Primary Myelofibrosis
Janus Kinase 2
Thrombocytémie Hémorragique
Polyglobulie Primitive Essentielle
Syndromes Myéloprolifératifs
Récepteurs
Myelodysplastic-Myeloproliferative Diseases
Thrombocytose
Syndrome De L'Histiocyte Bleu Outremer
Moelle Osseuse
Mutation
Pronostic
Hématopoïèse Extramédullaire
Numération Plaquettes
World Health Organization
Mutation Faux Sens
Réticuline
Substitution D'Un Acide Aminé
Disease Progression
Survival Rate
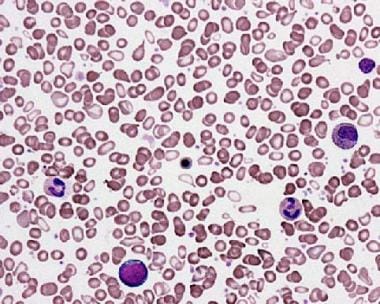
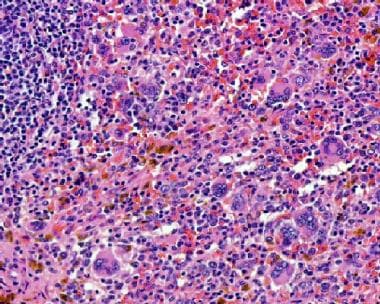
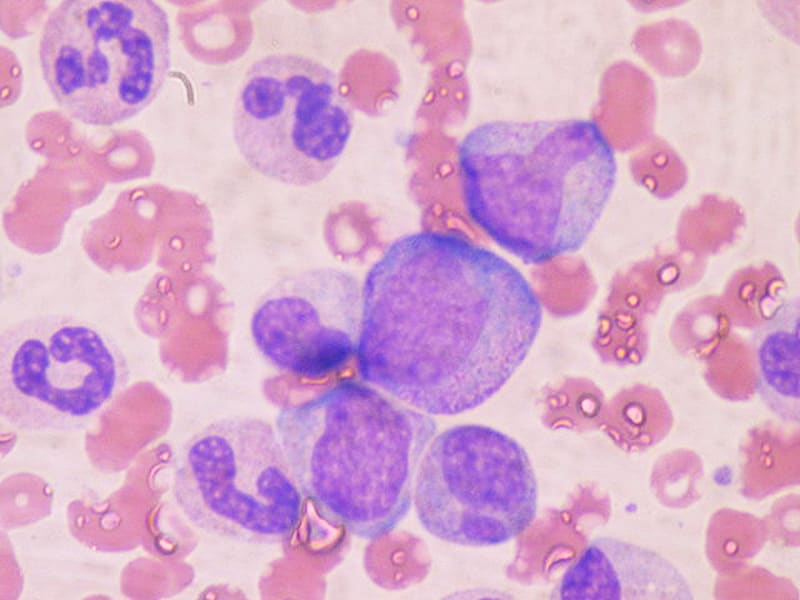
La principale myélofibrose (PMF) est une forme rare et progressive de cancer des cellules souches sanguines qui commence dans la moelle osseuse. Dans la PMF, la moelle osseuse devient rigide et fibreuse (myélofibrose), ce qui entrave sa capacité à produire des cellules sanguines saines.
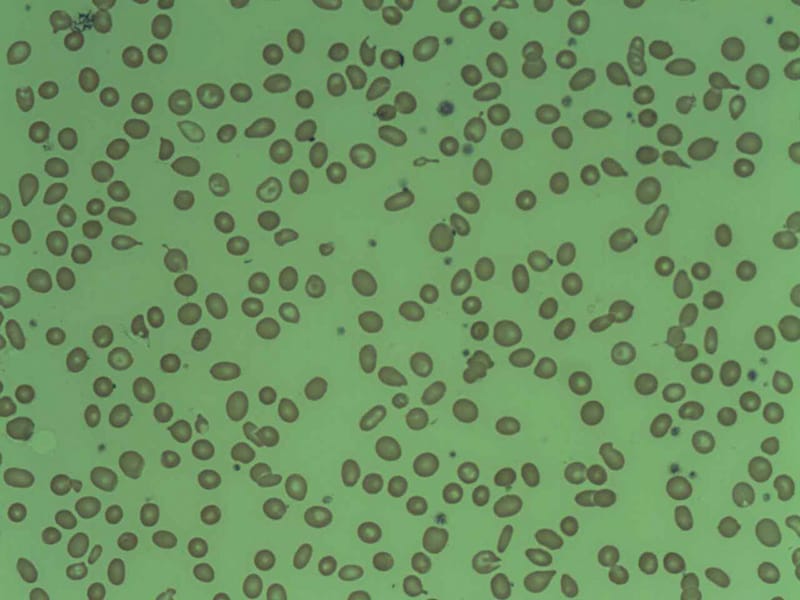
Au fil du temps, le nombre de globules rouges, de globules blancs et de plaquettes dans le sang peut diminuer, entraînant une anémie, une neutropénie (diminution du nombre de neutrophiles) et une thrombocytopénie (diminution du nombre de plaquettes).
La PMF peut également provoquer une augmentation du volume de la rate (splénomégalie), qui peut entraîner des douleurs abdominales, une sensation de satiété précoce et, dans certains cas, une rupture de la rate. Les complications supplémentaires peuvent inclure des transformations en leucémie aiguë myéloïde (LAM) et un risque accru de thrombose et d'hémorragie.
Bien que la cause exacte de la PMF ne soit pas claire, certains cas soient associés à des mutations génétiques spécifiques, telles que JAK2V617F, MPL et CALR. Le diagnostic de PMF est généralement posé par une biopsie de moelle osseuse et un examen du sang.
Le traitement de la PMF vise à soulager les symptômes et à ralentir la progression de la maladie. Les options de traitement peuvent inclure des médicaments pour stimuler la production de cellules sanguines, des agents antithrombotiques pour réduire le risque de thrombose, des immunomodulateurs et des thérapies ciblées contre les mutations génétiques spécifiques. Dans certains cas, une greffe de moelle osseuse peut être considérée comme un traitement potentiellement curatif.
La Janus Kinase 2 (JAK2) est une protéine qui joue un rôle crucial dans la transduction des signaux intracellulaires pour plusieurs cytokines et facteurs de croissance. Elle est nommée d'après la divinité romaine Janus, car elle possède deux domaines tyrosine kinase qui peuvent être actifs simultanément.
La protéine JAK2 est associée à des récepteurs de cytokines à la surface cellulaire. Lorsqu'un ligand se lie à ces récepteurs, il active la JAK2, ce qui entraîne une cascade de phosphorylation et l'activation d'autres protéines intracellulaires, y compris les facteurs de transcription STAT (Signal Transducers and Activators of Transcription). Cela conduit finalement à la régulation de l'expression des gènes.
Des mutations dans le gène JAK2 ont été associées à certaines maladies, telles que la polycythémie vraie (PV), une forme de cancer du sang caractérisée par une production excessive de globules rouges. La mutation la plus courante est appelée V617F, où la valine en position 617 est remplacée par la phénylalanine. Cette mutation entraîne une activation constitutive de JAK2, ce qui conduit à une prolifération cellulaire incontrôlée et à la maladie.
La thrombocytémie hémorragique est un trouble rare de la coagulation sanguine caractérisé par une production excessive de plaquettes (thrombocytes) dans la moelle osseuse. Cette condition peut entraîner la formation de caillots sanguins (thromboses) ou, paradoxalement, des saignements excessifs (hémorragies) en raison d'un déséquilibre dans la régulation de l'hémostase.
Il existe deux types principaux de thrombocytémie hémorragique: la forme essentielle et la forme secondaire. La forme essentielle, également appelée thrombocytose essentielle, est une affection primaire qui n'est associée à aucune autre maladie sous-jacente. Elle est souvent asymptomatique mais peut présenter des symptômes tels que maux de tête, vertiges, acouphènes, vision trouble, fatigue et épisodes thrombotiques ou hémorragiques.
La forme secondaire de thrombocytémie hémorragique est liée à une maladie sous-jacente, comme une infection, une inflammation, une intervention chirurgicale récente, un traumatisme, une carence en fer ou une affection myéloproliférative chronique (comme la polycythémie vraie, la myélofibrose primitive ou la leucémie myéloïde chronique).
Le diagnostic de thrombocytémie hémorragique repose sur des tests sanguins qui montrent un nombre élevé de plaquettes (généralement supérieur à 450 000-600 000 par microlitre) et l'exclusion d'autres causes possibles de thrombocytose. Le traitement dépend du type de thrombocytémie hémorragique et des symptômes présentés par le patient. Dans les cas légers, aucun traitement n'est nécessaire. Cependant, dans les cas sévères ou lorsque la maladie sous-jacente est identifiée, un traitement spécifique peut être recommandé, comme l'aspirine à faible dose, l'hydroxyurée ou l'interféron alpha. Dans certains cas graves, une phlébotomie ou une greffe de moelle osseuse peuvent être envisagées.
La polyglobulie primitive essentielle, également connue sous le nom de polycythémie vraie ou maladie de Vaquez, est un trouble myéloprolifératif caractérisé par une prolifération excessive et autonome des érythroblastes dans la moelle osseuse, entraînant une production accrue d'érythrocytes (globules rouges), de leucocytes (globules blancs) et de plaquettes. Cela conduit à un volume globulaire élevé (HCT) et à une concentration élevée en hémoglobine (Hb).
Contrairement aux autres types de polyglobulie, la polyglobulie primitive essentielle n'est pas causée par une réponse secondaire à une hypoxie ou à une autre maladie sous-jacente. Au lieu de cela, il s'agit d'une affection chronique qui résulte généralement d'une mutation acquise dans le gène JAK2 (janus kinase 2), bien que des mutations dans d'autres gènes puissent également être responsables dans certains cas.
Les symptômes de la polyglobulie primitive essentielle peuvent inclure des maux de tête, une fatigue, une sensation d'étourdissement, une rougeur du visage, une vision trouble, des acouphènes et une augmentation du risque de thrombose (caillots sanguins) en raison de la viscosité élevée du sang. Le diagnostic est généralement posé sur la base d'une combinaison d'anomalies sanguines, d'un examen physique et d'études d'imagerie, ainsi que de tests génétiques pour détecter une mutation JAK2.
Le traitement de la polyglobulie primitive essentielle vise généralement à réduire le volume globulaire et la viscosité du sang afin de minimiser le risque de complications thrombotiques. Cela peut inclure des procédures telles que la phlébotomie (prélèvement de sang) ou l'utilisation de médicaments pour abaisser les taux d'hématocrite et d'hémoglobine. D'autres traitements peuvent être nécessaires pour gérer les symptômes spécifiques ou les complications associées à la maladie.
Les syndromes myéloprolifératifs (SMP) représentent un groupe de troubles hématologiques caractérisés par la prolifération clonale d'un ou plusieurs types de cellules myéloïdes dans la moelle osseuse. Cela conduit à une production excessive de globules blancs, de plaquettes et/ou d'érythrocytes immatures. Les SMP comprennent :
1. La polycythémie vraie (PV): Une augmentation du nombre de globules rouges dans le sang, souvent accompagnée d'une surproduction de globules blancs et/ou de plaquettes.
2. La thrombocytose essentielle (TE): Une élévation anormale du nombre de plaquettes dans le sang sans cause sous-jacente évidente.
3. La myélofibrose primitive (MFP): Un trouble caractérisé par une prolifération excessive de cellules myéloïdes immatures, entraînant une fibrose de la moelle osseuse et une cytopénie.
4. La leucémie myélomonocytaire chronique (LMMC): Une maladie rare dans laquelle il y a une prolifération excessive de cellules myéloïdes immatures, entraînant une augmentation du nombre de globules blancs dans le sang.
5. La néoplasie myéloproliférative atypique (NMPA): Un diagnostic utilisé lorsqu'un patient présente des caractéristiques de SMP mais ne répond pas aux critères diagnostiques spécifiques d'aucune des catégories ci-dessus.
Les SMP peuvent entraîner une augmentation du risque de thrombose, d'hémorragie et de transformation en leucémie aiguë myéloïde (LAM). Le traitement dépend du type et de la gravité de la maladie et peut inclure des médicaments, une thérapie ciblée, une greffe de cellules souches ou une surveillance attentive.
La splénomégalie est un terme médical qui décrit l'élargissement anormal de la rate. La rate est un organe situé dans le côté supérieur gauche de l'abdomen, près de l'estomac et du diaphragme. Elle joue un rôle important dans le système immunitaire en aidant à combattre les infections et en éliminant les globules rouges usés.
Normalement, la rate ne devrait pas être palpable, c'est-à-dire qu'on ne devrait pas pouvoir la sentir lors d'un examen physique. Cependant, lorsqu'elle est hypertrophiée ou élargie, elle peut devenir assez grande pour être ressentie pendant un examen médical.
La splénomégalie peut être causée par diverses affections, y compris les infections (comme la mononucléose infectieuse), les maladies du sang (telles que la leucémie ou l'anémie falciforme), les maladies hépatiques (comme la cirrhose), les troubles inflammatoires (comme le lupus érythémateux disséminé) et certains cancers.
Les symptômes associés à la splénomégalie dépendent de sa cause sous-jacente. Dans certains cas, la personne peut ne présenter aucun symptôme autre que la sensation d'une masse dans le côté supérieur gauche de l'abdomen. Cependant, si la rate est fortement élargie, elle peut provoquer une sensation de satiété précoce, des douleurs abdominales, une fatigue accrue et parfois même une gêne respiratoire en raison de la pression sur le diaphragme.
Il est important de noter que la splénomégalie elle-même n'est pas une maladie mais plutôt un signe d'une affection sous-jacente. Par conséquent, il est crucial de consulter un médecin si vous ressentez une masse dans votre abdomen ou présentez des symptômes associés à la splénomégalie afin de diagnostiquer et de traiter correctement la cause sous-jacente.
Les Maladies Myélodysplasiques-Myéloprolifératives (MMD-MP) représentent un groupe hétérogène de troubles myéloprolifératifs caractérisés par la coexistence de dysplasie et de prolifération dans la moelle osseuse. Ces maladies sont considérées comme des entités nosologiques intermédiaires entre les syndromes myélodysplasiques (MDS) et les néoplasies myéloprolifératives (NMP).
Les MMD-MP se manifestent par une production anormale de cellules sanguines immatures et fonctionnellement altérées, entraînant des cytopénies dans un ou plusieurs lignages cellulaires. Les patients peuvent présenter une thrombocytose, une leucocytose ou une anémie, selon la maladie sous-jacente.
Les diagnostics différentiels incluent les MDS, les NMP et d'autres affections hématologiques. Le diagnostic de MMD-MP repose sur l'examen clinique, l'histopathologie de la moelle osseuse, la cytométrie en flux, le caryotype et la génétique moléculaire.
Les traitements peuvent inclure des thérapies ciblées, une chimiothérapie, une greffe de cellules souches hématopoïétiques ou un suivi expectant, en fonction du stade et du sous-type de la maladie. La prise en charge doit être individualisée pour chaque patient, en tenant compte de ses comorbidités, de son âge et de ses préférences personnelles.
La thrombocytose est un terme utilisé en médecine pour décrire une augmentation anormale du nombre de plaquettes (thrombocytes) dans le sang. Les plaquettes sont des petites cellules sanguines qui jouent un rôle crucial dans la coagulation sanguine et l'arrêt des saignements.
Une thrombocytose peut être classée comme réactive ou essentielle. La thrombocytose réactive est souvent observée en réponse à une maladie sous-jacente, telle qu'une infection, une inflammation, une intervention chirurgicale, une hémorragie aiguë, une maladie néoplasique ou une carence en fer. Dans ces cas, le nombre de plaquettes revient généralement à la normale une fois que la cause sous-jacente est traitée.
D'un autre côté, la thrombocytose essentielle, également appelée thrombocytose primaire, est une affection myéloproliférative chronique caractérisée par une production excessive de plaquettes dans la moelle osseuse. Cette forme de thrombocytose n'est pas directement liée à une autre maladie et peut entraîner des complications telles que des saignements ou des risques accrus de formation de caillots sanguins (thrombose).
Il est important de noter qu'une thrombocytose légère à modérée peut ne pas provoquer de symptômes et être détectée lors d'examens sanguins de routine. Cependant, des niveaux élevés de plaquettes peuvent entraîner des complications graves telles que des accidents vasculaires cérébraux, des crises cardiaques ou des embolies pulmonaires. Par conséquent, il est essentiel d'identifier et de traiter la cause sous-jacente de la thrombocytose pour prévenir ces complications potentiellement mortelles.
Le syndrome de l'histiocyte bleu outremer, également connu sous le nom de maladie de Niemann-Pick type B, est une maladie héréditaire rare liée à un déficit en enzyme spécifique appelée sphingomyélinase acide. Cette enzyme est responsable du métabolisme d'un lipide appelé sphingomyéline, qui est abondant dans les membranes cellulaires. En l'absence de cette enzyme, la sphingomyéline s'accumule dans certaines cellules, y compris les histiocytes (cellules du système immunitaire), entraînant une augmentation de leur taille et les faisant apparaître bleues lorsqu'elles sont observées au microscope.
Les symptômes du syndrome de l'histiocyte bleu outremer peuvent varier considérablement d'une personne à l'autre, mais ils comprennent souvent une hypertrophie du foie et de la rate, une augmentation du tissu adipeux dans le visage et le cou, une insuffisance respiratoire, une dégénérescence pulmonaire et neurologique, ainsi qu'une augmentation des niveaux de cholestérol et de triglycérides dans le sang.
Le diagnostic du syndrome de l'histiocyte bleu outremer repose généralement sur une combinaison d'examens cliniques, d'imagerie médicale et de tests génétiques et enzymatiques spécifiques. Il n'existe actuellement aucun traitement curatif pour cette maladie, bien que des thérapies symptomatiques et de soutien puissent être proposées pour améliorer la qualité de vie des personnes atteintes.
La moelle osseuse est la substance molle et grasse contenue dans les cavités des os longs et plats. Elle est composée de cellules souches hématopoïétiques, de matrice extracellulaire, de vaisseaux sanguins et nerveux. La moelle osseuse rouge est responsable de la production de cellules sanguines telles que les globules rouges, les globules blancs et les plaquettes. La moelle osseuse jaune contient principalement des graisses. La moelle osseuse joue un rôle crucial dans le maintien de la fonction immunitaire, du transport de l'oxygène et de la coagulation sanguine. Des maladies telles que la leucémie, l'anémie aplastique et les myélodysplasies peuvent affecter la moelle osseuse.
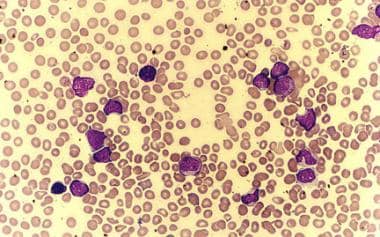
Les mégacaryocytes sont des cellules géantes trouvées dans la moelle osseuse. Ils sont responsables de la production et de la sécrétion de plaquettes, qui sont des fragments cellulaires essentiels à la coagulation sanguine. Les mégacaryocytes subissent une série de processus complexes appelés maturation thrombopoïétique, au cours de laquelle ils s'élargissent et se remplissent de granules contenant des facteurs de coagulation. Ensuite, ils éclatent pour libérer des plaquettes dans la circulation sanguine. Les troubles qui affectent la production ou la fonction des mégacaryocytes peuvent entraîner une thrombopénie (un nombre insuffisant de plaquettes), ce qui peut augmenter le risque de saignement.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
En médecine, le terme "pronostic" se réfère à la prévision du résultat ou de l'issue attendue d'une maladie ou d'une blessure dans le corps humain. Il s'agit essentiellement d'une estimation de la probabilité du rétablissement complet, de l'amélioration continue, de l'évolution vers une invalidité permanente ou du décès d'un patient atteint d'une certaine maladie ou blessure.
Le pronostic est généralement fondé sur les antécédents médicaux du patient, les résultats des tests diagnostiques, l'étendue de la maladie ou de la lésion, la réponse au traitement et d'autres facteurs pertinents. Il peut être exprimé en termes généraux ou spécifiques, tels qu'un pronostic favorable, défavorable ou incertain.
Il est important de noter que le pronostic n'est pas une garantie et ne doit pas être considéré comme tel. Il s'agit simplement d'une estimation basée sur des données probantes et l'expérience clinique, qui peut varier d'un patient à l'autre. Les médecins doivent communiquer clairement le pronostic aux patients et à leur famille, en s'assurant qu'ils comprennent les risques, les avantages et les incertitudes associés au traitement et à la maladie sous-jacente.
Un caryotype est une représentation standardisée de l'ensemble des chromosomes d'une cellule, organisme ou espèce donnée. Il s'agit d'un outil diagnostique important en génétique médicale pour identifier d'éventuelles anomalies chromosomiques.
Un caryotype humain typique se compose de 46 chromosomes, répartis en 23 paires. Chaque paire est constituée d'un chromosome d'origine maternelle et d'un chromosome d'origine paternelle, à l'exception des chromosomes sexuels X et Y. Les femmes ont deux chromosomes X (XX), tandis que les hommes en ont un X et un Y (XY).
Pour réaliser un caryotype, on prélève généralement des cellules du sang ou des tissus. Ensuite, ces cellules sont cultivées en laboratoire pour parvenir à la phase de division cellulaire appelée métaphase. À ce stade, les chromosomes sont le plus condensés et donc les plus faciles à visualiser.
Les chromosomes sont ensuite colorés avec des teintures spécifiques qui permettent de distinguer visuellement chaque paire. Ils sont ensuite disposés en fonction de leur taille, du centromère (point de jonction entre les bras courts et longs) et des bandes caractéristiques propres à chaque chromosome.
Un caryotype anormal peut révéler divers types d'anomalies chromosomiques, telles que des délétions, des duplications, des translocations ou des inversions partielles ou totales de certains segments chromosomiques. Ces anomalies peuvent être responsables de maladies génétiques, de retards de développement, d'anomalies congénitales et d'autres problèmes de santé.
L'hématopoïèse extramédullaire est un processus dans lequel la production de cellules sanguines a lieu en dehors de la moelle osseuse, qui est le site habituel de l'hématopoïèse. Normalement, tous les types de cellules sanguines sont produits dans la moelle osseuse à l'intérieur des os plats comme l'os iliaque, le sternum, les vertèbres et les côtes.
Cependant, en raison de certaines affections médicales telles que des maladies hématologiques malignes (leucémie, lymphome), une insuffisance médullaire ou une irradiation, la moelle osseuse peut ne plus être capable de produire suffisamment de cellules sanguines. Dans ces cas, le processus d'hématopoïèse peut se déplacer vers d'autres organes et tissus, tels que le foie, la rate et les poumons, pour assurer la production continue de cellules sanguines.
L'hématopoïèse extramédullaire peut être bénigne ou maligne. Dans sa forme bénigne, elle est souvent observée chez les nouveau-nés prématurés et les nourrissons, où la moelle osseuse n'est pas encore complètement développée pour assurer une production adéquate de cellules sanguines. Dans sa forme maligne, elle peut être associée à des tumeurs malignes telles que le myélome multiple ou les lymphomes.
Il est important de noter que l'hématopoïèse extramédullaire peut entraîner une augmentation de la taille des organes affectés, ce qui peut provoquer une compression des structures voisines et entraîner divers symptômes cliniques. Par conséquent, il est essentiel de diagnostiquer et de traiter rapidement cette condition pour prévenir les complications potentielles.
La numération plaquettaire, également connue sous le nom de compte de plaquettes ou test de plaquettes, est un examen de laboratoire utilisé pour évaluer le nombre de plaquettes dans un échantillon de sang. Les plaquettes, qui sont des fragments cellulaires produits dans la moelle osseuse, jouent un rôle crucial dans la coagulation sanguine et l'arrêt des saignements.
Un compte de plaquettes normal se situe généralement entre 150 000 et 450 000 plaquettes par microlitre de sang. Un nombre inférieur à 150 000 plaquettes par microlitre est considéré comme une thrombocytopénie, ce qui peut augmenter le risque de saignement. À l'inverse, un nombre supérieur à 450 000 plaquettes par microlitre est considéré comme une thrombocytose, ce qui peut accroître le risque de formation de caillots sanguins.
Des numérations plaquettaires anormales peuvent être liées à diverses affections médicales, telles que des maladies infectieuses, des troubles immunitaires, des maladies hématologiques, des cancers, des effets secondaires de médicaments ou une carence en vitamine B12 ou en acide folique. Par conséquent, un compte de plaquettes anormalement bas ou élevé doit être évalué plus avant par un professionnel de la santé pour déterminer la cause sous-jacente et élaborer un plan de traitement approprié.
La World Health Organization (WHO) est l'organisation des Nations Unies chargée de la santé publique internationale. Fondée en 1948, la WHO travaille dans plus de 150 pays à améliorer la santé et à réduire les souffrances. Elle définit les normes et les directives mondiales en matière de santé, renforce les capacités nationales et soutient les systèmes de santé.
La WHO mène des initiatives pour prévenir et contrôler les maladies transmissibles telles que le VIH/SIDA, la tuberculose et le paludisme, ainsi que les maladies non transmissibles telles que les maladies cardiovasculaires, le cancer et le diabète. Elle travaille également sur des questions de santé publique telles que la nutrition, la sécurité sanitaire des aliments, la santé mentale, la préparation et la réponse aux situations d'urgence sanitaire, y compris les catastrophes naturelles et les crises humanitaires.
La WHO est dirigée par un directeur général élu par les États membres pour une période de cinq ans. Le siège de l'organisation est à Genève, en Suisse, et elle dispose de bureaux régionaux dans le monde entier.
Une mutation « faux sens » (ou missense mutation) est un type de mutation génétique où une seule paire de bases dans l'ADN est modifiée, ce qui entraîne le remplacement d'un acide aminé par un autre dans la protéine codée par ce gène. Cela peut altérer la fonction, la structure ou la stabilité de la protéine, dépendant de la position et de l'importance de l'acide aminé remplacé. Dans certains cas, ces mutations peuvent entraîner des maladies génétiques ou prédisposer à certaines conditions médicales. Toutefois, il est important de noter que toutes les mutations faux sens ne sont pas nécessairement pathogènes et que leur impact sur la santé dépend du contexte dans lequel elles se produisent.
Je suis désolé, mais il semble y avoir une certaine confusion dans votre question. "Réticuline" n'est pas un terme médical communément utilisé. Cependant, la réticuline est une protéine qui peut être mentionnée dans des contextes scientifiques ou médico-scientifiques.
La réticuline est une protéine structurelle importante du cytosquelette et joue un rôle crucial dans la formation de la matrice extracellulaire. Elle est particulièrement abondante dans les tissus conjonctifs, nerveux et musculaires. Dans le système nerveux central, elle participe à la structure des gaines de myéline entourant les axones des neurones.
Si vous faisiez référence à un terme médical similaire ou associé, pouvez-vous s'il vous plaît me fournir plus d'informations ou clarifier votre question? Je serais heureux de vous aider davantage.
La «substitution d'un acide aminé» est un terme utilisé en biologie moléculaire et en médecine pour décrire le processus de remplacement d'un acide aminé spécifique dans une protéine ou dans une chaîne polypeptidique par un autre acide aminé. Cette substitution peut être due à des mutations génétiques, des modifications post-traductionnelles ou à des processus pathologiques tels que les maladies neurodégénératives et les cancers.
Les substitutions d'acides aminés peuvent entraîner des changements dans la structure et la fonction de la protéine, ce qui peut avoir des conséquences importantes sur la santé humaine. Par exemple, certaines substitutions d'acides aminés peuvent entraîner une perte de fonction de la protéine, tandis que d'autres peuvent conduire à une activation ou une inhibition anormale de la protéine.
Les substitutions d'acides aminés sont souvent classées en fonction de leur impact sur la fonction de la protéine. Les substitutions conservatives sont celles où l'acide aminé substitué a des propriétés chimiques et physiques similaires à l'acide aminé d'origine, ce qui entraîne généralement une faible impact sur la fonction de la protéine. En revanche, les substitutions non conservatives sont celles où l'acide aminé substitué a des propriétés chimiques et physiques différentes, ce qui peut entraîner un impact plus important sur la fonction de la protéine.
Dans certains cas, les substitutions d'acides aminés peuvent être bénéfiques, comme dans le cadre de thérapies de remplacement des enzymes pour traiter certaines maladies héréditaires rares. Dans ces situations, une protéine fonctionnelle est produite en laboratoire et administrée au patient pour remplacer la protéine défectueuse ou absente.
L'ostéosclérose est un terme médical qui décrit un épaississement et une densification anormaux du tissu osseux. Dans des conditions normales, l'os est constamment remodelé grâce à un processus impliquant la résorption de l'os vieillissant par les ostéoclastes et la formation de nouvel os par les ostéoblastes. Cependant, dans l'ostéosclérose, il y a une augmentation de la quantité d'os produit par rapport à la quantité résorbée, entraînant une densification osseuse accrue.
L'ostéosclérose peut être focale (limitée à une petite zone) ou généralisée (affectant de larges zones ou l'ensemble du squelette). Elle peut être asymptomatique et découverte fortuitement sur des radiographies ou des examens d'imagerie, ou elle peut entraîner des symptômes tels que des douleurs osseuses, une raideur articulaire et une réduction de la mobilité en raison de l'augmentation de la densité osseuse.
L'ostéosclérose peut être secondaire à plusieurs conditions sous-jacentes, notamment des maladies hématologiques telles que la thalassémie et la drépanocytose, des troubles métaboliques tels que l'hypoparathyroïdie et le rachitisme, des infections osseuses chroniques, des tumeurs malignes telles que les myélomes multiples et les ostéosarcomes, ainsi qu'une exposition à des radiations. Dans certains cas, l'ostéosclérose peut également être idiopathique, ce qui signifie qu'elle n'est associée à aucune autre condition sous-jacente connue.
La progression d'une maladie, également appelée évolution de la maladie, se réfère à la manifestation temporelle des stades ou étapes d'une maladie chez un patient. Il s'agit essentiellement de la détérioration continue ou de l'aggravation d'un trouble médical au fil du temps, qui peut entraîner une augmentation de la gravité des symptômes, une déficience accrue, une invalidité et, éventuellement, la mort. La progression de la maladie est généralement mesurée en termes de déclin fonctionnel ou de dommages aux organes affectés. Elle peut être influencée par divers facteurs, notamment l'âge du patient, la durée de la maladie, le traitement et les comorbidités sous-jacentes. Le suivi de la progression de la maladie est crucial pour évaluer l'efficacité des interventions thérapeutiques et pour la planification des soins futurs.
Taux de survie est un terme médical utilisé pour décrire la proportion de patients qui survivent à une certaine maladie ou condition pendant un intervalle de temps spécifique. Il est généralement exprimé comme le pourcentage de personnes qui sont encore en vie après un, trois ou cinq ans suivant le diagnostic ou le traitement. Le taux de survie peut être influencé par divers facteurs, tels que l'âge du patient, le stade et le grade de la maladie au moment du diagnostic, ainsi que les options de traitement disponibles. Les taux de survie sont souvent utilisés pour évaluer l'efficacité des différents traitements et pour aider les médecins à prendre des décisions concernant les soins aux patients.
 William Vainchenker
William Vainchenker dermatoses papulosquameuses; dermatoses papulo-squameuses
dermatoses papulosquameuses; dermatoses papulo-squameuses Myélofibrose primitive - Hématologie et oncologie - Édition professionnelle du Manuel MSD
Myélofibrose primitive - Hématologie et oncologie - Édition professionnelle du Manuel MSD Réalisations
Réalisations Les cellules souches ont-elles l'âge de leur niche ? - À la recherche d'un sérum de jouvence… | médecine/sciences
Les cellules souches ont-elles l'âge de leur niche ? - À la recherche d'un sérum de jouvence… | médecine/sciences DeCS
DeCS