avec peu d'effets secondaires graves signalés. Cependant, il est important de noter que l'utilisation du Piracétam doit être supervisée par un professionnel de la santé qualifié, car il peut interagir avec d'autres médicaments et conditions médicales préexistantes.
Les spasmes infantiles, également connus sous le nom de syndrome de West ou d'épilepsie myoclonique du nourrisson, sont un type rare mais grave de convulsions et d'épilepsie chez les nourrissons. Ils se manifestent généralement entre 3 et 12 mois après la naissance.
Les spasmes infantiles se caractérisent par des mouvements soudains, rapides et brusques du corps ou des membres, qui durent moins de deux secondes. Ces mouvements ressemblent à ceux d'un chat qui étire ses pattes arrière. Les spasmes peuvent se produire en salves ou en séries, souvent après le réveil ou pendant le sommeil.
Les nourrissons atteints de cette condition peuvent également présenter un retard du développement et une perte des acquis précédemment atteints, tels que la capacité à s'asseoir, à ramper ou à parler. Les spasmes infantiles sont souvent associés à des anomalies cérébrales structurelles ou fonctionnelles, telles qu'une lésion cérébrale, une malformation congénitale ou une encéphalopathie.
Le traitement des spasmes infantiles implique généralement l'utilisation de médicaments anticonvulsivants, tels que le vigabatrin ou le cannabidiol, ainsi qu'une prise en charge multidisciplinaire pour soutenir le développement du nourrisson. Dans certains cas, une intervention chirurgicale peut être recommandée pour traiter les lésions cérébrales sous-jacentes.
Le valproate est un médicament utilisé dans le traitement des troubles neurologiques et psychiatriques. Il est disponible sous différentes formes, notamment l'acide valproïque, le valproate de sodium et le valpromidate. Le valproate est principalement prescrit pour le traitement des crises d'épilepsie, car il peut aider à réduire la fréquence et l'intensité des convulsions. Il est également utilisé dans le traitement du trouble bipolaire et de la migraine.
Le valproate agit en augmentant les niveaux de neurotransmetteurs inhibiteurs dans le cerveau, tels que le GABA (acide gamma-aminobutyrique), ce qui aide à réguler l'activité électrique anormale des neurones. Les effets secondaires courants du valproate peuvent inclure des nausées, des vomissements, des douleurs abdominales, des étourdissements, des maux de tête et des troubles de la coordination.
Il est important de noter que le valproate peut avoir des effets tératogènes importants pendant la grossesse, entraînant un risque accru de malformations congénitales graves chez le fœtus. Par conséquent, il est généralement recommandé d'éviter l'utilisation du valproate pendant la grossesse si possible, et les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement.
La « épilepsie, forme partielle complexe » est un type spécifique d'épilepsie qui implique des crises récurrentes. Pendant ces crises, une personne peut avoir des hallucinations visuelles ou auditives, des expériences déformées de la réalité, des émotions intenses et involontaires, ainsi qu'une perte partielle ou complète de conscience. Ces crises peuvent également inclure des mouvements corporels incontrôlables, tels que des mâchonnements ou des mouvements des membres.
Les personnes atteintes d'épilepsie, forme partielle complexe, ont souvent des difficultés à se souvenir de ce qui s'est passé pendant la crise et peuvent se sentir fatiguées ou désorientées après celle-ci. Ce type d'épilepsie est généralement plus difficile à traiter que les autres types et peut être causé par des lésions cérébrales, des malformations congénitales ou une épilepsie symptomatique.
Il est important de noter qu'une personne atteinte d'épilepsie, forme partielle complexe, a besoin d'un traitement médical approprié pour gérer ses crises et prévenir les complications à long terme. Un diagnostic précis et un plan de traitement individualisé sont essentiels pour assurer une qualité de vie optimale pour ces personnes.
Les crises convulsives fébriles sont des convulsions qui surviennent chez les enfants en raison d'une fièvre élevée. Elles sont relativement courantes et concernent environ 2 à 5% des enfants avant l'âge de 5 ans. Ces crises ne sont généralement pas le signe d'un problème plus grave, surtout si l'enfant est en bonne santé autrement.
Les crises convulsives fébriles se produisent le plus souvent entre les âges de 6 mois et 5 ans, lorsque les enfants sont incapables de réguler leur température corporelle aussi bien que les adultes. La plupart des enfants qui en font une fois ou deux n'en auront plus jamais.
Les symptômes comprennent la rigidité musculaire, les secousses répétitives des membres, la perte de conscience et parfois la miction involontaire ou la défécation. Les crises durent généralement de quelques secondes à quelques minutes.
La cause exacte des crises convulsives fébriles n'est pas claire, mais elles sont associées à une infection qui provoque une fièvre élevée. Les infections courantes qui peuvent déclencher ces crises comprennent les infections de l'oreille, la varicelle, la rougeole et les infections respiratoires.
Il est important de consulter un médecin si votre enfant a une crise convulsive fébrile pour écarter toute autre cause possible de convulsion, comme l'épilepsie ou une infection plus grave. Le traitement se concentre généralement sur la réduction de la fièvre et l'administration de soins de soutien.
Je suis désolé, mais il semble y avoir une confusion dans votre question. Le terme "Pentétrazole" ne correspond à aucun terme médical connu. Il est possible que vous ayez fait une erreur d'orthographe ou que vous vous référiez à un autre composé ou médicament. Veuillez vérifier l'orthographe et me fournir la bonne information afin que je puisse vous aider correctement.
L'épilepsie du lobe frontal est un type d'épilepsie focale qui prend origine dans le lobe frontal du cerveau. Cette région du cerveau est responsable de fonctions telles que le mouvement volontaire, l'expression faciale, le langage et la pensée complexe.
Les crises d'épilepsie du lobe frontal peuvent être très variées, allant de mouvements anormaux subtils à des convulsions généralisées. Les symptômes courants incluent des mouvements saccadés ou rythmiques des membres, des cris soudains, une rigidité ou des secousses musculaires, une perte de conscience partielle ou totale, et des changements soudains d'humeur ou de comportement.
Les crises peuvent survenir à tout moment de la journée, mais sont souvent plus fréquentes pendant le sommeil. Les personnes atteintes d'épilepsie du lobe frontal peuvent également présenter des symptômes interictaux tels que des changements de personnalité, des problèmes de mémoire et des difficultés de concentration.
Le diagnostic de l'épilepsie du lobe frontal repose sur l'histoire clinique, les résultats de l'examen neurologique et les enregistrements vidéo-EEG. Le traitement peut inclure des médicaments antiépileptiques, la chirurgie ou d'autres thérapies telles que la stimulation du nerf vague.
La épilepsie de Rolando, également connue sous le nom d'épilepsie benigne centrotemporale de l'enfance (CBTE), est un type focal de épilepsie qui se produit principalement chez les enfants. Il tire son nom de la région du cerveau où il commence généralement, appelée gyrus central de Rolando ou sillon central.
Les crises associées à l'épilepsie de Rolando impliquent typiquement des contractions unilatérales (un seul côté) des muscles du visage, de la langue et de la bouche, qui peuvent entraîner des sensations désagréables telles que des picotements ou des engourdissements dans ces régions. Les crises peuvent également inclure des mouvements involontaires d'un côté du corps, tels qu'une jambe ou un bras. Ces crises sont généralement brèves, durent moins de deux minutes et se produisent le plus souvent pendant le sommeil ou juste après s'être endormi.
L'épilepsie de Rolando est généralement diagnostiquée pendant l'enfance, entre les âges de 3 et 13 ans, avec un pic d'apparition autour de 8 à 10 ans. Les garçons sont plus susceptibles d'être touchés que les filles. Dans la plupart des cas, cette forme d'épilepsie disparaît spontanément à l'adolescence.
Le traitement de l'épilepsie de Rolando implique généralement des médicaments anticonvulsivants pour contrôler les crises. Dans certains cas, le médecin peut recommander une surveillance attentive sans traitement, en particulier si les crises sont rares et ne perturbent pas la vie quotidienne de l'enfant. Les enfants atteints d'épilepsie de Rolando ont généralement un bon pronostic et peuvent s'attendre à une vie normale et productive après la disparition des crises.
Le statut épileptique est une condition médicale grave caractérisée par une crise épileptique prolongée ou deux crises successives sans que la personne ne retrouve un état de conscience normal entre les deux. Il s'agit d'une urgence médicale qui nécessite une prise en charge rapide, car elle peut entraîner des dommages cérébraux permanents ou même la mort si elle n'est pas traitée rapidement.
Les crises épileptiques sont généralement définies comme étant prolongées si elles durent plus de cinq minutes, bien que certaines sources puissent considérer qu'une crise qui dure plus de trois minutes est déjà anormale et nécessite une intervention médicale urgente.
Les causes du statut épileptique peuvent être variées, allant de facteurs déclenchants tels que le stress, la fièvre ou l'absence de traitement adéquat d'une crise épileptique préexistante, à des causes plus graves telles qu'un traumatisme crânien, une infection cérébrale, un accident vasculaire cérébral ou une tumeur cérébrale.
Le traitement du statut épileptique dépend de sa cause sous-jacente et peut inclure des médicaments anticonvulsivants, une oxygénothérapie, une surveillance étroite de la fonction cardiorespiratoire et, dans les cas graves, une intervention chirurgicale. Dans tous les cas, il est essentiel de consulter rapidement un médecin en cas de suspicion de statut épileptique.
La épilepsie post-traumatique (EPT) est un type de épilepsie qui se développe después de una lesión cerebral traumática (LCT). La LCT puede ser causada por una variedad de eventos, como un accidente automovilístico, una caída, un trauma contuso o penetrante al cerebro, o un shock eléctrico.
Para que se diagnostique la EPT, generalmente se requiere que la primera convulsión ocurra dentro de los siete días posteriores a la lesión cerebral. Sin embargo, algunos estudios sugieren que las convulsiones que ocurren más tarde también pueden estar relacionadas con la lesión cerebral y, por lo tanto, también se consideran parte de la EPT.
La EPT puede presentarse como convulsiones recurrentes, pero también puede manifestarse como deterioro cognitivo, cambios de personalidad o problemas emocionales. El tratamiento de la EPT generalmente implica el uso de medicamentos antiepilépticos para controlar las convulsiones y la rehabilitación para ayudar a mejorar los déficits cognitivos y funcionales resultantes de la lesión cerebral. En algunos casos, la cirugía puede ser una opción de tratamiento si los medicamentos no son efectivos en el control de las convulsiones.
Le clonazépam est un médicament appartenant à une classe de médicaments appelés benzodiazépines. Il agit en augmentant l'activité d'un neurotransmetteur inhibiteur dans le cerveau appelé GABA (acide gamma-aminobutyrique). Cela a un effet calmant et sédatif sur le système nerveux central.
Le clonazépam est principalement utilisé pour le traitement des crises d'épilepsie, en particulier dans les formes graves ou difficiles à contrôler. Il peut également être prescrit hors AMM pour le traitement de l'anxiété sévère et des troubles du sommeil.
Comme avec tous les médicaments de cette classe, le clonazépam comporte un risque de dépendance et d'abus, en particulier lorsqu'il est utilisé à long terme ou à des doses élevées. Par conséquent, il doit être prescrit avec prudence et sous surveillance médicale étroite. Les effets secondaires courants du clonazépam comprennent la somnolence, l'étourdissement, la faiblesse musculaire et la coordination altérée.
L'ataxie est un terme médical utilisé pour décrire une perte de coordination et de contrôle des mouvements musculaires volontaires, ce qui peut entraîner une démarche instable, des tremblements, des mouvements désordonnés et une mauvaise coordination des mains. Cette condition peut affecter les bras, les jambes, les yeux et la parole.
L'ataxie est souvent causée par des dommages au cervelet, qui est la partie du cerveau responsable de la coordination des mouvements, ou aux voies nerveuses qui vont du cervelet au reste du corps. Les causes courantes de l'ataxie comprennent les maladies dégénératives du cerveau telles que la sclérose en plaques, la maladie de Parkinson et la maladie de Charcot-Marie-Tooth, ainsi que les traumatismes crâniens, les tumeurs cérébrales, l'alcoolisme chronique et certains médicaments.
Le traitement de l'ataxie dépend de la cause sous-jacente et peut inclure des médicaments pour contrôler les symptômes, une thérapie physique pour aider à améliorer la coordination et la force musculaire, et des modifications de style de vie pour réduire les facteurs de risque. Dans certains cas, l'ataxie peut être progressive et entraîner une invalidité permanente.
Je suis désolé, "convulsivants" est un terme qui peut être interprété de différentes manières dans le contexte médical. Il pourrait se référer à des médicaments ou substances utilisés pour traiter ou prévenir les convulsions, telles que les benzodiazépines et l'acide valproïque. Alternativement, il peut également faire référence aux mouvements musculaires involontaires et rythmiques qui se produisent pendant une crise convulsive ou un spasme. Pourrait-il préciser ce que vous voulez savoir sur "convulsivants" pour fournir une réponse plus exacte?
Une remnographie est un type d'examen d'imagerie médicale qui utilise une faible dose de radiation pour produire des images détaillées des structures internes du corps. Contrairement à une radiographie standard, une remnographie implique l'utilisation d'un milieu de contraste, comme un produit de contraste à base d'iode, qui est ingéré ou injecté dans le patient avant l'examen.
Le milieu de contraste permet aux structures internes du corps, telles que les vaisseaux sanguins, les organes creux ou les tissus mous, d'être plus visibles sur les images radiographiques. Cela peut aider les médecins à diagnostiquer une variété de conditions médicales, y compris les maladies gastro-intestinales, les maladies rénales et les troubles vasculaires.
Les remnographies sont généralement considérées comme sûres, bien que comme avec toute procédure médicale qui utilise des radiations, il existe un risque minimal de dommages aux tissus ou au matériel génétique. Les avantages potentiels d'un diagnostic précis et opportun sont généralement considérés comme dépassant ce faible risque.
Il est important de noter que les remnographies ne doivent être effectuées que lorsqu'elles sont médicalement nécessaires, car l'exposition répétée aux radiations peut augmenter le risque de dommages à long terme. Les médecins et les technologues en imagerie médicale prennent des précautions pour minimiser l'exposition aux radiations pendant les procédures de remnographie.
La maladie de Lafora, également connue sous le nom de épilepsie myoclonique progressives de type 2 (EPM2), est une maladie neurodégénérative héréditaire rare. Elle est causée par des mutations dans les gènes EPM2A et NHLRC1, qui entraînent l'accumulation anormale de granules de glycogène dans les neurones du cerveau.
Ces granules, appelés corps de Lafora, perturbent le fonctionnement normal des cellules nerveuses et conduisent à une dégénérescence progressive du cerveau. Les symptômes de la maladie de Lafora commencent généralement à l'adolescence ou au début de l'âge adulte et comprennent des crises myocloniques, une perte de conscience, des convulsions, une déficience intellectuelle progressive, des problèmes de vision et une mobilité réduite.
Actuellement, il n'existe pas de traitement curatif pour la maladie de Lafora et le traitement se concentre sur la gestion des symptômes.
Le syndrome de MELAS est un rare trouble mitochondrial héréditaire qui affecte plusieurs parties du corps. L'abréviation "MELAS" signifie "Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes" (Encéphalomyopathie mitochondriale, acidose lactique et accidents vasculaires cérébraux comme).
Ce syndrome se manifeste généralement pendant l'enfance ou l'adolescence et est caractérisé par des crises récurrentes d'apparence similaire à un accident vasculaire cérébral, une acidose lactique, une intolérance à l'exercice, une perte progressive de la fonction musculaire (myopathie), une épilepsie, une migration anormale des cheveux sur le cuir chevelu (poliosis), une perte auditive et une démence.
Le syndrome de MELAS est causé par des mutations dans l'ADN mitochondrial, qui est hérité de la mère. Il n'existe actuellement aucun traitement curatif pour cette maladie, mais des symptômes spécifiques peuvent être gérés médicalement.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
L'ARN de transfert de lysine (tRNA-Lys) est un type spécifique d'acide ribonucléique (ARN) qui joue un rôle crucial dans la traduction du code génétique en protéines. Plus précisément, il s'agit d'un ARN de transfert (tRNA) qui transporte l'acide aminé lysine vers le site de fixation de l'ARN ribosomal pendant la synthèse des protéines.
Les tRNAs sont des molécules d'ARN non codantes qui se lient à des acides aminés spécifiques et les transportent vers le ribosome, où ils sont incorporés dans une chaîne polypeptidique en croissance pendant la traduction de l'ARN messager (ARNm) en protéines. Chaque tRNA possède une séquence d'anticodon à trois nucléotides qui s'apparie avec un codon spécifique sur l'ARNm, ce qui permet la bonne correspondance entre les acides aminés et les codons appropriés.
Le tRNA-Lys est responsable du transport de l'acide aminé lysine vers le site d'incorporation de l'ARN ribosomal pendant la synthèse des protéines. Il existe plusieurs isoformes de tRNA-Lys dans les cellules, chacune avec un anticodon différent qui s'apparie avec l'un des codons pour la lysine sur l'ARNm. Ces isoformes sont essentiels pour assurer une traduction précise et efficace du code génétique en protéines fonctionnelles.
Des mutations ou des altérations dans les gènes qui codent pour le tRNA-Lys peuvent entraîner des maladies génétiques graves, telles que la maladie mitochondriale de MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) et la neuropathie sensorimotrice autosomique dominante 4 (HSMN4). Ces maladies sont caractérisées par une variété de symptômes neurologiques, tels que des crises d'épilepsie, des troubles cognitifs, des faiblesses musculaires et des problèmes sensoriels.
Je suis désolé, mais "Enregistrement Vidéo" en soi n'est pas une définition médicale spécifique. Cependant, l'enregistrement vidéo peut être utilisé dans un contexte médical pour documenter des procédures médicales, des consultations, des entretiens ou des symptômes visibles du patient. Ces enregistrements sont utilisés à des fins de surveillance, d'enseignement, de recherche et de documentation médico-légale. Ils doivent être réalisés avec le consentement éclairé du patient, sauf dans certaines circonstances spécifiques où la loi l'autorise sans ce consentement.
La photodermatose est un terme général utilisé en dermatologie pour décrire diverses réactions cutanées anormales qui sont déclenchées par une exposition à la lumière, en particulier la lumière ultraviolette (UV) du soleil. Ces réactions peuvent varier considérablement, allant de légères rougeurs et des démangeaisons à des éruptions cutanées graves, des cloques ou des changements pigmentaires. Certaines photodermatoses peuvent également affaiblir le système immunitaire et augmenter le risque de cancer de la peau.
Les photodermatoses peuvent être classées en deux catégories principales : les réactions photoallergiques et les réactions phototoxiques.
1. Les réactions photoallergiques se produisent lorsque la lumière UV modifie une substance chimique, telle qu'un médicament ou un produit cosmétique, de sorte qu'elle devienne capable de déclencher une réponse immunitaire. Lorsque cette substance chimique modifiée est absorbée par la peau et exposée à la lumière UV, elle peut provoquer une éruption cutanée allergique.
2. Les réactions phototoxiques se produisent lorsque la lumière UV interagit directement avec certaines substances chimiques dans la peau, entraînant des dommages cellulaires et une inflammation. Ces réactions peuvent provoquer des rougeurs, des gonflements, des cloques et des douleurs sur les zones de la peau exposées au soleil.
Les photodermatoses peuvent également être classées en fonction de leur cause sous-jacente. Certaines sont causées par une exposition excessive au soleil, tandis que d'autres sont liées à des maladies sous-jacentes ou à la prise de certains médicaments. Par exemple, certaines maladies auto-immunes, telles que le lupus érythémateux disséminé, peuvent augmenter la sensibilité de la peau au soleil et entraîner des éruptions cutanées photodermatoses. De même, certains médicaments, tels que les antibiotiques tétracyclines et les antifongiques, peuvent rendre la peau plus sensible au soleil et augmenter le risque de développer une photodermatose.
Le traitement des photodermatoses dépend de leur cause sous-jacente. Dans certains cas, l'évitement de l'exposition au soleil ou l'utilisation d'écrans solaires à large spectre peut être suffisant pour prévenir les éruptions cutanées. Dans d'autres cas, des médicaments topiques ou systémiques peuvent être nécessaires pour soulager les symptômes et traiter la maladie sous-jacente. Il est important de consulter un médecin si vous pensez avoir une photodermatose, car certains types peuvent évoluer vers des conditions plus graves s'ils ne sont pas traités correctement.
En médecine, les facteurs déclenchant sont des stimuli ou des situations spécifiques qui provoquent ou exacerbent la manifestation des symptômes d'une certaine condition médicale préexistante. Ces facteurs peuvent être environnementaux, comportementaux, hormonaux, psychologiques ou liés à l'infection. Ils ne causent pas directement la maladie, mais ils peuvent déclencher une réponse physiologique qui entraîne une augmentation de la gravité des symptômes.
Par exemple, dans les maladies auto-immunes telles que la sclérose en plaques, certains facteurs déclenchant peuvent inclure le stress, l'infection ou l'exposition à des températures extrêmes, ce qui peut provoquer une poussée de la maladie. Dans l'asthme, les facteurs déclenchant courants peuvent comprendre les allergènes environnementaux, la fumée de tabac, l'exercice ou l'exposition à des irritants chimiques.
Il est important pour les personnes atteintes de maladies chroniques d'identifier et d'éviter autant que possible ces facteurs déclenchant afin de minimiser la fréquence et la gravité des poussées ou des exacerbations de leur maladie.
L'encéphale est la structure centrale du système nerveux situé dans la boîte crânienne. Il comprend le cerveau, le cervelet et le tronc cérébral. L'encéphale est responsable de la régulation des fonctions vitales telles que la respiration, la circulation sanguine et la température corporelle, ainsi que des fonctions supérieures telles que la pensée, la mémoire, l'émotion, le langage et la motricité volontaire. Il est protégé par les os de la boîte crânienne et recouvert de trois membranes appelées méninges. Le cerveau et le cervelet sont floating dans le liquide céphalo-rachidien, qui agit comme un coussin pour amortir les chocs et les mouvements brusques.
Les triazines sont un type de composé heterocyclique qui contient trois atomes d'azote et trois atomes de carbone dans leur structure cyclique. Ils forment un sous-groupe des diazines et sont largement utilisés dans l'industrie chimique pour la synthèse de divers produits, y compris les pesticides et les colorants.
Dans le contexte médical, certaines triazines ont été étudiées pour leurs propriétés thérapeutiques potentielles. Par exemple, certaines triazines ont montré des activités antimicrobiennes, antifongiques et antivirales dans des études de laboratoire. Cependant, il convient de noter que la plupart des triazines ne sont pas directement utilisées comme médicaments et nécessitent souvent d'être modifiées chimiquement pour être efficaces à des fins thérapeutiques.
En outre, certaines triazines peuvent également avoir des propriétés toxiques ou cancérigènes, il est donc important de les étudier soigneusement avant de les considérer comme des candidats potentiels pour une utilisation en médecine.
L'âge d'apparition, également connu sous le nom d'âge de début ou d'âge de survenue, fait référence à l'âge auquel une personne développe pour la première fois les symptômes ou manifestations d'une maladie, d'un trouble de santé mentale, d'un handicap ou d'autres conditions médicales. Il peut être exprimé en années, mois ou même semaines après la naissance, selon le type et la gravité de la condition concernée.
L'âge d'apparition est un aspect important du diagnostic et de la prise en charge médicale, car il peut fournir des indices sur les causes sous-jacentes de la maladie, influencer le choix des traitements et des interventions, et aider à prévoir l'évolution et le pronostic de la condition.
Par exemple, certaines maladies génétiques ou congénitales peuvent se manifester dès la naissance ou dans les premiers mois de vie, tandis que d'autres troubles, tels que la maladie d'Alzheimer ou la démence, ne se développent généralement qu'à un âge plus avancé. De même, certains troubles mentaux, comme l'autisme et le trouble déficitaire de l'attention avec hyperactivité (TDAH), peuvent présenter des signes avant-coureurs dès la petite enfance, tandis que d'autres, comme la schizophrénie, peuvent ne se manifester qu'à l'adolescence ou à l'âge adulte.
Il est important de noter que l'âge d'apparition peut varier considérablement d'une personne à l'autre, même au sein d'une même famille ou d'un même groupe de population. Des facteurs tels que les antécédents familiaux, l'environnement, le mode de vie et d'autres facteurs de risque peuvent influencer le moment où une personne développe les symptômes d'une condition donnée.
La sclérose est un terme médical qui décrit le processus de durcissement et d'épaississement des tissus conjonctifs dans le corps. Cela se produit lorsque les fibres de collagène dans ces tissus deviennent excessives ou anormalement organisées, ce qui entraîne une perte de flexibilité et de fonction. La sclérose peut affecter divers organes et tissus du corps, y compris la peau, les muscles, les tendons, les os, les vaisseaux sanguins et les organes internes.
Dans certains cas, la sclérose est une réponse normale à une blessure ou à une maladie sous-jacente. Cependant, dans d'autres cas, cela peut être le résultat d'une maladie auto-immune ou dégénérative, telle que la sclérose en plaques (SEP) ou la sclérodermie. Dans ces conditions, le système immunitaire du corps attaque et endommage les tissus conjonctifs, entraînant une sclérose progressive.
Les symptômes de la sclérose dépendent de l'emplacement et de la gravité de la maladie. Ils peuvent inclure des douleurs articulaires, des raideurs musculaires, des engourdissements, des picotements, une fatigue extrême et des problèmes respiratoires ou digestifs. Le traitement dépend également de la cause sous-jacente de la sclérose et peut inclure des médicaments, des thérapies physiques ou des changements de style de vie.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
La carbamazépine est un médicament anticonvulsivant utilisé pour traiter l'épilepsie et certains types de douleurs neuropathiques. Il fonctionne en réduisant les saisies nerveuses excessives dans le cerveau. Dans certains cas, il peut également être prescrit hors indication pour traiter d'autres conditions telles que les troubles bipolaires.
Le médicament est disponible sous forme de comprimés ou de suspension orale et doit être pris selon les directives d'un professionnel de la santé. Les effets secondaires courants peuvent inclure des étourdissements, des nausées, des vomissements, des maux de tête, des éruptions cutanées et une somnolence. Dans de rares cas, il peut provoquer des réactions allergiques graves ou affecter le nombre de cellules sanguines dans l'organisme.
Il est important de suivre attentivement les instructions posologiques et de signaler tout effet indésirable à un professionnel de la santé. La carbamazépine peut interagir avec d'autres médicaments, y compris certains antidépresseurs, antibiotiques et contraceptifs oraux, il est donc essentiel d'informer le médecin de tous les autres médicaments pris.
En résumé, la carbamazépine est un médicament anticonvulsivant utilisé pour traiter l'épilepsie et certains types de douleurs neuropathiques. Il doit être pris sous surveillance médicale stricte en raison des risques potentiels d'effets secondaires graves et d'interactions médicamenteuses.
Les encéphalomyopathies mitochondriales sont un groupe de troubles métaboliques héréditaires caractérisés par des lésions des mitochondries, qui sont les structures responsables de la production d'énergie dans les cellules. Ces maladies affectent principalement le cerveau et les muscles squelettiques, entraînant une variété de symptômes neurologiques et musculaires.
Les encéphalomyopathies mitochondriales peuvent être causées par des mutations dans l'ADN mitochondrial ou dans l'ADN nucléaire. Les mutations dans l'ADN mitochondrial sont héréditaires de la mère à l'enfant, tandis que les mutations dans l'ADN nucléaire peuvent être héréditaires de manière autosomique récessive ou autosomique dominante.
Les symptômes des encéphalomyopathies mitochondriales varient considérablement d'une personne à l'autre, mais peuvent inclure une faiblesse musculaire progressive, des crises épileptiques, des problèmes de vision et d'ouïe, des difficultés de coordination, des retards de développement, des problèmes cardiaques et respiratoires, et un retard de croissance.
Le diagnostic des encéphalomyopathies mitochondriales peut être difficile en raison de la grande variété de symptômes et de l'absence de tests diagnostiques spécifiques. Le diagnostic repose souvent sur une évaluation clinique approfondie, des antécédents familiaux détaillés, des tests de laboratoire spécialisés et des biopsies musculaires ou cérébrales.
Le traitement des encéphalomyopathies mitochondriales vise à soulager les symptômes et à prévenir les complications. Les options de traitement peuvent inclure des médicaments pour contrôler les crises épileptiques, des suppléments nutritionnels pour aider à améliorer la fonction mitochondriale, des thérapies de réadaptation pour aider à gérer la faiblesse musculaire et d'autres symptômes, et une gestion proactive des complications cardiaques et respiratoires.
La cyclopentolate est un médicament utilisé dans l'ophtalmologie comme mydriatique et cycloplégique, ce qui signifie qu'il dilate la pupille (mydriase) et paralyse les muscles de l'accommodation du cristallin de l'œil (cycloplégie). Il s'agit d'un anticholinergique qui agit en bloquant les récepteurs muscariniques de l'acétylcholine dans les muscles oculaires.
Ce médicament est souvent utilisé avant un examen ophtalmologique pour faciliter l'examen du fond de l'œil, ou avant certaines interventions chirurgicales oculaires. Il est disponible sous forme de gouttes ophtalmiques et la posologie dépend généralement de l'âge, du poids et de l'état de santé général du patient.
Les effets secondaires courants de la cyclopentolate comprennent une vision floue, une sensibilité accrue à la lumière, des rougeurs oculaires, une sécheresse buccale et une dilatation pupillaire. Dans de rares cas, il peut provoquer des effets systémiques tels que des vertiges, des maux de tête, une accélération du rythme cardiaque, une augmentation de la pression artérielle et des convulsions, en particulier chez les jeunes enfants ou lorsqu'il est utilisé à fortes doses.
L'hyperglycinémie non cétosique est une maladie métabolique rare et héréditaire, caractérisée par un taux élevé de glycine dans le sang (hyperglycinémie). Cette condition est causée par une mutation du gène d'une enzyme appelée l'aminotransférase de la sérine, qui joue un rôle crucial dans le métabolisme de la glycine. En raison de cette déficience enzymatique, la glycine s'accumule dans l'organisme, entraînant une variété de symptômes, tels que des convulsions, des retards de développement, des problèmes neurologiques et des crises pouvant évoluer vers le coma.
Contrairement à d'autres types d'hyperglycinémie, cette forme particulière ne s'accompagne pas d'une production accrue de corps cétoniques dans l'organisme. Les corps cétoniques sont des composés organiques produits lorsque le corps dégrade les graisses pour obtenir de l'énergie, un processus qui se produit généralement en cas de manque de glucose (sucre) dans l'organisme.
Il est important de noter que cette maladie peut être très grave et entraîner des complications à long terme, telles qu'une déficience intellectuelle et des problèmes neurologiques permanents, si elle n'est pas diagnostiquée et traitée rapidement et de manière adéquate. Le traitement de l'hyperglycinémie non cétosique implique généralement un régime alimentaire restrictif en glycine et des suppléments de certaines vitamines et minéraux qui peuvent aider à améliorer le métabolisme de la glycine.
La pilocarpine est un agent parasympathomimétique, ce qui signifie qu'il imite l'action de l'acétylcholine, un neurotransmetteur dans le corps qui stimule certaines cellules nerveuses et muscles.
Plus spécifiquement, la pilocarpine active les récepteurs muscariniques de la jonction neuromusculaire et des glandes exocrines, entraînant une augmentation de la sécrétion de sueur, de salive et d'autres sécrétions.
Elle est souvent utilisée dans le traitement du glaucome pour réduire la pression intraoculaire en augmentant le flux d'humeur aqueuse hors de l'œil. Elle peut également être utilisée pour traiter la bouche sèche sévère (xérostomie) causée par des médicaments ou des radiations dans la tête et le cou.
Les effets secondaires courants de la pilocarpine comprennent une sudation excessive, des nausées, des vomissements, des douleurs abdominales, des maux de tête et des troubles visuels. Dans de rares cas, elle peut provoquer des réactions allergiques graves.
Une lobectomie temporale antérieure est une procédure chirurgicale dans laquelle une partie ou la totalité du lobe temporal du cerveau est enlevée. Cette région du cerveau se trouve derrière l'os situé à la tempe et contient des structures importantes telles que l'hippocampus, qui joue un rôle crucial dans la formation de la mémoire.
Dans le cas d'une lobectomie temporale antérieure, c'est généralement une partie du lobe temporal qui est retirée, souvent comme traitement pour des crises d'épilepsie réfractaires (qui ne répondent pas aux autres formes de traitement). Cette procédure vise à éliminer la source des crises épileptiques en retirant le tissu cérébral endommagé ou mal fonctionnant.
Il est important de noter que cette intervention comporte des risques, tels que des changements possibles dans les capacités cognitives et émotionnelles, car le lobe temporal contient également des zones liées au langage, à l'audition et aux émotions. Par conséquent, une évaluation approfondie et un plan de traitement personnalisé sont nécessaires avant d'entreprendre cette procédure.
Le thalamus est une structure en forme de noix dans le cerveau qui joue un rôle central dans la perception sensorielle et la conscience. Il sert essentiellement comme un relais pour les signaux sensoriels en provenance du corps et des yeux, avant qu'ils ne soient transmis vers les régions appropriées du cortex cérébral pour une analyse plus poussée et une perception consciente. Le thalamus est également impliqué dans le contrôle de la vigilance, du sommeil et de l'éveil, ainsi que dans divers processus cognitifs tels que la mémoire et l'apprentissage. Il est composé de deux parties symétriques appelées thalamus gauche et droit, situées de chaque côté du troisième ventricule, un petit espace rempli de liquide dans le cerveau.
L'intelligence désigne les capacités d'une personne à apprendre, à raisonner, à résoudre des problèmes, à faire preuve de jugement et de pensée abstraite. Un handicap intellectuel, également connu sous le nom de déficience intellectuelle ou retard mental, est un trouble du développement qui affecte ces capacités intellectuelles et la capacité d'une personne à fonctionner de manière indépendante dans la vie quotidienne.
Il est généralement diagnostiqué avant l'âge de 18 ans et peut varier de léger à sévère. Les personnes atteintes de handicap intellectuel peuvent avoir des difficultés à acquérir et à appliquer de nouvelles connaissances, à communiquer efficacement, à prendre soin d'elles-mêmes, à établir des relations sociales et à faire face aux situations stressantes.
Les causes du handicap intellectuel peuvent être génétiques, environnementales ou résulter de complications pendant la grossesse ou la naissance. Il est important de noter que les personnes atteintes de handicap intellectuel ont des capacités et des besoins uniques, et qu'un diagnostic précoce et une intervention appropriée peuvent améliorer considérablement leur qualité de vie et leurs perspectives d'avenir.
Un tremblement est un mouvement rythmique oscillatoire, involontaire et rhythmique d'une partie du corps. Il s'agit d'un symptôme commun et peut être causé par plusieurs facteurs, y compris les conditions neurologiques, le stress, l'usage de certains médicaments, la consommation d'alcool ou de drogues, ou des problèmes liés au métabolisme. Les tremblements peuvent affecter les mains, les bras, la tête, les voix, et d'autres parties du corps. Ils peuvent se produire à différents moments, comme au repos, pendant l'action ou avec des mouvements intentionnels. La gravité et la fréquence des tremblements peuvent varier d'une personne à l'autre.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
Les chromosomes humains de la paire 16, également connus sous le nom de chromosomes 16, sont une partie importante du matériel génétique d'un être humain. Chaque personne a deux chromosomes 16, une copie héritée de chaque parent. Les chromosomes 16 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps et contiennent des milliers de gènes responsables de la détermination de nombreuses caractéristiques physiques et fonctionnelles d'un individu.
Les chromosomes 16 sont l'une des 23 paires de chromosomes humains, ce qui signifie qu'il y a en tout 46 chromosomes dans chaque cellule du corps humain (à l'exception des spermatozoïdes et des ovules, qui n'en contiennent que 23). Les chromosomes 16 sont relativement grands et se situent au milieu de la gamme de taille des chromosomes humains.
Les gènes contenus dans les chromosomes 16 jouent un rôle important dans divers processus biologiques, notamment le développement du cerveau, le métabolisme des lipides et des glucides, la réponse immunitaire et la régulation de l'expression génétique. Les mutations dans ces gènes peuvent entraîner un certain nombre de maladies génétiques rares, telles que la neurofibromatose de type 1, le syndrome de Prader-Willi et le syndrome d'Angelman.
En résumé, les chromosomes humains de la paire 16 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps humain et contiennent des milliers de gènes responsables de nombreuses caractéristiques physiques et fonctionnelles d'un individu. Les mutations dans ces gènes peuvent entraîner un certain nombre de maladies génétiques rares.
L'ARN de transfert de leucine (tRNA-Leu) est un type spécifique d'acide ribonucléique qui joue un rôle crucial dans la traduction du code génétique en protéines. Il s'agit d'un ARN non codant, ce qui signifie qu'il ne code pas directement pour une protéine spécifique. Au lieu de cela, il fonctionne comme un adaptateur moléculaire qui facilite la liaison des acides aminés spécifiques aux chaînes polypeptidiques en croissance pendant le processus de traduction.
Dans le cas de l'ARN de transfert de leucine, il s'agit plus précisément d'un ARN qui transporte l'acide aminé leucine vers le ribosome, où se déroule la synthèse des protéines. Le tRNA-Leu possède une extrémité 3' auquel est attachée l'acide aminé leucine, et une extrémité 5' qui contient une séquence d'anticodon complémentaire à un codon spécifique sur l'ARN messager (mRNA). Lorsque les deux se lient, la leucine est ajoutée à la chaîne polypeptidique en croissance.
Il existe plusieurs isoformes de tRNA-Leu dans une cellule, chacune avec un anticodon différent mais complémentaire aux codons spécifiques pour la leucine sur l'ARN messager. Cela permet d'assurer que les acides aminés sont correctement incorporés dans la bonne séquence pendant la synthèse des protéines.
Les troubles dystoniques sont un groupe de mouvements anormaux et de postures causés par des dysfonctionnements dans le système nerveux qui contrôle les muscles. Dans ces troubles, les muscles se contractent involontairement, entraînant des répétitions de mouvements involontaires, des postures tordues ou des contorsions. Ces mouvements peuvent être douloureux et affecter un seul muscle ou un groupe de muscles. Les troubles dystoniques peuvent affecter n'importe quelle partie du corps, y compris le visage, la langue, le cou, les bras, les mains, les jambes et les pieds.
Les exemples courants de troubles dystoniques comprennent la dystonie cervicale (torticolis spasmodique), dans laquelle les muscles du cou se contractent involontairement, entraînant une tête penchée ou tournée; la dystonie focale, qui affecte un muscle ou un groupe de muscles spécifiques; et la dystonie généralisée, qui affecte tout le corps.
Les causes des troubles dystoniques peuvent être génétiques, liées à une lésion cérébrale ou à une maladie neurologique sous-jacente, ou être d'origine inconnue. Le traitement peut inclure des médicaments, la physiothérapie, la thérapie occupationnelle, les injections de toxine botulique et, dans certains cas, la chirurgie.
Les canaux sodiques sont des protéines membranaires qui forment des pores spécifiques à travers la membrane cellulaire, permettant au ion sodium (Na+) de se déplacer vers l'intérieur de la cellule. Ils jouent un rôle crucial dans la génération et la transmission des potentiels d'action dans les neurones et les muscles, y compris le cœur.
Les canaux sodiques sont composés de plusieurs sous-unités qui s'assemblent pour former une structure complexe avec un filtre sélectif qui permet uniquement aux ions sodium de passer à travers. Ces canaux peuvent être régulés par divers mécanismes, tels que la voltage-dépendance, la liaison de ligands et la phosphorylation.
Les anomalies des canaux sodiques peuvent entraîner diverses maladies, y compris des troubles neuromusculaires et cardiovasculaires. Par exemple, certaines mutations dans les gènes codant pour les sous-unités des canaux sodiques peuvent entraîner une hyperactivité des canaux, ce qui peut conduire à des maladies telles que l'épilepsie, la migraine ou des arythmies cardiaques. D'autres mutations peuvent entraîner une hypoactivité des canaux, ce qui peut causer des faiblesses musculaires ou une paralysie.
Les récepteurs GABA-benzodiazépines sont des sites de liaison protéiques complexes qui se trouvent sur les neurones dans le cerveau et le système nerveux central. Ils forment une partie importante du système de transmission de signaux inhibiteurs dans le cerveau.
Le GABA (acide gamma-aminobutyrique) est un neurotransmetteur qui inhibe l'activité neuronale en réduisant la probabilité de dépolarisation et de déclenchement d'un potentiel d'action dans les neurones. Les benzodiazépines sont une classe de médicaments psychoactifs qui se lient aux récepteurs GABA-benzodiazépines et potentialisent l'activité du GABA en augmentant la fréquence des canaux chlorures ouverts, ce qui entraîne une hyperpolarisation de la membrane neuronale et une diminution de l'excitabilité.
Les récepteurs GABA-benzodiazépines sont composés de cinq sous-unités protéiques différentes (α, β, γ, δ, ε) qui s'assemblent pour former un complexe fonctionnel. Les benzodiazépines se lient spécifiquement aux sous-unités γ du récepteur, ce qui entraîne une modification de la conformation du récepteur et une augmentation de l'affinité du GABA pour son site de liaison.
Les benzodiazépines sont utilisées dans le traitement de diverses affections médicales, notamment l'anxiété, l'insomnie, les convulsions et les troubles musculaires squelettiques. Cependant, leur utilisation à long terme peut entraîner une dépendance physique et une tolérance, ce qui peut compliquer le sevrage et entraîner des symptômes de sevrage graves.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
La mort subite est un décès soudain et inattendu, généralement survenant dans les heures qui suivent l'apparition des symptômes ou au cours d'une activité apparemment sans danger. Dans la plupart des cas, elle est causée par une insuffisance cardiaque aiguë due à des arythmies ventriculaires malignes, telles que la fibrillation ventriculaire ou la torsade de pointes. Cependant, d'autres causes peuvent également être à l'origine d'une mort subite, notamment un infarctus du myocarde massif, une embolie pulmonaire massive, une hypertension maligne, une rupture aortique ou certaines maladies neurologiques.
Il est important de noter que la mort subite ne doit pas être confondue avec le décès soudain d'une personne qui était précédemment en bonne santé et n'avait aucun antécédent médical connu. Ce dernier scénario peut également être qualifié de décès subit, mais il ne relève pas nécessairement de la définition strictement médicale de la mort subite.
En outre, il existe des différences entre la mort subite cardiaque et la mort subite d'origine non cardiaque. La première est spécifiquement liée à des troubles cardiovasculaires, tandis que la seconde peut être due à une variété de causes, y compris respiratoires, neurologiques, infectieuses ou autres.
Dans le domaine médical, la mort subite fait souvent référence à la mort subite cardiaque, qui est un problème de santé publique majeur et la principale cause de décès chez les personnes souffrant de maladies cardiovasculaires. Des stratégies de prévention et de traitement sont mises en œuvre pour tenter de réduire le risque de mort subite, telles que l'utilisation d'un défibrillateur automatisé externe (DAE) et la mise en place de programmes de réanimation cardio-pulmonaire (RCP) dans les lieux publics.
Les troubles de la motricité, également connus sous le nom de troubles du mouvement, sont des conditions médicales qui affectent la capacité d'une personne à contrôler, coordonner et effectuer des mouvements volontaires et involontaires. Ces troubles peuvent affecter un seul muscle ou groupe musculaire, un membre ou une partie du corps, ou tout le corps.
Les causes sous-jacentes des troubles de la motricité peuvent varier considérablement et dépendent du type spécifique de trouble. Les causes courantes comprennent les lésions cérébrales, les maladies neurologiques, les troubles musculaires et les affections congénitales.
Les symptômes des troubles de la motricité peuvent inclure des mouvements anormaux, tels que des tremblements, des secousses, des spasmes ou des rigidités musculaires; une coordination et une équilibre altérés; des difficultés à initier ou à maintenir des mouvements volontaires; et une fatigue ou une faiblesse musculaire.
Les troubles de la motricité peuvent être classés en deux catégories principales: les troubles du mouvement hyperkinétiques, qui sont caractérisés par des mouvements excessifs ou involontaires, tels que les tremblements et les tics; et les troubles du mouvement hypokinétiques, qui sont caractérisés par une réduction ou une absence de mouvement, tels que la maladie de Parkinson.
Le traitement des troubles de la motricité dépend du type spécifique de trouble et peut inclure des médicaments, une thérapie physique, une chirurgie ou une combinaison de ces options.
Le lobe occipital est la région située à l'extrémité postérieure (vers l'arrière) du cerveau. Il fait partie des quatre lobes principaux de chaque hémisphère cérébral, avec les lobes frontal, pariétal et temporal. Le lobe occipital est principalement associé à la vision et contient la majorité des aires visuelles primaires et secondaires du cortex cérébral.
Le cortex visuel primaire, également connu sous le nom de V1 ou striatum de Brodmann, se trouve dans le lobe occipital et est responsable du traitement des informations visuelles les plus basiques, telles que la détection des bords, des formes, des couleurs et des mouvements. Les aires visuelles secondaires et supplémentaires situées dans le lobe occipital sont responsables de fonctions visuelles plus complexes, comme la reconnaissance des objets, la perception de la profondeur et la compréhension des scènes visuelles.
Le lobe occipital est délimité par les sillons latéraux (ou sulcus de Silvius) et le sillon pariéto-occipital qui le séparent respectivement du lobe temporal et du lobe pariétal. La partie postérieure du lobe occipital, située près de la ligne médiane du cerveau, est appelée la région occipitale interhémisphérique.
Les lésions ou dommages au lobe occipital peuvent entraîner des troubles visuels, tels que des hallucinations visuelles, une perte de vision partielle ou complète (appelée hémianopsie) dans un ou les deux champs visuels, des difficultés à reconnaître des objets familiers (appelées agnosies visuelles) et d'autres problèmes de perception visuelle.
La neuroimagerie est un domaine de la médecine qui se spécialise dans l'utilisation d'techniques d'imagerie pour étudier la structure et le fonctionnement du système nerveux central, y compris le cerveau et la moelle épinière. Cela peut inclure une variété de techniques, telles que l'imagerie par résonance magnétique (IRM), la tomographie par émission de positrons (TEP), la tomographie par cohérence optique (TCO) et l'électroencéphalographie (EEG).
L'IRM est une technique d'imagerie non invasive qui utilise un champ magnétique puissant et des ondes radio pour produire des images détaillées de la structure interne du cerveau. La TEP, d'autre part, est une technique d'imagerie moléculaire qui mesure l'activité métabolique dans le cerveau en suivant la distribution d'un marqueur radioactif.
La neuroimagerie est utilisée dans un large éventail d'applications cliniques et de recherche, y compris le diagnostic et le traitement des maladies neurologiques et psychiatriques, l'étude du développement et du vieillissement du cerveau, et la compréhension des bases neurales du comportement et de la cognition.
Les interventions neurochirurgicales sont des procédures médico-chirurgicales complexes qui impliquent l'intervention directe sur le système nerveux central (cerveau et moelle épinière) ou périphérique (nerfs crâniens, racines nerveuses et plexus nerveux). Ces interventions sont généralement effectuées par des médecins spécialisés appelés neurochirurgiens.
Elles peuvent être réalisées pour diverses raisons telles que le traitement de tumeurs cérébrales ou spinaux, la décompression des nerfs comprimés, la stabilisation de la colonne vertébrale dans les cas de fractures ou de maladies dégénératives, l'épilepsie, les anévrismes et autres malformations vasculaires cérébrales, les mouvements anormaux comme la maladie de Parkinson, etc.
Les techniques utilisées dans ces interventions varient considérablement, allant de procédures minimales invasives telles que la rhizotomie percutanée et la stimulation cérébrale profonde, à des opérations plus complexes nécessitant une craniotomie ou une laminectomie. Récemment, avec l'avancée de la technologie, certaines interventions neurochirurgicales peuvent également être effectuées à l'aide de robots chirurgicaux contrôlés par le médecin.
Comme toute intervention chirurgicale, les interventions neurochirurgicales comportent des risques et des complications potentielles, y compris, mais sans s'y limiter, l'hémorragie, l'infection, la réaction adverse à l'anesthésie, les dommages aux structures nerveuses environnantes et les effets indésirables liés à la pathologie sous-jacente. Par conséquent, une évaluation approfondie et un consentement éclairé sont essentiels avant de procéder à ces interventions.
Les épilepsies partielles, également connues sous le nom d'épilepsie focale ou épilepsie localisée, sont un type d'épilepsie dans laquelle les crises surviennent à partir d'une région spécifique du cerveau. Contrairement aux crises généralisées qui impliquent tout le cerveau, les crises partielles affectent initialement une seule partie du cerveau, bien qu'elles puissent s'étendre à d'autres régions ou évoluer en crises généralisées.
Les épilepsies partielles peuvent être classées en deux catégories principales : les crises simples partielles et les crises complexes partielles.
1. Crises simples partielles : Durant ces crises, la personne reste consciente et alerte, bien qu'elle puisse présenter des symptômes variés tels que des mouvements involontaires d'un membre, des sensations étranges (paresthésies), des hallucinations visuelles ou auditives, des modifications de l'odorat ou du goût, une augmentation de la fréquence cardiaque ou de la respiration, ou un sentiment de dépersonnalisation. Ces crises durent généralement moins de deux minutes et ne causent pas de confusion ou de perte de mémoire après la crise.
2. Crises complexes partielles : Pendant ces crises, la personne peut présenter une altération de la conscience ou de la vigilance, ce qui signifie qu'elle peut être désorientée, confuse ou incapable de répondre aux questions ou d'interagir avec son environnement. Ces crises peuvent inclure des automatismes stéréotypés, tels que des mouvements répétitifs de la bouche, des mains ou des yeux, et peuvent durer plus de deux minutes. Après une crise complexe partielle, la personne peut présenter une amnésie pour l'événement et peut se sentir fatiguée ou désorientée.
Les crises complexes partielles et les crises tonico-cloniques généralisées sont souvent associées à l'épilepsie du lobe temporal, qui est la forme d'épilepsie la plus courante chez les adultes. Les crises peuvent être déclenchées par des stimuli spécifiques, tels que le stress, les émotions fortes, les odeurs ou les sons inhabituels, et peuvent être difficiles à contrôler avec un traitement médical standard. Dans ces cas, une intervention chirurgicale peut être envisagée pour retirer la zone cérébrale responsable des crises.
Les malformations du développement cortical (MDC) représentent un groupe hétérogène d'anomalies congénitales du cerveau qui affectent la structure et la fonction du cortex cérébral. Elles résultent de perturbations survenant pendant la période critique du développement neuronal, qui s'étend de la neuvième semaine de gestation à la fin de la vingt-quatrième semaine.
Les MDC peuvent être classées en trois catégories principales :
1. Malformations focales : Elles sont dues à des anomalies localisées dans une région spécifique du cortex cérébral. Les exemples incluent la schizencéphalie, l'ectopie péricentrique et l'hétérotopie sous-corticale.
2. Malformations diffuses : Elles concernent des anomalies plus largement réparties dans le cortex cérébral. Les exemples comprennent la lissencéphalie, la pachygyrie et la polymicrogyrie.
3. Malformations mixtes : Elles combinent à la fois des caractéristiques focales et diffuses.
Les MDC peuvent être causées par des facteurs génétiques, environnementaux ou une combinaison des deux. Les conséquences cliniques varient considérablement en fonction de l'étendue et de la localisation des anomalies, allant d'un retard mental léger à une déficience intellectuelle sévère, ainsi qu'à des troubles moteurs, épileptiques et sensoriels. Le diagnostic repose sur l'imagerie médicale, telle que l'IRM, et peut être confirmé par des tests génétiques moléculaires. Le traitement est généralement symptomatique et dépend de la présentation clinique spécifique de chaque patient.
L'hippocampus est une structure du cerveau en forme de cheval de mer, située dans la région médiale temporale du lobe temporal. Il joue un rôle crucial dans le processus de formation de la mémoire à long terme, en particulier pour les souvenirs déclaratifs et spatiaux. Les neurones de l'hippocampus sont également importants pour la navigation et la reconnaissance des environnements. Des anomalies ou des dommages à cette région peuvent entraîner des troubles de la mémoire, tels que ceux observés dans la maladie d'Alzheimer.
Le lobe temporal est une région de l'encéphale (cerveau) situé dans la partie inférieure et latérale de chaque hémisphère cérébral. Il est délimité par les sillons latéraux et occipitaux. Le lobe temporal contient plusieurs structures importantes, dont le gyrus hippocampique et l'amygdale, qui sont associées à la mémoire et aux émotions. Cette région du cerveau est également responsable de l'audition et du traitement des informations auditives complexes, y compris la compréhension du langage parlé. Des dommages au lobe temporal peuvent entraîner des troubles de la mémoire, des difficultés d'apprentissage, des problèmes de langage et des changements dans le comportement émotionnel.
La psychochirurgie, également connue sous le nom de neurochirurgie fonctionnelle ou de chirurgie cérébrale stéréotaxique, est un type de procédure neurochirurgicale qui implique l'altération délibérée des structures cérébrales pour traiter certaines conditions mentales et neurologiques graves et réfractaires. Historiquement, la psychochirurgie a été pratiquée dans le passé pour traiter une variété de troubles mentaux, tels que la schizophrénie et la dépression sévère, mais elle est actuellement largement limitée à des indications très spécifiques et restreintes.
Les procédures de psychochirurgie consistent généralement à créer de petites lésions dans certaines régions ciblées du cerveau en utilisant diverses techniques, telles que la thermocoagulation par radiofréquence, la stimulation ultrasonique focalisée des faisceaux ou le traitement au gamma-knife. Ces interventions sont conçues pour modifier de manière sélective la connectivité et la fonctionnalité des circuits neuronaux impliqués dans les processus pathologiques sous-jacents aux troubles mentaux ciblés.
Actuellement, les indications approuvées pour la psychochirurgie comprennent principalement le traitement de certaines affections neurologiques et psychiatriques graves et réfractaires, telles que:
1. Troubles obsessionnels compulsifs (TOC) sévères et réfractaires: Dans ces cas, des lésions ciblées peuvent être créées dans les noyaux gris centraux ou dans le cortex cingulaire antérieur pour soulager les symptômes invalidants.
2. Épilepsie réfractaire: Lorsque les médicaments et d'autres traitements ne parviennent pas à contrôler efficacement les crises épileptiques, une chirurgie ciblée peut être envisagée pour éliminer ou désactiver la zone du cerveau responsable des crises.
3. Douleur chronique réfractaire: Dans certains cas, des lésions ciblées peuvent être créées dans les voies de la douleur pour soulager les symptômes chez les patients atteints de douleurs chroniques sévères et réfractaires.
4. Syndrome de Gilles de la Tourette (SGT) sévère et réfractaire: Dans ces cas, des lésions ciblées peuvent être créées dans le thalamus ou dans le cortex cingulaire antérieur pour soulager les symptômes invalidants.
5. Maladie de Parkinson avancée: Lorsque les médicaments ne parviennent pas à contrôler efficacement les symptômes moteurs de la maladie de Parkinson, une chirurgie ciblée peut être envisagée pour éliminer ou désactiver la zone du cerveau responsable des symptômes.
Il est important de noter que ces interventions sont considérées comme des traitements de dernier recours et ne sont envisagées qu'après l'échec d'autres options thérapeutiques moins invasives. De plus, les risques et les avantages doivent être soigneusement évalués au cas par cas pour chaque patient.
Les études rétrospectives, également connues sous le nom d'études de cohorte rétrospectives ou d'études cas-témoins rétrospectives, sont un type d'étude observationnelle dans laquelle les chercheurs examinent et analysent des données recueillies à partir de dossiers médicaux, de questionnaires ou d'autres sources préexistantes pour tenter de découvrir des relations de cause à effet ou des associations entre des facteurs de risque et des résultats de santé.
Dans ces études, les chercheurs identifient et sélectionnent des participants en fonction de leur exposition à un facteur de risque spécifique ou d'un résultat de santé particulier dans le passé, puis examinent les antécédents médicaux et les données de ces participants pour déterminer si des associations significatives existent entre l'exposition et le résultat.
Les études rétrospectives présentent plusieurs avantages, notamment leur faible coût, la rapidité de réalisation et la possibilité d'inclure un grand nombre de participants. Cependant, elles peuvent également être limitées par des biais potentiels dans la collecte et l'enregistrement des données, ainsi que par l'absence de contrôle sur les variables confondantes qui peuvent affecter les résultats.
En raison de ces limites, les études rétrospectives sont généralement considérées comme moins robustes que les études prospectives, dans lesquelles les participants sont suivis activement au fil du temps pour évaluer l'incidence et la progression des maladies ou des résultats de santé. Néanmoins, elles peuvent fournir des informations précieuses sur les associations entre les facteurs de risque et les résultats de santé, en particulier dans les situations où la réalisation d'études prospectives est difficile ou impossible.
Benigne Neonatal Epilepsy (BNE) est un type rare d'épilepsie qui se produit chez les nouveau-nés pendant les premiers mois de leur vie. Le terme "benign" est utilisé ici pour décrire le fait que l'état est généralement autolimité et a tendance à disparaître spontanément au cours de la première ou deuxième année de vie, sans causer de dommages neurologiques permanents.
Les crises associées à BNE sont souvent caractérisées par des mouvements cloniques (des contractions rythmiques) d'un seul groupe musculaire ou d'un côté du corps. Ces crises peuvent être déclenchées par le réveil, l'alimentation ou d'autres stimuli sensoriels.
BNE est souvent causé par des mutations génétiques spécifiques et peut se produire dans le cadre de certaines syndromes neurogénétiques. Cependant, dans certains cas, la cause sous-jacente peut rester inconnue. Le diagnostic repose généralement sur l'observation des crises, l'historique clinique et les résultats d'examens complémentaires tels que l'EEG (électroencéphalogramme) et l'IRM (imagerie par résonance magnétique).
Il est important de noter que, bien que BNE soit considéré comme bénin en raison de son caractère autolimité, une prise en charge appropriée des crises est nécessaire pour prévenir les complications à court terme et assurer le confort et le bien-être du nouveau-né.
La leucoencéphalite sclérosante subaiguë (LES) est une maladie rare et généralement progressive du système nerveux central qui implique une inflammation et une dégénération des gaines de myéline entourant les fibres nerveuses dans le cerveau blanc. La cause exacte de la LES est inconnue, mais elle est souvent associée à une infection virale préalable ou à une réaction auto-immune.
Les symptômes de la LES peuvent varier considérablement d'une personne à l'autre et dépendent de la région du cerveau affectée. Les premiers symptômes peuvent inclure des maux de tête, une fatigue extrême, des changements de comportement ou de personnalité, des problèmes de coordination et d'équilibre, des difficultés de concentration et de mémoire, ainsi que des problèmes de vision.
La LES est généralement diagnostiquée en utilisant une combinaison de tests d'imagerie cérébrale, tels qu'une imagerie par résonance magnétique (IRM), et des analyses de liquide céphalo-rachidien pour détecter les marqueurs inflammatoires et infectieux.
Le traitement de la LES vise à gérer les symptômes et à ralentir la progression de la maladie. Les options de traitement peuvent inclure des corticostéroïdes pour réduire l'inflammation, des immunosuppresseurs pour supprimer le système immunitaire, ainsi que des médicaments antiviraux et antibiotiques pour traiter toute infection sous-jacente.
La LES peut être une maladie débilitante et dans certains cas, elle peut entraîner une invalidité permanente ou même la mort. Cependant, avec un diagnostic et un traitement précoces, il est possible de ralentir la progression de la maladie et d'améliorer la qualité de vie des patients atteints de LES.
Une diète cétogène est un type de régime alimentaire à très faible teneur en glucides et riche en graisses qui est conçu pour imiter les effets métaboliques du jeûne. Lorsqu'une personne suit un régime cétogène, son corps passe à un état de cétose, où il brûle les graisses pour l'énergie au lieu des glucides.
Ce processus entraîne la production accrue de molécules appelées cétones, qui peuvent être utilisées comme source d'énergie alternative pour le cerveau et d'autres organes. Les régimes cétogènes sont souvent utilisés dans le traitement de certaines conditions médicales, telles que l'épilepsie réfractaire chez les enfants, ainsi que dans la gestion du poids et le contrôle de la glycémie chez les personnes atteintes de diabète de type 2.
Les régimes cétogènes sont généralement très restrictifs en termes de glucides et peuvent inclure des aliments tels que des viandes, des poissons, des œufs, des produits laitiers gras, des noix et des légumes à faible teneur en glucides. Les aliments riches en glucides tels que les céréales, les fruits, les légumineuses et les sucres ajoutés sont généralement évités.
Il est important de noter que les régimes cétogènes ne sont pas appropriés pour tout le monde et peuvent entraîner des effets secondaires indésirables tels que la constipation, la fatigue, les nausées et les maux de tête. Avant de commencer un régime cétogène, il est recommandé de consulter un professionnel de la santé pour déterminer si cela convient à vos besoins et à votre état de santé général.
Une mutation « faux sens » (ou missense mutation) est un type de mutation génétique où une seule paire de bases dans l'ADN est modifiée, ce qui entraîne le remplacement d'un acide aminé par un autre dans la protéine codée par ce gène. Cela peut altérer la fonction, la structure ou la stabilité de la protéine, dépendant de la position et de l'importance de l'acide aminé remplacé. Dans certains cas, ces mutations peuvent entraîner des maladies génétiques ou prédisposer à certaines conditions médicales. Toutefois, il est important de noter que toutes les mutations faux sens ne sont pas nécessairement pathogènes et que leur impact sur la santé dépend du contexte dans lequel elles se produisent.
La santé familiale est un domaine de la médecine qui se concentre sur la santé et le bien-être des membres d'une famille pris dans leur ensemble, ainsi que sur les facteurs sociaux, économiques et environnementaux qui peuvent influencer leur santé. Elle met l'accent sur la prévention des maladies et des blessures, la promotion de la santé et le traitement des problèmes de santé existants.
La santé familiale peut inclure des soins primaires pour tous les membres de la famille, ainsi que des services spécialisés pour les membres qui ont des besoins de santé spécifiques. Les prestataires de soins de santé familiaux peuvent offrir une gamme de services, y compris des examens physiques, des vaccinations, des contrôles de la tension artérielle et du cholestérol, des dépistages de maladies chroniques telles que le diabète et l'hypertension, des soins de maternité et de nouveau-né, et des services de santé mentale.
La santé familiale peut également inclure des services d'éducation et de counseling pour aider les familles à adopter des modes de vie sains, tels que une alimentation équilibrée, l'exercice régulier, la gestion du stress et la prévention du tabagisme et de la consommation excessive d'alcool.
En outre, les prestataires de soins de santé familiaux peuvent travailler en étroite collaboration avec d'autres professionnels de la santé, tels que des spécialistes, des infirmières praticiennes, des travailleurs sociaux et des thérapeutes, pour fournir des soins coordonnés et complets à tous les membres de la famille.
Dans l'ensemble, la santé familiale vise à promouvoir la santé et le bien-être des familles dans leur ensemble, en prenant en compte les facteurs qui influencent leur santé et en travaillant avec eux pour atteindre leurs objectifs de santé.
En médecine, l'embrasement (ou conflagration en termes plus formels) est un terme utilisé pour décrire une situation où il y a une propagation rapide et incontrôlable d'une maladie, d'une infection ou d'une réaction inflammatoire dans tout le corps. Cela peut se produire lorsqu'un processus pathologique local s'étend rapidement à travers la circulation sanguine, entraînant une défaillance potentiellement mortelle de plusieurs organes.
Un exemple courant est l'embrasement septique, qui survient lorsque le système immunitaire réagit de manière excessive à une infection, libérant une cascade de cytokines et d'autres molécules inflammatoires dans la circulation sanguine. Cela peut entraîner une inflammation généralisée, une hypotension, une coagulation intravasculaire disséminée (CIVD) et d'autres complications graves.
Le traitement de l'embrasement nécessite une prise en charge agressive et rapide, y compris des soins intensifs, une antibiothérapie à large spectre, une thérapie de support et souvent des interventions telles que la dialyse rénale ou la ventilation mécanique.
Je suis désolé, mais la phrase "Maladies Expérimentales" ne correspond pas à une définition médicale spécifique. Le terme "maladie expérimentale" peut faire référence à des études ou recherches scientifiques dans lesquelles des maladies sont simulées ou créées dans un laboratoire, généralement sur des animaux de laboratoire, pour mieux comprendre la pathogenèse, le développement et les traitements possibles d'une maladie. Cependant, il ne s'agit pas d'un terme médical standardisé. Si vous cherchez des informations sur une procédure ou un concept médical spécifique, veuillez me fournir plus de détails afin que je puisse vous aider au mieux.
 Syndrome de Dravet
Syndrome de Dravet Anomalies de l'ADN mitochondrial - Sujets spéciaux - Édition professionnelle du Manuel MSD
Anomalies de l'ADN mitochondrial - Sujets spéciaux - Édition professionnelle du Manuel MSD DeCS
DeCS Myopathies (hors Duchenne/Becker) - Moteurline
Myopathies (hors Duchenne/Becker) - Moteurline