Malignant Atrophic Papulosis
Dermatoses Papulosquameuses
Papulose Lymphomatoïde
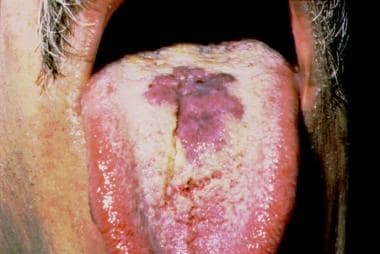
Malignant atrophic papulosis (MAP), également connue sous le nom de maladie de Kohlmeier-Degos, est une maladie rare et grave des vaisseaux sanguins de nature auto-immune. Elle affecte la peau, les articulations, le tube digestif, le système nerveux et d'autres organes.
La caractéristique distinctive de cette maladie sont des lésions cutanées atrophiques, souvent décrites comme «en forme de cible», qui évoluent en papules puis en plaques déprimées avec une surface atrophique et une bordure pigmentée. Ces lésions peuvent être douloureuses ou prurigineuses.
Dans les cas graves, MAP peut affecter le système digestif, entraînant des douleurs abdominales, des nausées, des vomissements, une diarrhée sévère et même une perforation intestinale. L'implication du système nerveux central peut provoquer des accidents vasculaires cérébraux ischémiques et d'autres complications neurologiques.
La maladie est causée par une inflammation et une destruction anormales des petits vaisseaux sanguins (vasculopathie), entraînant une mauvaise circulation sanguine dans les organes affectés. Bien que la cause exacte de cette réaction auto-immune soit inconnue, il existe certainement un lien avec des anomalies du système immunitaire.
Le diagnostic de MAP repose sur l'observation des symptômes typiques, les antécédents médicaux et les résultats d'examens complémentaires tels que des biopsies cutanées et des analyses sanguines spécifiques. Il n'existe actuellement aucun traitement curatif pour MAP, mais une prise en charge multidisciplinaire peut aider à gérer les symptômes et à prévenir les complications graves.
Les dermatoses papulo-squameuses sont un groupe de troubles cutanés caractérisés par l'apparition de lésions cutanées en forme de papules (petites bosses sur la peau) et de squames (écailles). Ces lésions peuvent être sèches, squameuses, prurigineuses (démangeaisons) ou non prurigineuses. Ce groupe comprend plusieurs affections cutanées telles que le psoriasis, la dermatite séborrhéique, la pityriasis rosea, le lichen plan et la maladie de Bowen, entre autres. Le traitement dépend du diagnostic spécifique de l'affection sous-jacente.
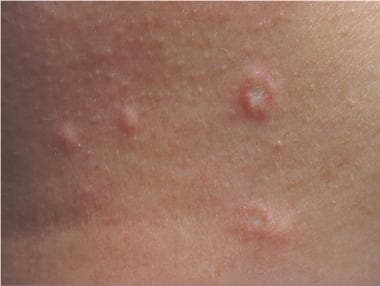
La papulose lymphomatoïde est une maladie cutanée rare et progressive, caractérisée par des lésions papulo-squameuses persistantes ou récurrentes. Elle est considérée comme un stade précoce de la maladie lymphoproliférative cutanée, qui peut évoluer vers un lymphome à cellules T.
Les lésions cutanées se présentent sous forme de papules ou de plaques rouges, prurigineuses, pouvant apparaître sur le tronc, les membres et parfois sur le visage. Elles peuvent être accompagnées d'une inflammation locale et d'un infiltrat lymphocytaire dans la peau.
L'étiologie de cette maladie est inconnue, mais elle peut être associée à des infections virales telles que le virus d'Epstein-Barr ou à une réaction immunitaire anormale. Le diagnostic repose sur l'examen histopathologique de la biopsie cutanée, qui montre un infiltrat lymphocytaire atypique dans la peau.
Le traitement peut inclure des corticostéroïdes topiques ou systémiques, de la photothérapie, des médicaments immunosuppresseurs ou des agents chimiques tels que la chlorambucil ou le méthotrexate. Dans les cas graves ou réfractaires, une thérapie ciblée ou une greffe de cellules souches peut être envisagée.
 Maladie de Degos
Maladie de Degos
Maladie de Degos - Wikipedia
 DeCS
DeCSDegos1
- en) Katz SK, Mudd LJ, Roenigk HH Jr, « Malignant atrophic papulosis (Degos' disease) involving three generations of a family » J Am Acad Dermatol. (wikipedia.org)