Perivascular Epithelioid Cell Neoplasms
Cellules
Angiomyolipome
Adenomyosis
Neoplasms, Plasma Cell
Tumeurs Du Tissu Conjonctif Et Des Tissus Mous
Ligament Rond De L'Utérus
Sclérose Tubéreuse De Bourneville
Cellules
Les tumeurs épithélioïdes périvasculaires (PEComes) forment un groupe hétérogène de néoplasmes mésoencéphaliques rares qui partagent une association étroite avec les vaisseaux sanguins et lymphatiques et expriment des marqueurs myomélanocytaires, tels que l'HMB45 et la melan A. Les PEComes peuvent se développer dans divers organes, y compris les reins, le poumon, le foie, le pancréas, le tractus gastro-intestinal, le système nerveux central et la peau.
Les PEComes comprennent un large spectre de lésions, allant des tumeurs bénignes aux sarcomes malins agressifs. Les exemples les plus courants de PEComes incluent les angiomyolipomes rénaux et pulmonaires, qui sont généralement considérés comme des néoplasmes à faible potentiel malin. Cependant, certaines sous-catégories de PEComes, telles que les sarcomes épithélioïdes périvasculaires (PES), les léiomyosarcomes épithélioïdes périvasculaires (LPS-PE) et les tumeurs épithélioïdes périvasculaires à cellules claires indifférenciées (PECCT), sont considérées comme des sarcomes malins agressifs avec un potentiel élevé de récidive locale, de métastases et de décès.
Le diagnostic de PEComes repose sur l'histopathologie et l'immunohistochimie. Les caractéristiques histopathologiques typiques comprennent des cellules épithélioïdes à noyau atypique, une architecture périvasculaire et une expression immunohistochimique positive pour les marqueurs myomélanocytaires tels que l'HMB45 et la melan A.
Le traitement des PEComes dépend du stade de la maladie, de l'agressivité histopathologique et de la localisation anatomique. Les options thérapeutiques comprennent la chirurgie, la radiothérapie et la chimiothérapie. Dans les cas de sarcomes épithélioïdes périvasculaires (PES), de léiomyosarcomes épithélioïdes périvasculaires (LPS-PE) et de tumeurs épithélioïdes périvasculaires à cellules claires indifférenciées (PECCT), une approche multimodale comprenant la chirurgie, la radiothérapie et la chimiothérapie est recommandée en raison de leur potentiel élevé de récidive locale et de métastases.
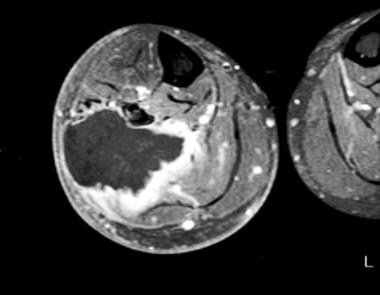
Un angiomyolipome est un type rare et généralement bénin de tumeur qui se développe dans les reins. Il s'agit d'une tumeur composée de vaisseaux sanguins anormaux (angio-), de cellules musculaires lisses (myo-) et de graisse (lipome).
Les angiomyolipomes peuvent être asymptomatiques ou provoquer des symptômes tels que des douleurs abdominales, des saignements dans les urines ou une masse palpable dans le bas du dos. Dans de rares cas, ils peuvent se transformer en tumeurs malignes et se propager à d'autres parties du corps.
Les angiomyolipomes sont souvent associés à une maladie génétique appelée sclérose tubéreuse de Bourneville, qui affecte environ 1 personne sur 6 000. Cependant, ils peuvent également se produire spontanément chez les personnes sans antécédents familiaux de la maladie.
Le traitement des angiomyolipomes dépend de leur taille, de leur localisation et de la présence ou non de symptômes. Les petites tumeurs asymptomatiques peuvent être surveillées par imagerie régulière, tandis que les tumeurs plus grandes ou symptomatiques peuvent nécessiter une intervention chirurgicale ou une ablation par radiofréquence pour prévenir les complications telles que des saignements importants.
Adenomyosis est une condition dans laquelle le tissu qui tapisse l'intérieur de l'utérus (l'endomètre) s'infiltre dans les muscles de l'utérus (le myomètre). Cette infiltration peut provoquer des douleurs pelviennes, des crampes menstruelles sévères, des saignements menstruels abondants et une augmentation du volume de l'utérus.
Les symptômes peuvent varier considérablement d'une femme à l'autre, allant de légers à graves. Certaines femmes atteintes d'adénomyose ne présentent aucun symptôme. Les facteurs de risque comprennent l'âge (elle est plus fréquente chez les femmes de plus de 30 ans), les antécédents de chirurgie utérine, les accouchements par voie basse et certaines conditions médicales telles que l'endométriose.
Le diagnostic d'adénomyose peut être difficile car elle présente des symptômes similaires à d'autres affections gynécologiques. L'imagerie par résonance magnétique (IRM) est considérée comme le gold standard pour le diagnostic de l'adénomyose.
Le traitement dépend de la gravité des symptômes et peut inclure des analgésiques, des contraceptifs hormonaux ou un traitement hormonal, une intervention chirurgicale ou une hystérectomie (ablation complète de l'utérus). Il est important de consulter un professionnel de la santé pour établir un diagnostic et un plan de traitement appropriés.
Les antigènes des mélanomes spécifiques sont des protéines ou des molécules qui se trouvent principalement sur les cellules de mélanome, une forme agressive et mortelle de cancer de la peau. Ces antigènes ne sont pas normalement présents dans les cellules saines ou sont présentés à des niveaux beaucoup plus faibles. Les mélanomes spécifiques antigènes peuvent être utilisés pour aider le système immunitaire du corps à reconnaître et à combattre les cellules cancéreuses.
Il existe deux principales catégories de mélanome-spécifiques antigènes : les antigènes tumoraux spécifiques et les antigènes tumoraux associés aux tissus. Les antigènes tumoraux spécifiques sont uniques au cancer du mélanome et ne se trouvent pas dans les cellules saines. Les antigènes tumoraux associés aux tissus, en revanche, peuvent également être présents dans les cellules normales à des niveaux plus faibles.
Les antigènes des mélanomes spécifiques sont souvent utilisés comme cibles pour le développement de thérapies immunitaires contre le cancer, telles que les vaccins thérapeutiques et les thérapies cellulaires adoptives. Ces traitements visent à stimuler la réponse immunitaire du corps contre les cellules cancéreuses en leur permettant de mieux reconnaître et d'attaquer les antigènes spécifiques au mélanome.
Cependant, il est important de noter que les tumeurs peuvent évoluer et devenir plus hétérogènes au fil du temps, ce qui peut entraîner une perte de l'expression des antigènes spécifiques au mélanome ou l'acquisition de nouvelles mutations qui permettent aux cellules cancéreuses d'échapper à la reconnaissance immunitaire. Par conséquent, les thérapies ciblant un seul antigène peuvent ne pas être suffisamment efficaces pour traiter tous les types de mélanomes ou toutes les étapes de la maladie.
Les néoplasmes des cellules plasmatiques, également connus sous le nom de gammapathies monoclonales, sont des affections caractérisées par la prolifération anormale et excessive de cellules plasmatiques dans la moelle osseuse et/ou les tissus extra-médullaires. Les cellules plasmatiques sont un type de globule blanc qui produit des anticorps ou des immunoglobulines pour aider à combattre les infections.
Dans les néoplasmes des cellules plasmatiques, ces cellules se multiplient de manière incontrôlable et peuvent produire un seul type d'immunoglobuline anormale, appelée paraprotéine. Cette prolifération anormale peut entraîner une variété de symptômes, tels qu'une fatigue excessive, des douleurs osseuses, des infections fréquentes, une insuffisance rénale et des saignements anormaux.
Le type le plus courant de néoplasme des cellules plasmatiques est le myélome multiple, qui se caractérise par la présence de lésions osseuses multiples et la production de grandes quantités de paraprotéines. D'autres types de néoplasmes des cellules plasmatiques comprennent le plasma cellulaires solitaires (ou monoclonaux), qui se produisent généralement dans les tissus extra-médullaires et sont souvent asymptomatiques, et l'amylose à chaînes légères, qui se caractérise par l'accumulation de chaînes légères d'immunoglobulines dans divers organes et tissus.
Le diagnostic des néoplasmes des cellules plasmatiques repose sur une combinaison de tests de laboratoire, d'imagerie et de biopsie de la moelle osseuse. Le traitement dépend du type et de la gravité de la maladie et peut inclure une chimiothérapie, une radiothérapie, une greffe de cellules souches et des thérapies ciblées.
Les tumeurs du tissu conjonctif et des tissus mous sont des growths anormales qui se développent dans les tissus conjonctifs ou les tissus mous du corps. Les tissus conjonctifs sont les structures de soutien du corps qui relient et entourent d'autres tissus, telles que les articulations, les muscles, les tendons et les ligaments. Les tissus mous comprennent les muscles, les tendons, les ligaments, le tissu adipeux et les vaisseaux sanguins.
Les tumeurs du tissu conjonctif et des tissus mous peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes sont généralement lentes à se développer et ne se propagent pas à d'autres parties du corps. Cependant, ils peuvent encore causer des problèmes en exerçant une pression sur les structures voisines ou en devenant suffisamment grands pour limiter la fonction.
Les tumeurs malignes, en revanche, sont agressives et ont le potentiel de se développer rapidement et de se propager à d'autres parties du corps. Ces tumeurs peuvent endommager les structures voisines et entraîner des complications graves, telles que la douleur, la perte de fonction et même la mort si elles ne sont pas traitées.
Les types courants de tumeurs du tissu conjonctif et des tissus mous comprennent le sarcome des tissus mous, qui peut se développer dans n'importe quel type de tissu mou, y compris les muscles, les tendons et les ligaments ; le sarcome d'Ewing, qui affecte principalement les os; et le fibrosarcome, qui se développe à partir des cellules du tissu conjonctif appelées fibroblastes.
Le traitement de ces tumeurs dépend de leur type, de leur emplacement et de leur étendue. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces approches. Dans certains cas, des thérapies ciblées ou des essais cliniques peuvent également être envisagés.
Le ligament rond de l'utérus, également connu sous le nom de ligament utéro-ovarien, est un ligament pair et robuste qui relie l'utérus à la paroi abdominale latérale et aux ovaires dans l'anatomie humaine. Il joue un rôle crucial dans le maintien de la position et de la stabilité de l'utérus et des ovaires dans la cavité pelvienne.
Le ligament rond de l'utérus est composé de deux couches de tissu conjonctif dense contenant des fibres musculaires lisses. Il s'étend du fond de l'utérus, près de l'isthme, vers l'extérieur et se divise en deux parties : une partie antérieure qui s'insère sur la ligne blanche de la paroi abdominale latérale et une partie postérieure qui s'insère sur le ligament large.
Ce ligament a également des propriétés vasculaires importantes, car il contient des artères et des veines qui fournissent du sang aux ovaires et à l'utérus. Les artères utéro-ovariennes, branches de l'artère ovarienne, se trouvent dans le ligament rond de l'utérus et sont responsables de l'apport sanguin vers les ovaires.
La fonction principale du ligament rond de l'utérus est de soutenir la structure pelvienne et de maintenir l'utérus et les ovaires en place. Pendant la grossesse, ce ligament contribue à la mobilité relative de l'utérus et participe aux mouvements de balancement pendant la marche, aidant ainsi à réduire l'impact des chocs sur le fœtus en développement.
Des modifications du ligament rond de l'utérus peuvent être observées dans certaines conditions gynécologiques, telles que les fibromes utérins ou l'endométriose, et peuvent contribuer à des symptômes tels que la douleur pelvienne. Des interventions chirurgicales peuvent parfois être nécessaires pour traiter ces affections et préserver la fonction du ligament rond de l'utérus.
La sclérose tubéreuse de Bourneville, également connue sous le nom de maladie de Bourneville-Pringle ou simplement de sclérose tubéreuse, est une maladie génétique rare et complexe qui affecte plusieurs organes du corps. Elle est causée par des mutations dans deux gènes spécifiques, TSC1 et TSC2, qui codent pour des protéines régulatrices de la croissance cellulaire.
Les manifestations cliniques de cette maladie peuvent varier considérablement d'un individu à l'autre, mais les signes les plus courants comprennent :
1. Lésions cutanées : apparition de taches cutanées pigmentées (naevus flammeus ou angiomes plans) et de plaques cutanées blanches (leucinocytoclastic vasculopathy).
2. Tumeurs bénignes : développement de tumeurs bénignes dans divers organes, telles que les reins (angiomyolipomes), le cerveau (tubers corticaux et sous-épendymaires) et le cœur (rhabdomyomes).
3. Épilepsie : présence d'une épilepsie réfractaire, souvent associée à des retards de développement intellectuel et moteur.
4. Troubles du comportement : apparition de troubles du comportement, tels que l'autisme ou le syndrome d'Asperger.
5. Autres complications : risque accru de développer des complications rénales, cardiaques, pulmonaires et ophtalmologiques.
Le diagnostic de la sclérose tubéreuse de Bourneville repose sur les critères cliniques, radiologiques et génétiques. Le traitement est multidisciplinaire et vise à prévenir et gérer les complications associées à cette maladie. Les options thérapeutiques comprennent des médicaments anticonvulsivants, une surveillance régulière des organes touchés, une intervention chirurgicale si nécessaire et un soutien psychologique pour les patients et leur famille.
 Tumeurs Du Fémur; Tumeurs fémorales
Tumeurs Du Fémur; Tumeurs fémorales