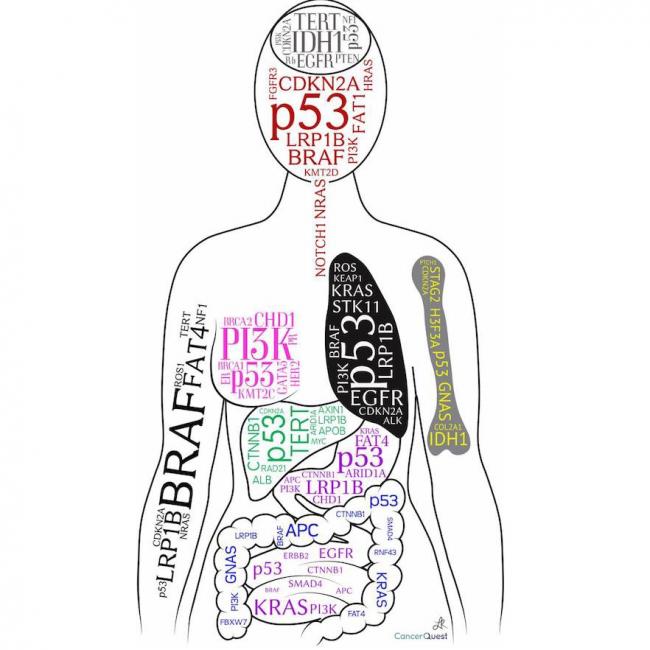
Sarcoma, Ewing
Sarcomes
Sarcome Synovial
Sarcome De Kaposi
Protéine De Liaison
Protéine Du Proto-Oncogène C-Fli-1
Virus Du Sarcome Aviaire
Sarcome Expérimental
Sarcome 180 De Crocker
Virus De Sarcome Murin
Tumeurs Des Tissus Mous
Sarcome Aviaire
Protéine Oncogène P65(Gag-Jun)
Histiocytic Sarcoma
Sarcoma, Myeloid
Sarcome Du Stroma Endométrial
Sarcome
Tumeurs Neuroectodermiques Primitives Périphériques
Tumeurs Neuroectodermiques Primitives
Sarcome De Yoshida
Kirsten Murine Sarcoma Virus
Ostéosarcome
Moloney Murine Sarcoma Virus
Virus De Sarcome Félin
Rous Sarcoma Virus
Ifosfamide
Sarcome
Rhabdomyosarcome
Sarcome Alvéolaire Des Parties Molles
Léiomyosarcome
Human Herpesvirus 8
Translocation, Genetic
Alpharetrovirus
Tumeurs
Chromosomes Humains De La Paire 22
Transformation Cellulaire D'Origine Virale
Harvey Murine Sarcoma Virus
Cell Transformation, Neoplastic
Gammarétrovirus
Histiocytome Fibreux Malin
Chondrosarcome
Doxorubicine
Liposarcome
Protéines De Liaison
Régulation Expression Génique Tumorale
Nanodiamonds
Sarcome 37
Traitement Combiné
Immunohistochimie
Histiocytome Fibreux Bénin
Réaction Polymérisation En Chaîne Par Transcriptase Inverse
Séquence Nucléotidique
Tumeurs De L'Abdomen
Pronostic
Kératine-17
Langerhans Cell Sarcoma
Tumeurs Vasculaires
Tumeurs Du Rétropéritoine
Embryon De Poulet
Seconde Tumeur Primitive
Treatment Outcome
Dactinomycine
Récepteur Igf De Type 1
Dendritic Cell Sarcoma, Follicular
Virus Auxiliaire
Récidive Tumorale Locale
Protocoles Polychimiothérapie Antinéoplasique
Chromosome Breakpoints
Tumeurs Musculaires
Cellules Cancéreuses En Culture
Transplantation Tumorale
Facteurs De Transcription
Avian Leukosis Virus
Souris Nude
Etoposide
Marqueur Biologique Tumeur
Hémangiosarcome
Rétroviridae
Retrospective Studies
Fibrosarcome
Ribonucléoprotéines Nucléaires Hétérogènes
Le sarcome d'Ewing est un type rare de cancer qui se développe dans les tissus mous et osseux du corps. Il tire son nom du pathologiste américain James Ewing, qui l'a décrit pour la première fois en 1921. Ce sarcome affecte principalement les enfants, les adolescents et les jeunes adultes, bien qu'il puisse survenir à tout âge.
Le sarcome d'Ewing se forme à partir de cellules appelées primitives neuroectodermiques, qui sont des cellules souches indifférenciées capables de se différencier en plusieurs types de tissus. Dans le cas du sarcome d'Ewing, ces cellules deviennent cancéreuses et forment une tumeur maligne.
Les symptômes courants du sarcome d'Ewing comprennent une douleur osseuse ou articulaire, un gonflement ou une masse dans la zone touchée, des ecchymoses faciles, une fatigue excessive et une perte de poids involontaire. Le diagnostic repose généralement sur une biopsie de la tumeur suspecte, suivie d'examens d'imagerie tels que la résonance magnétique (IRM), la tomodensitométrie (TDM) et la scintigraphie osseuse pour évaluer l'étendue de la maladie.
Le traitement du sarcome d'Ewing dépend du stade et de l'emplacement de la tumeur, ainsi que de l'âge et de l'état général du patient. Les options thérapeutiques comprennent la chirurgie pour enlever la tumeur, la radiothérapie pour détruire les cellules cancéreuses à l'aide de rayonnements ionisants et la chimiothérapie pour tuer les cellules cancéreuses avec des médicaments cytotoxiques. Dans certains cas, une greffe de moelle osseuse peut être proposée après une chimiothérapie intensive pour reconstituer le système immunitaire du patient.
Le pronostic dépend du stade et de l'emplacement de la tumeur, ainsi que de l'âge et de l'état général du patient au moment du diagnostic. Les taux de survie à cinq ans varient considérablement en fonction du stade de la maladie : environ 70 % pour les patients atteints d'une maladie localisée, contre seulement 15 % pour ceux présentant une maladie métastatique au moment du diagnostic.
Les sarcomes sont un type rare de cancer qui développe dans les tissus mous du corps, y compris les muscles, les tendons, les graisses, les vaisseaux sanguins, les nerfs, et les membranes qui entourent les articulations. Ils peuvent se produire n'importe où dans le corps, mais sont plus fréquents dans les bras, les jambes, la tête, le cou, l'abdomen et le dos.
Les sarcomes se développent à partir de cellules souches adultes qui se spécialisent pour former différents types de tissus mous. Lorsqu'une cellule souche subit des changements génétiques anormaux, elle peut devenir cancéreuse et se multiplier de manière incontrôlable, formant une masse tumorale.
Il existe plus de 70 sous-types différents de sarcomes, chacun ayant des caractéristiques uniques et des traitements spécifiques. Les symptômes dépendent du type et de l'emplacement du sarcome, mais peuvent inclure des douleurs, des gonflements ou des bosses sur le corps, une perte de poids inexpliquée, et une fatigue excessive.
Le traitement des sarcomes dépend du stade et du type de cancer, ainsi que de l'âge et de la santé générale du patient. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie, l'immunothérapie ou une combinaison de ces traitements. Dans certains cas, une greffe de moelle osseuse peut être recommandée pour traiter les sarcomes avancés.
Un sarcome synovial est un type rare et spécifique de cancer des tissus mous qui se développe généralement dans les membranes synoviales qui tapissent les articulations. Bien que son nom puisse prêter à confusion, ce type de sarcome ne dérive pas directement des cellules synoviales normales. Il s'agit plutôt d'une tumeur maligne qui présente certaines caractéristiques et une apparence similaires aux cellules synoviales lorsqu'elle est vue au microscope.
Les sarcomes synoviaux se produisent le plus souvent dans les extenseurs des membres inférieurs, en particulier près du genou, mais ils peuvent également se développer dans d'autres parties du corps, comme les bras, les mains, les pieds, la tête et le tronc. Ils ont tendance à survenir chez les jeunes adultes, avec un pic d'incidence entre 30 et 40 ans, mais ils peuvent affecter des personnes de tous âges.
Les symptômes du sarcome synovial peuvent inclure une masse ou un gonflement dans la région touchée, souvent accompagnés de douleur, de raideur articulaire et d'une limitation de la mobilité. La tumeur peut également provoquer des douleurs nocturnes et une fatigue générale. Le diagnostic repose sur l'examen clinique, les images médicales (telles que les radiographies, les IRM ou les TDM) et la biopsie de la tumeur pour analyse histopathologique.
Le traitement du sarcome synovial implique généralement une combinaison de chirurgie, de chimiothérapie et de radiothérapie. L'objectif principal est d'enlever complètement la tumeur tout en préservant autant que possible la fonction articulaire et musculaire. Les taux de survie à long terme pour le sarcome synovial dépendent du stade au moment du diagnostic, de l'emplacement de la tumeur et de la réponse au traitement.
Le sarcome de Kaposi est un type rare de cancer qui affecte les vaisseaux sanguins sous la forme de tumeurs cutanées. Il peut également se propager aux organes internes. Cette condition tire son nom du dermatologue hongrois Moritz Kaposi, qui l'a décrite pour la première fois en 1872.
Il existe quatre principaux types de sarcome de Kaposi :
1. Classique ou sarcome de Kaposi méditerranéen : Ce type affecte principalement les hommes d'origine méditerranéenne, juive ashkénaze ou africaine subsaharienne âgés de plus de 60 ans. Les tumeurs se développent généralement sur les jambes et progressent lentement.
2. Sarcome de Kaposi associé au sida (SKS) : Ce type est le plus agressif et affecte les personnes atteintes du virus d'immunodéficience humaine (VIH). Les tumeurs peuvent se développer rapidement sur la peau, les muqueuses et les organes internes.
3. Sarcome de Kaposi iatrogénique : Ce type est associé à l'utilisation de médicaments immunosuppresseurs après une transplantation d'organe. Les tumeurs se développent généralement dans les premiers mois suivant la transplantation et peuvent être agressives.
4. Sarcome de Kaposi endémique africain : Ce type affecte principalement les enfants et les jeunes adultes en Afrique subsaharienne. Il existe deux formes : une forme nodulaire qui ressemble au sarcome de Kaposi classique et une forme disseminée plus agressive qui se propage rapidement aux organes internes.
Les symptômes du sarcome de Kaposi comprennent des lésions cutanées rouge-violettes, des ulcères, des gonflements et des saignements dans les zones touchées. Le diagnostic repose généralement sur une biopsie cutanée et peut inclure des tests d'imagerie pour évaluer l'étendue de la maladie. Le traitement dépend du type et de l'étendue de la maladie, mais peut inclure la chimiothérapie, la radiothérapie et l'arrêt des médicaments immunosuppresseurs si possible.
La protéine du proto-oncogène C-Fli-1, également connue sous le nom de protéine ETS transcriptionnelle Fli-1, est une protéine qui en conditions normales joue un rôle important dans la régulation des processus cellulaires tels que la prolifération, l'apoptose et la différenciation. Elle agit comme facteur de transcription, se liant à l'ADN pour activer ou réprimer l'expression des gènes.
Cependant, dans certaines circonstances, des mutations ou des anomalies chromosomiques peuvent entraîner une activation anormale ou une surproduction de la protéine C-Fli-1, ce qui peut conduire à une transformation maligne des cellules et contribuer au développement de divers types de cancer, tels que les leucémies et les sarcomes. En raison de son rôle potentialisé dans la cancérogenèse, la protéine C-Fli-1 est également classée comme une oncoprotéine.
Le virus du sarcome aviaire, également connu sous le nom de virus de l'avian sarcoma (ASV), est un rétrovirus qui cause des tumeurs malignes chez les oiseaux. Il s'agit d'un oncovirus qui appartient à la famille des Retroviridae et au genre Alpharetrovirus. Ce virus est étroitement lié au virus de la leucose aviaire (ALV), mais il se distingue par sa capacité à induire des sarcomes chez les oiseaux infectés.
Le virus du sarcome aviaire est capable d'intégrer son matériel génétique dans le génome de l'hôte, ce qui peut entraîner une transformation maligne des cellules et la formation de tumeurs. Les tumeurs induites par l'ASV peuvent se développer dans divers tissus, y compris les muscles, les os, les vaisseaux sanguins et le tissu conjonctif.
L'infection par l'ASV est courante chez les oiseaux d'élevage tels que les poulets et les canards, ainsi que chez certains oiseaux sauvages. La transmission du virus se produit généralement par contact direct avec des sécrétions ou des excrétions infectées, telles que la salive, le sang, les larmes ou les fientes.
Il est important de noter que le virus du sarcome aviaire ne présente aucun risque pour la santé humaine, car il est spécifique aux oiseaux et ne peut pas infecter les mammifères, y compris les humains.
Je suis désolé, mais la phrase que vous avez fournie est incorrecte. "Sarcome Expérimental" ne devrait pas être écrit comme ça dans un contexte médical. Les termes médicaux doivent être en lettres minuscules et séparés par une espace, sauf si c'est un nom propre. De plus, "sarcome" est déjà un type de cancer qui affecte les tissus mous du corps, donc il n'est pas correct d'ajouter "expérimental" directement après sans une explication ou contexte supplémentaire.
Si vous voulez parler d'un sarcome qui est étudié dans des expériences de recherche, la façon correcte de l'exprimer serait "sarcome (type spécifique) expérimental". Par exemple, un sarcome des tissus mous expérimental ou un sarcome d'Ewing expérimental.
En général, un sarcome est un type rare de cancer qui se développe dans les tissus conjonctifs du corps, tels que les os, les muscles, les tendons, les ligaments, le cartilage et les graisses. Les différents types de sarcomes sont nommés d'après le type de tissu où ils se développent et leur apparence au microscope.
Dans un contexte expérimental, des chercheurs peuvent étudier ces types de cancers pour comprendre leurs causes, les facteurs de risque, la façon dont ils se propagent et répondent aux traitements, dans le but de développer de nouveaux traitements plus efficaces.
Je suis désolé, mais il y a apparemment une confusion dans votre question. "Sarcome 180 de Crocker" ne semble pas être une terminologie médicale reconnue ou un nom spécifique pour un type de sarcome. Il est possible qu'il y ait une erreur ou une omission dans la formulation de votre question.
Un sarcome, en général, est un type de cancer qui se développe dans les tissus conjonctifs du corps, comme le tissu adipeux, les muscles, les tendons, les ligaments, les nerfs et les vaisseaux sanguins ou lymphatiques. Il existe plus de 70 sous-types différents de sarcomes, chacun ayant des caractéristiques uniques et des options de traitement spécifiques.
Si vous cherchez des informations sur un type particulier de sarcome ou une situation médicale spécifique, veuillez fournir plus de détails pour que je puisse vous aider au mieux.
Les tumeurs osseuses sont des croissances anormales qui se forment dans les os. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent pas à d'autres parties du corps et ont tendance à croître lentement. Dans de nombreux cas, elles ne causent aucun symptôme et peuvent être découvertes par hasard lors d'examens médicaux ou radiologiques effectués pour d'autres raisons. Cependant, certaines tumeurs bénignes peuvent devenir assez grandes et affaiblir l'os, ce qui peut entraîner des fractures.
Les tumeurs malignes, en revanche, ont le potentiel de se propager à d'autres parties du corps. Elles sont souvent plus agressives que les tumeurs bénignes et peuvent croître rapidement. Les symptômes associés aux tumeurs osseuses malignes dépendent de la taille et de l'emplacement de la tumeur, mais peuvent inclure des douleurs osseuses, des gonflements, des fractures osseuses spontanées, une fatigue excessive et une perte de poids involontaire.
Le traitement des tumeurs osseuses dépend du type, de la taille, de l'emplacement et du stade de la tumeur. Les options de traitement peuvent inclure la surveillance attentive, la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces approches. Dans certains cas, des thérapies ciblées ou des immunothérapies peuvent également être utilisées pour traiter les tumeurs osseuses malignes.
Le virus du sarcome de Moloney (MSM-TV) est un type de rétrovirus qui cause des tumeurs malignes chez les souris. Il s'agit d'un oncovirus, ce qui signifie qu'il a la capacité de transformer les cellules normales en cellules cancéreuses. Le MSM-TV est un virus endogène murin, ce qui signifie qu'il est présent dans le génome de certaines souches de souris et peut être transmis verticalement de génération en génération.
Le virus du sarcome de Moloney appartient au genre de rétrovirus des Betaretrovirus et est étroitement lié au virus du sarcome de Harvey, qui cause également des tumeurs chez les souris. Le MSM-TV code pour deux protéines virales principales : la protéine d'enveloppe (env) et la protéine Gag. La protéine d'enveloppe est responsable de la liaison du virus aux récepteurs des cellules hôtes, tandis que la protéine Gag est importante pour l'assemblage et la libération du virus.
L'infection par le MSM-TV se produit généralement pendant la période périnatale et peut entraîner une leucémie ou un sarcome chez les souris infectées. Le virus se réplique dans les cellules hématopoïétiques et peut provoquer une transformation maligne de divers types de cellules, notamment des lymphocytes T, des monocytes/macrophages et des fibroblastes. Les souris infectées par le MSM-TV développent généralement des tumeurs malignes dans les 3 à 6 mois suivant l'infection.
Le virus du sarcome de Moloney est un outil important dans la recherche sur le cancer, car il permet aux chercheurs d'étudier les mécanismes moléculaires de la transformation maligne et de tester de nouvelles thérapies anticancéreuses. Cependant, il est important de noter que le MSM-TV n'est pas considéré comme un modèle pertinent pour l'étude des cancers humains, car les mécanismes moléculaires de la transformation maligne sont très différents entre les souris et les humains.
Les tumeurs des tissus mous sont des growths anormales qui se développent dans les muscles, les tendons, les ligaments, les nerfs, les graisses et les fibres conjonctives qui forment la majorité du corps humain. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent généralement pas à d'autres parties du corps et sont moins susceptibles de réapparaître après le traitement. D'autre part, les tumeurs malignes peuvent envahir les tissus voisins et se propager à d'autres parties du corps, ce qui rend le traitement plus difficile.
Les symptômes des tumeurs des tissus mous dépendent de la taille et de l'emplacement de la tumeur. Certaines personnes peuvent ne présenter aucun symptôme, tandis que d'autres peuvent ressentir une masse ou un gonflement dans la région affectée, des douleurs, des picotements, des faiblesses ou une limitation de la mobilité.
Le traitement dépend du type et du stade de la tumeur. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces méthodes. Dans certains cas, une surveillance attentive peut être recommandée si la tumeur est bénigne et ne provoque aucun symptôme.
Il est important de noter que les tumeurs des tissus mous peuvent se développer n'importe où dans le corps, y compris dans les zones difficiles à atteindre ou à traiter. Par conséquent, il est crucial de consulter un médecin dès que possible si vous remarquez des symptômes suspects, tels qu'une masse ou une grosseur inhabituelle, des douleurs ou une limitation de la mobilité.
Je suis désolé, mais la phrase que vous me avez demandé de définir semble contenir une erreur. Il n'existe pas de condition médicale connue sous le nom de "sarcome aviaire". Les sarcomes sont des tumeurs malignes qui peuvent se développer dans divers tissus conjonctifs du corps, mais il n'y a pas de variante spécifique à une espèce d'oiseaux.
Si vous vouliez parler d'une autre condition ou si j'ai mal compris votre question, veuillez me fournir plus d'informations.
L'histiocytose maligne ou l'histiocytome malin, communément appelé sarcome histiocytaire, est un type rare et agressif de cancer qui affecte les cellules du système immunitaire appelées histiocytes. Les histiocytes sont des cellules trouvées dans les tissus conjonctifs et sont responsables de la phagocytose (processus d'engloutissement et de destruction des bactéries, des cellules mortes et d'autres particules étrangères).
Le sarcome histiocytaire peut survenir n'importe où dans le corps, mais il a une préférence pour les organes internes comme la rate, le foie, les poumons et les ganglions lymphatiques. Les symptômes varient en fonction de la localisation du cancer et peuvent inclure des douleurs, des gonflements, une fatigue, une perte de poids et une diminution de l'appétit.
Le diagnostic de sarcome histiocytaire est posé après avoir effectué une biopsie d'un échantillon de tissu suspect et analysé les cellules au microscope. Le traitement dépend du stade et de la localisation du cancer, mais peut inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses à l'aide de rayonnements, et une chimiothérapie pour tuer les cellules cancéreuses à l'aide de médicaments anticancéreux. Dans certains cas, une greffe de moelle osseuse peut également être recommandée.
Le pronostic dépend du stade et de la localisation du cancer au moment du diagnostic, ainsi que de la réponse au traitement. Les sarcomes histiocytaires sont généralement des cancers agressifs avec un pronostic relativement sombre, en particulier s'ils se sont propagés à d'autres parties du corps. Cependant, certains patients peuvent vivre plusieurs années après le diagnostic et le traitement réussi de leur cancer.
Le sarcome myéloïde est un type rare et agressif de cancer qui prend naissance dans les cellules souches immatures des tissus mous ou des os. Il s'agit d'un sous-type de sarcome qui se développe à partir de cellules myéloïdes, qui sont des cellules sanguines immatures qui peuvent devenir des globules blancs, des plaquettes ou des globules rouges.
Le sarcome myéloïde peut survenir n'importe où dans le corps, mais il est le plus souvent diagnostiqué dans les bras, les jambes, le torse ou derrière la cavité abdominale. Les symptômes peuvent varier en fonction de l'emplacement et de la taille du sarcome, mais ils peuvent inclure des douleurs osseuses ou articulaires, des gonflements ou des masses, une fatigue, une fièvre ou une perte de poids inexpliquée.
Le traitement du sarcome myéloïde dépend généralement de l'emplacement et de l'étendue de la tumeur, ainsi que de l'âge et de l'état de santé général du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, une chimiothérapie pour tuer les cellules cancéreuses ou une thérapie ciblée pour perturber la croissance et la propagation des cellules cancéreuses.
Il est important de noter que le sarcome myéloïde est un cancer rare et agressif qui nécessite un traitement spécialisé par une équipe de médecins expérimentés dans le traitement des sarcomes. Si vous ou un membre de votre famille êtes préoccupé par les symptômes du sarcome myéloïde, il est important de consulter un médecin dès que possible pour obtenir un diagnostic et un plan de traitement appropriés.
Le sarcome du stroma endométrial est un type rare et agressif de cancer qui se développe dans les cellules du stroma, le tissu conjonctif qui remplit l'utérus et supporte l'endomètre. Bien que la plupart des cancers de l'endomètre commencent dans les cellules glandulaires de l'endomètre, dans le cas du sarcome du stroma endométrial, il s'agit d'une forme de cancer qui prend naissance dans les cellules du stroma.
Ce type de sarcome est généralement diagnostiqué chez des femmes ménopausées ou post-ménopausées, avec un âge moyen au diagnostic d'environ 55 ans. Les symptômes peuvent inclure des saignements vaginaux anormaux, des douleurs pelviennes et une sensation de pression dans le bas-ventre. Le sarcome du stroma endométrial a tendance à se propager rapidement et peut métastaser vers d'autres parties du corps, comme les poumons, le foie ou les os.
Le traitement du sarcome du stroma endométrial dépend généralement de l'étendue de la maladie au moment du diagnostic. Les options thérapeutiques peuvent inclure une chirurgie pour enlever l'utérus, les ovaires et d'autres organes touchés, ainsi qu'une radiothérapie et/ou une chimiothérapie pour détruire les cellules cancéreuses restantes. Malheureusement, le pronostic pour ce type de sarcome est généralement défavorable, avec un taux de survie à cinq ans inférieur à 20%. Des recherches sont en cours pour développer de nouvelles options de traitement et améliorer les résultats pour les patientes atteintes de sarcome du stroma endométrial.
Les Tumeurs Neuroectodermiques Primaires Périphériques (TNEPP) sont des tumeurs malignes rares et agressives qui se développent à partir des cellules nerveuses situées en dehors du cerveau et de la moelle épinière. Ces tumeurs sont appelées "neuroectodermiques" car elles dérivent des cellules du neuroectoderme, un tissu embryonnaire qui donne naissance aux structures nerveuses du corps.
Les TNEPP se forment le plus souvent dans les tissus mous profonds des membres, mais peuvent également apparaître dans d'autres parties du corps telles que la cavité abdominale ou thoracique. Elles sont caractérisées par une croissance rapide et une tendance à se propager rapidement vers d'autres parties du corps (métastases).
Les TNEPP peuvent toucher les personnes de tous âges, mais elles sont plus fréquentes chez les enfants et les jeunes adultes. Les symptômes dépendent de la localisation de la tumeur et peuvent inclure des douleurs, des gonflements, des faiblesses musculaires, des engourdissements ou des picotements dans les membres, ainsi que des difficultés à respirer ou à avaler si la tumeur se trouve dans la cavité thoracique ou abdominale.
Le diagnostic de TNEPP repose sur l'analyse histopathologique d'un échantillon de tissu prélevé par biopsie. Le traitement dépend du stade et de l'emplacement de la tumeur, mais peut inclure une chirurgie pour enlever la tumeur, suivie d'une chimiothérapie et/ou d'une radiothérapie pour détruire les cellules cancéreuses restantes. Malgré le traitement, les TNEPP ont un pronostic généralement défavorable en raison de leur agressivité et de leur tendance à se propager rapidement dans tout le corps.
Les Tumeurs Neuroectodermiques Primaires (TNEP) sont un groupe hétérogène de tumeurs malignes cérébrales qui se développent à partir des cellules neuroectodermiques du système nerveux central. Elles sont caractérisées par une croissance rapide, une forte tendance à l'invasion locale et une propension à la métastase vers d'autres parties du cerveau ou de la moelle épinière, ainsi que dans des cas plus rares, vers d'autres organes.
Les TNEP comprennent plusieurs sous-types histologiques, dont les plus courants sont le médulloblastome, l'astrocytome primitif à grande cellule, l'ectoméningeome et le pineoblastome. Chaque sous-type a ses propres caractéristiques cliniques, radiologiques et moléculaires.
Le traitement des TNEP implique généralement une combinaison de chirurgie, de radiothérapie et de chimiothérapie. Le pronostic dépend du sous-type de tumeur, de l'âge du patient, de l'étendue de la maladie au moment du diagnostic et de la réponse au traitement. Les TNEP sont généralement des tumeurs agressives avec un mauvais pronostic, en particulier pour les patients atteints de médulloblastomes de haut grade ou d'astrocytomes primitifs à grande cellule. Cependant, certains sous-types, comme les ectoméningeomes, peuvent avoir un meilleur pronostic avec un traitement approprié.
Le sarcome de Yoshida est un type rare de tumeur des tissus mous qui se développe généralement dans l'abdomen. Il s'agit d'une forme de tumeur myxosarcomateuse à cellules rondes, qui a été initialement décrite en 1964 par le pathologiste japonais Yoshida et ses collègues.
Ce sarcome se caractérise par la présence de deux types de cellules tumorales : des cellules myxoides et des cellules rondes. Les cellules myxoides produisent une substance mucoïde qui donne à la tumeur un aspect gélatineux, tandis que les cellules rondes ont un noyau arrondi et un cytoplasme basophile.
Le sarcome de Yoshida se développe le plus souvent chez les jeunes adultes, bien qu'il puisse affecter des personnes de tous âges. Les symptômes peuvent varier en fonction de la localisation et de la taille de la tumeur, mais ils peuvent inclure une douleur abdominale, une distension abdominale, une sensation de satiété précoce, des nausées, des vomissements et une perte de poids.
Le diagnostic du sarcome de Yoshida repose sur l'analyse histopathologique d'une biopsie de la tumeur. Le traitement standard consiste en une chirurgie pour enlever la tumeur, suivie d'une radiothérapie et/ou d'une chimiothérapie pour réduire le risque de récidive. Malgré un traitement agressif, le pronostic du sarcome de Yoshida reste incertain en raison de sa rareté et de la possibilité de récidives à distance.
Le virus du sarcome de Kirsten Murine (KMSV) est un rétrovirus qui appartient au genre des Betaretrovirus. Il a été initialement découvert dans un sarcome des tissus mous chez une souris et il est connu pour provoquer une tumeur chez les souris en laboratoire. Le KMSV n'est pas considéré comme un virus qui infecte les humains ou d'autres animaux que la souris.
Le génome du KMSV est composé d'ARN simple brin et contient des gènes typiques des rétrovirus, tels que gag, pol et env. Le virus se réplique en insérant son matériel génétique dans le génome de la cellule hôte, où il est transcrit en ARNm et traduit en protéines virales. Les protéines virales s'assemblent ensuite pour former de nouvelles particules virales qui peuvent infecter d'autres cellules.
Le KMSV est un modèle important pour l'étude des rétrovirus et de la pathogenèse des tumeurs. Les chercheurs utilisent ce virus pour étudier les mécanismes moléculaires de la transformation maligne des cellules et pour développer de nouvelles stratégies thérapeutiques contre le cancer.
L'ostéosarcome est un type agressif et rare de cancer des os. Il se développe généralement à partir des cellules qui forment l'os (les ostéoblastes), entraînant ainsi une production anormale d'os ou d'une masse tissulaire osseuse dans la moelle osseuse. Bien que ce cancer puisse se développer dans n'importe quel os, il est le plus souvent localisé près des articulations longues telles que les genoux et les hanches.
Les symptômes de l'ostéosarcome peuvent inclure une douleur osseuse ou articulaire, un gonflement autour de la zone touchée, des difficultés à bouger la partie affectée du corps, une fatigue générale et une perte de poids involontaire. Les ostéosarcomes sont généralement traités par une combinaison de chirurgie, de chimiothérapie et de radiothérapie, en fonction de l'étendue et de la localisation du cancer.
Le pronostic pour les personnes atteintes d'ostéosarcome dépend de plusieurs facteurs, tels que l'âge du patient, la taille et la localisation de la tumeur, le stade du cancer au moment du diagnostic et la réponse au traitement. Les taux de survie à cinq ans pour les patients atteints d'ostéosarcome peuvent varier considérablement, allant de 20% à 70%, selon ces facteurs.
Le Moloney Murine Sarcoma Virus (MMSV) est un type de rétrovirus qui provoque des tumeurs chez la souris. Il a été découvert par John Moloney en 1960. Le MMSV est un virus complexe qui contient deux types d'information génétique : l'ARN viral et une enzyme appelée reverse transcriptase. Cette enzyme permet au virus de créer une copie d'ADN à partir de son ARN, ce qui lui permet de s'intégrer dans le génome de la cellule hôte.
Le MMSV est capable d'induire la transformation maligne des cellules, c'est-à-dire qu'il les rend cancéreuses. Il le fait en activant certains gènes de la cellule hôte, appelés "oncogènes", qui favorisent la division cellulaire incontrôlée et l'inhibition de la mort cellulaire programmée (apoptose).
Le MMSV est un outil important dans la recherche sur le cancer et les rétrovirus. Il a été utilisé pour étudier les mécanismes moléculaires de la transformation maligne des cellules, ainsi que pour développer des modèles animaux de cancer. Cependant, il est important de noter que ce virus ne peut pas infecter les humains et ne pose donc pas de risque pour la santé publique.
Le virus de sarcome félin (FeSV) est un rétrovirus qui cause des sarcomes chez les chats. Il existe deux types principaux : le FeSV de type A, associé à la leucémie féline, et le FeSV de type B, qui est responsable du sarcome vasculaire félin. Ce virus se transmet généralement par contact direct avec des sécrétions infectées, telles que la salive, l'urine ou les matières fécales d'un chat infecté. Les symptômes peuvent inclure des gonflements cutanés ou sous-cutanés, une perte de poids, une faiblesse et une anémie. Le FeSV est considéré comme une maladie grave et souvent fatale chez les chats.
Le virus du sarcome de Rous, également connu sous le nom de RSV (de l'anglais Rous Sarcoma Virus), est un type de rétrovirus oncogène qui a été découvert en 1910 par Peyton Rous. Il est associé au développement de sarcomes chez les poulets, d'où il tire son nom. Le RSV est capable d'induire une transformation maligne des cellules, entraînant leur croissance et leur division incontrôlées, ce qui conduit à la formation de tumeurs malignes.
Le génome du RSV se compose d'ARN simple brin, qui est reverse-transcrit en ADN double brin après l'infection des cellules hôtes. Ce matériel génétique viral s'intègre ensuite dans le génome de la cellule hôte, où il peut être exprimé et entraîner une transformation maligne. Le gène oncogène clé du RSV est appelé src, qui code pour une tyrosine kinase impliquée dans la régulation de divers processus cellulaires tels que la prolifération, la différenciation et l'apoptose (mort cellulaire programmée).
L'isolement et la caractérisation du RSV ont été des étapes cruciales dans le développement de la virologie et de la compréhension générale des mécanismes moléculaires sous-jacents au cancer. Les recherches sur le RSV et d'autres rétrovirus oncogènes ont contribué à l'identification des gènes proto-oncogènes dans le génome humain, qui peuvent être activés ou surexprimés dans certaines conditions pour favoriser la tumorigenèse.
L'ifosfamide est un agent alkylant utilisé dans le traitement de divers types de cancer, y compris les tumeurs sarcome, les lymphomes et les cancers du poumon. Il s'agit d'un médicament chimiothérapeutique qui agit en interférant avec la capacité des cellules cancéreuses à se diviser et à se développer.
L'ifosfamide est un dérivé de la substance chimique moutarde azotée, qui a été initialement développée comme arme chimique pendant la Seconde Guerre mondiale. Il fonctionne en endommageant l'ADN des cellules cancéreuses, ce qui empêche leur croissance et leur division ultérieures.
Cependant, l'ifosfamide peut également affecter les cellules saines du corps, entraînant des effets secondaires tels que la nausée, la vomissement, la diarrhée, la fatigue, la perte de cheveux et une augmentation du risque d'infections. Il peut également endommager les reins et le système nerveux central, entraînant des effets secondaires graves tels que des lésions rénales et neurologiques.
Par conséquent, l'ifosfamide est généralement administré sous surveillance médicale stricte dans un cadre hospitalier, avec une hydratation adéquate pour protéger les reins et des mesures préventives pour minimiser les effets secondaires.
Le rhabdomyosarcome est un type rare et agressif de cancer qui se développe dans les cellules des muscles squelettiques. Ces cellules sont responsables du mouvement volontaire des os, ce qui rend le rhabdomyosarcome capable d'apparaître dans presque n'importe quelle partie du corps. Il est plus fréquent chez les enfants et les adolescents, bien que les adultes puissent également en être atteints.
Les symptômes varient en fonction de la localisation du rhabdomyosarcome dans le corps. Ils peuvent inclure des gonflements ou des masses douloureuses, des ecchymoses faciles, des maux de tête, des difficultés à avaler, des saignements du nez et des changements dans les habitudes urinaires. Le diagnostic repose généralement sur une biopsie pour confirmer la présence de cellules cancéreuses.
Le traitement dépend du stade et de la localisation du cancer. Il peut inclure une combinaison de chirurgie, de radiothérapie et de chimiothérapie. Le pronostic varie également en fonction des caractéristiques spécifiques du cancer et de la réponse au traitement. Les taux de survie à cinq ans sont généralement bons pour les cas diagnostiqués tôt et traités de manière agressive, mais ils diminuent considérablement pour les cancers avancés ou récurrents.
Le sarcome alvéolaire des parties moles est un type rare de cancer qui se développe dans les tissus mous du corps, tels que les muscles, les tendons, les nerfs, les graisses et les vaisseaux sanguins. Il tire son nom de la structure anormale des cellules cancéreuses, qui forment des cavités ressemblant à des alvéoles lorsqu'elles se développent dans le tissu affecté.
Ce type de sarcome est souvent agressif et a tendance à se propager rapidement vers d'autres parties du corps. Les symptômes peuvent varier en fonction de la localisation du sarcome, mais peuvent inclure des douleurs, des gonflements ou des nodules dans la région affectée, une limitation de la mobilité et une fatigue générale.
Le diagnostic de sarcome alvéolaire des parties moles est généralement posé à partir d'une biopsie de la tumeur suspecte, suivie d'examens d'imagerie pour évaluer l'étendue de la maladie. Le traitement dépend du stade et de la localisation de la tumeur, mais peut inclure une chirurgie pour enlever la tumeur, une radiothérapie pour aider à détruire les cellules cancéreuses restantes, et une chimiothérapie pour aider à prévenir la récidive de la maladie.
Il est important de noter que le sarcome alvéolaire des parties moles est un cancer rare et que les informations fournies ici peuvent ne pas être complètes ou entièrement exactes. Il est donc recommandé de consulter un médecin pour obtenir des informations plus détaillées et personnalisées sur cette maladie.
Le terme "léiomyosarcome" est utilisé en médecine pour désigner un type rare de cancer qui se développe dans les muscles lisses, qui sont les muscles involontaires trouvés dans les parois des vaisseaux sanguins, du tube digestif, de l'utérus et d'autres organes.
Les leiomyosarcomes peuvent survenir n'importe où dans le corps, mais ils sont le plus souvent trouvés dans le muscle lisse de l'abdomen ou du bassin. Ces tumeurs peuvent être très agressives et ont tendance à se développer rapidement.
Les symptômes du leiomyosarcome dépendent de la localisation de la tumeur, mais peuvent inclure des douleurs abdominales ou pelviennes, une sensation de masse dans l'abdomen, des saignements anormaux et une perte de poids inexpliquée.
Le diagnostic d'un leiomyosarcome est généralement posé après une biopsie de la tumeur suspecte, suivie d'une analyse histopathologique pour confirmer le type de cancer. Le traitement dépend du stade et de l'emplacement de la tumeur, mais peut inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie pour aider à détruire les cellules cancéreuses restantes.
Une lignée cellulaire tumorale, dans le contexte de la recherche en cancérologie, fait référence à une population homogène de cellules cancéreuses qui peuvent être cultivées et se diviser en laboratoire. Ces lignées cellulaires sont généralement dérivées de biopsies ou d'autres échantillons tumoraux prélevés sur des patients, et elles sont capables de se multiplier indéfiniment en culture.
Les lignées cellulaires tumorales sont souvent utilisées dans la recherche pour étudier les propriétés biologiques des cellules cancéreuses, tester l'efficacité des traitements anticancéreux et comprendre les mécanismes de progression du cancer. Cependant, il est important de noter que ces lignées cellulaires peuvent ne pas toujours se comporter ou réagir aux traitements de la même manière que les tumeurs d'origine dans le corps humain, ce qui peut limiter leur utilité en tant que modèles pour la recherche translationnelle.
La définition médicale de l'human herpesvirus 8 (HHV-8), également connu sous le nom de virus associé au sarcome de Kaposi (KSHV), est celui d'un type de virus à ADN de la famille des Herpesviridae. Il est classé dans le sous-groupe gammaherpesvirus, avec l'Epstein-Barr Virus (EBV). Le HHV-8 est l'agent étiologique du sarcome de Kaposi, une forme rare de cancer des vaisseaux sanguins. Ce virus peut également être associé à d'autres affections malignes telles que les lymphomes primitifs des tissus extrafolliculaires et le Castleman à cellules multicentriques.
Le HHV-8 se transmet principalement par contact direct avec des fluides corporels infectés, comme la salive, le sperme ou les sécrétions vaginales, lors de rapports sexuels, de transplantations d'organes ou de greffes de moelle osseuse. Chez certaines personnes immunodéprimées, telles que celles infectées par le VIH, le HHV-8 peut provoquer une maladie plus grave et plus répandue.
Le diagnostic du HHV-8 repose généralement sur la détection de l'ADN viral dans des échantillons tissulaires ou sanguins à l'aide de techniques de biologie moléculaire, telles que la PCR (polymerase chain reaction). Il n'existe actuellement aucun vaccin ni traitement spécifique pour prévenir ou guérir l'infection par le HHV-8. Cependant, des thérapies antivirales et immunosuppressives peuvent contribuer à gérer les complications liées aux maladies associées au virus.
La translocation génétique est un type d'anomalie chromosomique où des segments entiers de deux chromosomes différents changent de place. Il existe deux types principaux de translocations génétiques : les translocations réciproques et les translocations Robertsoniennes.
Les translocations réciproques se produisent lorsque des segments de deux chromosomes différents sont échangés l'un avec l'autre. Ces translocations peuvent être équilibrées, ce qui signifie qu'aucun matériel génétique n'est ni gagné ni perdu dans le processus, ou déséquilibrée, ce qui entraîne une perte ou un gain de matériel génétique.
Les translocations Robertsoniennes, quant à elles, se produisent lorsque la partie distale (la partie la plus éloignée du centromère) de deux chromosomes acrocentriques (qui comprennent les chromosomes 13, 14, 15, 21 et 22) est interchangée, entraînant la fusion des deux chromosomes à leur centromère commun. Cela entraîne la formation d'un seul chromosome avec deux bras courts (p) et aucun bras long (q). Les translocations Robertsoniennes sont le plus souvent équilibrées, mais lorsqu'elles ne le sont pas, elles peuvent entraîner des anomalies génétiques et des troubles du développement.
Les translocations génétiques peuvent être héritées ou spontanées (de novo). Lorsqu'elles sont héritées, elles peuvent être asymptomatiques ou causer des problèmes de santé dépendamment de la façon dont les gènes affectés sont exprimés. Cependant, lorsqu'elles sont spontanées, elles peuvent entraîner des anomalies chromosomiques telles que le syndrome de Down (translocation entre les chromosomes 21 et un autre chromosome) ou le syndrome de Patau (translocation entre les chromosomes 13 et un autre chromosome).
En résumé, les translocations génétiques sont des réarrangements chromosomiques qui peuvent entraîner des problèmes de santé et des anomalies du développement. Elles peuvent être héritées ou spontanées et peuvent affecter n'importe quel chromosome. Les translocations Robertsoniennes sont un type spécifique de translocation qui implique la fusion de deux chromosomes à leur centromère commun, entraînant la formation d'un seul chromosome avec deux bras courts et aucun bras long.
Les alpharétrovirus sont un type de rétrovirus qui peuvent infecter une variété d'hôtes, y compris les humains. Ils sont classés dans la famille des Retroviridae et le genre Alpharetrovirus. Les alpharétrovirus ont un génome à ARN simple brin et se répliquent en créant une copie ADN de leur génome à l'aide d'une enzyme appelée transcriptase inverse.
L'un des alpharétrovirus les plus connus est le virus de la leucose aviaire (ALV), qui infecte les oiseaux et peut causer une forme de cancer appelée leucose. Il existe plusieurs sous-types d'ALV, chacun avec un tropisme différent pour certains types de cellules hôtes.
Bien que les alpharétrovirus puissent infecter les humains, ils ne sont pas connus pour causer une maladie spécifique chez l'homme. Cependant, certaines preuves suggèrent qu'ils peuvent s'intégrer dans le génome humain et y rester sous forme de provirus latents.
Il est important de noter que les rétrovirus, y compris les alpharétrovirus, peuvent être dangereux pour certaines personnes, en particulier celles dont le système immunitaire est affaibli. Par conséquent, il est essentiel de prendre des précautions appropriées lors de la manipulation de ces virus dans un contexte de laboratoire ou clinique.
Les chromosomes humains de la paire 22, également connus sous le nom de chromosomes 22, sont une partie importante du génome humain. Ils font partie des 23 paires de chromosomes trouvés dans chaque cellule humaine. Chaque chromosome 22 contient des milliers de gènes qui fournissent les instructions pour la production de protéines et d'autres produits fonctionnels nécessaires au bon fonctionnement de l'organisme.
Les chromosomes 22 sont parmi les plus petits des chromosomes humains, avec une longueur d'environ 50 millions de paires de bases. Ils représentent environ 1,5 à 3% du génome humain total. Les chromosomes 22 contiennent un certain nombre de gènes importants qui sont associés à diverses maladies et conditions médicales.
L'un des gènes les plus connus sur le chromosome 22 est le gène NF2, qui code pour une protéine appelée mercaptopurine nucléotide synthase (MER). Les mutations dans ce gène sont associées à la neurofibromatose de type 2, une maladie génétique caractérisée par la croissance de tumeurs bénignes le long des nerfs.
Les chromosomes 22 sont également importants dans l'étude de la schizophrénie, une maladie mentale grave qui affecte environ 1% de la population mondiale. Des études ont montré que certaines variations génétiques sur le chromosome 22 peuvent augmenter le risque de développer cette condition.
En plus des gènes, les chromosomes 22 contiennent également une grande quantité d'ADN non codant qui ne code pas directement pour des protéines. Cet ADN peut jouer un rôle important dans la régulation de l'expression génique et peut être associé à diverses maladies et conditions médicales.
En résumé, les chromosomes 22 sont importants dans l'étude de diverses maladies et conditions médicales, y compris la neurofibromatose de type 2 et la schizophrénie. Les mutations dans les gènes du chromosome 22 peuvent augmenter le risque de développer ces conditions, tandis que l'ADN non codant sur le chromosome 22 peut également jouer un rôle important dans la régulation de l'expression génique et être associé à diverses maladies.
La "Transformation cellulaire d'origine virale" est un processus dans lequel un virus introduit du matériel génétique étranger dans les cellules hôtes, entraînant des changements fondamentaux dans la fonction et la structure de ces cellules. Ce phénomène peut conduire à une altération de la régulation de la croissance et de la division cellulaires, ce qui peut entraîner la transformation maligne des cellules et éventuellement provoquer le développement d'un cancer.
Les virus capables de provoquer une transformation cellulaire sont appelés "virus oncogènes" ou "virus transformants". Ils peuvent insérer leur propre matériel génétique, comme des gènes viraux ou des séquences d'ADN/ARN, dans le génome de la cellule hôte. Ces gènes viraux peuvent activer ou désactiver les gènes cellulaires régulateurs de la croissance et de la division, entraînant une prolifération cellulaire incontrôlée et la formation de tumeurs malignes.
Les exemples de virus oncogènes comprennent le virus du papillome humain (VPH), qui est associé au cancer du col de l'utérus, et le virus de l'hépatite B (VHB), qui peut provoquer un cancer du foie. Il est important de noter que tous les virus ne sont pas capables de transformer les cellules ; seuls certains virus présentent cette propriété oncogène.
Le Harvey Murine Sarcoma Virus (HMSV) est un rétrovirus oncogène qui a été découvert dans les années 1960. Il est associé à la formation de tumeurs malignes chez les souris, en particulier des sarcomes. Le HMSV est un virus complexe qui contient plusieurs gènes, dont certains sont capables d'induire la transformation maligne des cellules infectées.
Le génome du HMSV est composé d'ARN simple brin, qui est reverse-transcrit en ADN après l'infection de la cellule hôte. Cet ADN est ensuite intégré dans le génome de la cellule, où il peut être transmis à des cellules filles lors de la division cellulaire.
Le HMSV code pour plusieurs protéines qui jouent un rôle clé dans la transformation maligne des cellules. Parmi ces protéines, on trouve les protéines Src, qui sont des kinases activées par les tyrosines et qui peuvent activer d'autres voies de signalisation cellulaire impliquées dans la régulation de la croissance cellulaire et de la différenciation.
L'infection par le HMSV peut entraîner une série de changements dans les cellules infectées, y compris la activation de gènes qui favorisent la prolifération cellulaire et l'inhibition de gènes qui favorisent l'apoptose (mort cellulaire programmée). Ces changements peuvent conduire à la formation de tumeurs malignes, telles que des sarcomes.
Le HMSV est un modèle important pour l'étude des mécanismes moléculaires sous-jacents à la transformation maligne des cellules et à la cancérogenèse. Les découvertes faites sur ce virus ont contribué de manière significative à notre compréhension des processus impliqués dans le développement du cancer et ont conduit à l'identification de nouvelles cibles thérapeutiques pour le traitement de cette maladie.
La "transformation cellulaire néoplasique" est un processus biologique dans lequel une cellule normale et saine se transforme en une cellule cancéreuse anormale et autonome. Ce processus est caractérisé par des changements irréversibles dans la régulation de la croissance et de la division cellulaire, entraînant la formation d'une tumeur maligne ou d'un néoplasme.
Les facteurs qui peuvent contribuer à la transformation cellulaire néoplasique comprennent des mutations génétiques aléatoires, l'exposition à des agents cancérigènes environnementaux tels que les radiations et les produits chimiques, ainsi que certains virus oncogènes.
Les changements cellulaires qui se produisent pendant la transformation néoplasique comprennent des anomalies dans les voies de signalisation cellulaire, une régulation altérée de l'apoptose (mort cellulaire programmée), une activation anormale des enzymes impliquées dans la réplication de l'ADN et une augmentation de l'angiogenèse (croissance de nouveaux vaisseaux sanguins pour fournir de l'oxygène et des nutriments à la tumeur).
La transformation cellulaire néoplasique est un processus complexe qui peut prendre plusieurs années, voire plusieurs décennies, avant qu'une tumeur maligne ne se développe. Cependant, une fois que cela se produit, les cellules cancéreuses peuvent envahir les tissus environnants et se propager à d'autres parties du corps, ce qui peut entraîner des complications graves et même la mort.
Les gammaretrovirus sont un type de rétrovirus qui peuvent causer des maladies chez les animaux, y compris les humains. Ils ont un génome à ARN simple brin et se répliquent en créant une copie d'ADN de leur génome à l'aide de la transcriptase inverse. Cette copie d'ADN est ensuite intégrée dans le génome de l'hôte à l'aide de l'intégrase. Les gammaretrovirus sont associés à un certain nombre de maladies, notamment des cancers et des troubles dégénératifs du système nerveux central. Le virus de la leucémie féline (FeLV) est un exemple bien connu de gammaretrovirus.
Le histiocytose fibreuse maligne (HFM), également connu sous le nom de dermatofibrosarcome protuberans, est une tumeur rare des tissus mous qui se développe à partir des cellules dermiques ou musculaires lisses. Il s'agit d'un type agressif de sarcome des tissus mous, ce qui signifie qu'il a le potentiel de se propager à d'autres parties du corps.
Les lésions cutanées typiques de HFM sont souvent décrites comme étant indolores, fermes et de couleur chair ou brune. Elles peuvent apparaître comme des plaques surélevées, des papules ou des nodules sur la peau. Les lésions ont tendance à se développer lentement au fil du temps, bien que certaines puissent croître et s'étendre rapidement.
Le diagnostic de HFM est généralement posé par biopsie et examen histopathologique des tissus. Le traitement standard consiste en une chirurgie large avec des marges de sécurité adéquates pour minimiser le risque de récidive locale. La radiothérapie et la chimiothérapie peuvent également être utilisées dans certains cas, en particulier lorsque les marges chirurgicales sont inadéquates ou que la tumeur s'est propagée à d'autres parties du corps.
Il est important de noter que HFM est un type agressif de cancer et qu'un diagnostic et un traitement précoces sont essentiels pour améliorer les résultats. Les patients atteints de HFM doivent être surveillés de près pour détecter toute récidive locale ou métastase à d'autres parties du corps.
Le chondrosarcome est un type rare de cancer qui se développe dans le cartilage, un tissu conjonctif flexible qui recouvre et protège la plupart des extrémités osseuses. Ce cancer se produit généralement dans les os pelviens, la base du crâne, les côtes ou les membres longs.
Les chondrosarcomes sont classés en fonction de leur apparence au microscope et comprennent plusieurs sous-types, notamment:
1. Chondrosarcome conventionnel : C'est le type le plus courant et il se développe généralement dans les os des membres longs ou du bassin. Il est divisé en trois grades (I, II, III) basés sur son agressivité et sa vitesse de croissance.
2. Chondrosarcome dédifférencié : Ce sous-type se produit lorsqu'un chondrosarcome conventionnel devient plus agressif et envahit les tissus environnants. Il est considéré comme un cancer de haut grade et a un pronostic moins favorable.
3. Chondrosarcome mésenchymateux : C'est un type rare qui se produit principalement chez les enfants et les jeunes adultes. Il peut se développer dans n'importe quel os, mais il est plus fréquent dans la mâchoire inférieure (mandibule) et le bassin.
4. Chondrosarcome à cellules claires : Ce sous-type est également rare et a tendance à se produire chez les adultes plus âgés. Il peut affecter n'importe quel os, mais il est plus fréquent dans la colonne vertébrale et le bassin.
Les symptômes du chondrosarcome peuvent inclure une masse ou un gonflement douloureux dans la zone touchée, des douleurs articulaires, des fractures osseuses spontanées et une limitation de la mobilité. Le diagnostic est généralement posé après avoir effectué une biopsie pour examiner les tissus affectés. Le traitement dépend du stade et du type de cancer, mais il peut inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie.
La doxorubicine est un médicament de chimiothérapie utilisé pour traiter divers types de cancer, y compris les cancers du sein, des ovaires, des poumons, des reins et des tissus conjonctifs. Elle appartient à une classe de médicaments appelés anthracyclines qui interfèrent avec l'ADN des cellules cancéreuses pour empêcher leur croissance et leur division.
La doxorubicine fonctionne en se liant à l'ADN des cellules cancéreuses, ce qui empêche la synthèse de l'ADN et de l'ARN nécessaires à la réplication cellulaire. Elle peut également produire des radicaux libres qui endommagent les membranes cellulaires et d'autres structures cellulaires, entraînant la mort des cellules cancéreuses.
Ce médicament est généralement administré par injection dans une veine (voie intraveineuse) et peut être utilisé seul ou en association avec d'autres agents chimiothérapeutiques. Les effets secondaires courants de la doxorubicine comprennent des nausées, des vomissements, une perte d'appétit, une diarrhée, une fatigue, une alopécie (perte de cheveux) et une inflammation ou une douleur au site d'injection.
La doxorubicine peut également entraîner des effets secondaires cardiaques graves, tels qu'une insuffisance cardiaque congestive, en particulier lorsqu'elle est administrée à fortes doses ou sur une longue période. Par conséquent, les professionnels de la santé doivent surveiller attentivement la fonction cardiaque des patients recevant ce médicament.
Un liposarcome est un type rare de cancer qui se développe dans les cellules graisseuses (adipocytes) du tissu adipeux. Il s'agit d'un sarcome des tissus mous, ce qui signifie qu'il peut se former n'importe où dans le corps où il y a du tissu adipeux, mais il est le plus souvent trouvé dans l'abdomen, le bassin, les jambes et les bras.
Les liposarcomes sont généralement classés en plusieurs sous-types en fonction de la façon dont ils se présentent au microscope. Ces sous-types comprennent :
- Liposarcome bien différencié : C'est le type le plus courant et il ressemble beaucoup aux cellules graisseuses normales. Il a tendance à se développer lentement et est moins susceptible de se propager à d'autres parties du corps.
- Liposarcome myxoïde : Ce type est également relativement fréquent et se caractérise par un aspect gélatineux. Il a tendance à se développer dans les membres inférieurs et a un taux de récidive élevé, mais un faible potentiel de propagation à distance.
- Liposarcome pléomorphe : Ce type est moins courant et se caractérise par une grande variété de cellules anormales. Il a tendance à se développer rapidement et peut se propager à d'autres parties du corps.
- Liposarcome dédifférencié : Ce type est très agressif et se compose de cellules qui ne ressemblent plus aux cellules graisseuses normales. Il a un fort potentiel de propagation à distance.
Le traitement d'un liposarcome dépend du stade et du sous-type de la tumeur, ainsi que de l'âge et de l'état général de santé du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie pour aider à détruire les cellules cancéreuses restantes.
La régulation de l'expression génique tumorale dans un contexte médical se réfère aux mécanismes moléculaires et cellulaires qui contrôlent la manière dont les gènes s'expriment dans les cellules cancéreuses. Les changements dans l'expression des gènes peuvent entraîner une prolifération cellulaire accrue, une résistance à l'apoptose (mort cellulaire programmée), une angiogenèse (croissance de nouveaux vaisseaux sanguins) et une métastase, qui sont tous des processus clés dans le développement du cancer.
La régulation de l'expression génique tumorale peut être influencée par une variété de facteurs, y compris les mutations génétiques, les modifications épigénétiques (telles que la méthylation de l'ADN et l'acétylation des histones), les facteurs de transcription anormaux, les miARN (petits ARN non codants qui régulent l'expression des gènes) et les interactions entre les cellules tumorales et leur microenvironnement.
Comprendre la régulation de l'expression génique tumorale est crucial pour le développement de thérapies ciblées contre le cancer, car il permet d'identifier de nouvelles cibles thérapeutiques et de prédire la réponse des patients aux traitements existants. Des approches telles que l'édition du génome, la modulation épigénétique et l'interférence avec les miARN sont autant de stratégies prometteuses pour réguler l'expression des gènes dans le cancer et améliorer les résultats cliniques.
Les nanodiamants sont des nanoparticules de diamant avec une taille généralement inférieure à 1 micromètre. Ils présentent des propriétés uniques telles qu'une grande surface spécifique, une biocompatibilité élevée et une stabilité chimique, ce qui les rend intéressants pour diverses applications en médecine.
Dans le domaine médical, les nanodiamants sont étudiés pour leur potentiel dans des domaines tels que la délivrance de médicaments, l'imagerie médicale et la thérapie cellulaire. Par exemple, ils peuvent être utilisés pour transporter des médicaments directement vers les cellules cibles, améliorant ainsi l'efficacité du traitement et réduisant les effets secondaires.
Cependant, il convient de noter que l'utilisation des nanodiamants dans la médecine est encore en cours d'exploration et nécessite des recherches supplémentaires pour comprendre pleinement leurs avantages et leurs risques potentiels.
Je suis désolé, mais il n'existe pas de sarcome spécifiquement référencé sous le nom de "sarcome 37" dans la classification médicale standard. Les sarcomes sont un groupe de tumeurs malignes qui se développent à partir des tissus conjonctifs du corps, comme les os, les tendons, les ligaments, le tissu adipeux et les vaisseaux sanguins. Ils peuvent être classés par leur type histologique (par exemple, sarcome des tissus mous, ostéosarcome, etc.) et leur grade (de bas grade à haut grade, selon leur agressivité). Pour obtenir des informations précises sur un type de sarcome ou un état spécifique, il est préférable de fournir le nom correct du sarcome.
Un traitement combiné, dans le contexte médical, fait référence à l'utilisation simultanée de deux ou plusieurs thérapies différentes pour traiter une maladie, un trouble de santé ou une condition médicale spécifique. Cela peut inclure une combinaison de médicaments, de procédures chirurgicales, de thérapies de radiation, de thérapies comportementales ou d'autres formes de traitement.
L'objectif d'un traitement combiné est souvent de maximiser les avantages thérapeutiques pour le patient, en tirant parti des mécanismes d'action uniques de chaque thérapie pour attaquer la maladie sous différents angles. Cela peut entraîner une efficacité accrue, une réduction des effets secondaires et une amélioration globale des résultats cliniques.
Un exemple courant de traitement combiné est l'utilisation de plusieurs médicaments pour contrôler le VIH/sida. Dans ce cas, un cocktail de médicaments antirétroviraux est utilisé pour attaquer le virus à différentes étapes de son cycle de réplication, ce qui permet de réduire la charge virale et d'améliorer la fonction immunitaire du patient.
Cependant, il convient de noter que les traitements combinés peuvent également entraîner des risques accrus d'interactions médicamenteuses et d'effets secondaires, ce qui nécessite une surveillance étroite et un ajustement attentif des doses pour assurer la sécurité et l'efficacité du traitement.
La vincristine est un médicament anticancéreux utilisé dans le traitement de divers types de cancer, y compris la leucémie, le lymphome de Hodgkin et non hodgkinien, et les tumeurs solides telles que le sarcome des tissus mous. Elle appartient à une classe de médicaments appelés alcaloïdes de la vinca rosea, qui interfèrent avec la division cellulaire en inhibant la polymérisation des tubulines, ce qui entraîne l'arrêt de la mitose et la mort cellulaire.
La vincristine est administrée par voie intraveineuse et doit être utilisée sous surveillance médicale stricte en raison de ses effets secondaires potentiellement graves, tels que des neuropathies périphériques, des nausées et des vomissements sévères, une alopécie (perte de cheveux), et dans de rares cas, des réactions allergiques.
Il est important de noter qu'il s'agit d'une définition médicale générale et que l'utilisation de la vincristine doit être individualisée en fonction du type de cancer, de l'état général du patient et des autres traitements administrés.
L'immunohistochimie est une technique de laboratoire utilisée en anatomopathologie pour localiser les protéines spécifiques dans des tissus prélevés sur un patient. Elle combine l'utilisation d'anticorps marqués, généralement avec un marqueur fluorescent ou chromogène, et de techniques histologiques standard.
Cette méthode permet non seulement de déterminer la présence ou l'absence d'une protéine donnée dans une cellule spécifique, mais aussi de déterminer sa localisation précise à l'intérieur de cette cellule (noyau, cytoplasme, membrane). Elle est particulièrement utile dans le diagnostic et la caractérisation des tumeurs cancéreuses, en permettant d'identifier certaines protéines qui peuvent indiquer le type de cancer, son stade, ou sa réponse à un traitement spécifique.
Par exemple, l'immunohistochimie peut être utilisée pour distinguer entre différents types de cancers du sein en recherchant des marqueurs spécifiques tels que les récepteurs d'œstrogènes (ER), de progestérone (PR) et HER2/neu.
Le histiocytose fibreuse bénigne (HFB) est une affection cutanée rare et généralement bénigne caractérisée par la prolifération anormale de histiocytes, qui sont des cellules du système immunitaire qui aident à combattre l'infection. Ces histiocytes produisent une accumulation anormale de tissu cicatriciel dans la peau, formant une masse ou une papule ferme et souvent indolore.
Les lésions d'HFB sont généralement solitaires, mais peuvent parfois être multiples. Ils se produisent le plus souvent sur les membres inférieurs, en particulier autour des chevilles et des pieds, bien qu'ils puissent apparaître n'importe où sur la peau. Les lésions ont tendance à se développer lentement au fil du temps et peuvent atteindre jusqu'à plusieurs centimètres de diamètre.
Bien que l'HFB soit généralement bénin, il peut parfois être difficile de le distinguer d'autres affections cutanées plus graves, telles que les sarcomes ou les carcinomes. Par conséquent, une biopsie et un examen histopathologique sont souvent nécessaires pour confirmer le diagnostic.
Le traitement de l'HFB dépend généralement de la taille et de la localisation des lésions. Dans de nombreux cas, aucun traitement n'est nécessaire car les lésions peuvent rester stables ou même régresser spontanément avec le temps. Cependant, si les lésions sont grandes, ennuyeuses ou situées dans une zone esthétiquement sensible, des options de traitement telles que l'excision chirurgicale, la cryochirurgie ou le laser peuvent être envisagées.
La réaction de polymérisation en chaîne par transcriptase inverse (RT-PCR en anglais) est une méthode de laboratoire utilisée pour amplifier des fragments d'ARN spécifiques. Cette technique combine deux processus distincts : la transcription inverse, qui convertit l'ARN en ADN complémentaire (ADNc), et la polymérisation en chaîne, qui permet de copier rapidement et de manière exponentielle des millions de copies d'un fragment d'ADN spécifique.
La réaction commence par la transcription inverse, où une enzyme appelée transcriptase inverse utilise un brin d'ARN comme matrice pour synthétiser un brin complémentaire d'ADNc. Ce processus est suivi de la polymérisation en chaîne, où une autre enzyme, la Taq polymérase, copie le brin d'ADNc pour produire des millions de copies du fragment d'ADN souhaité.
La RT-PCR est largement utilisée dans la recherche médicale et clinique pour détecter et quantifier l'expression génétique, diagnostiquer les maladies infectieuses, détecter les mutations génétiques et effectuer des analyses de génome. Elle est également utilisée dans les tests de diagnostic COVID-19 pour détecter le virus SARS-CoV-2.
Une séquence nucléotidique est l'ordre spécifique et linéaire d'une série de nucléotides dans une molécule d'acide nucléique, comme l'ADN ou l'ARN. Chaque nucléotide se compose d'un sucre (désoxyribose dans le cas de l'ADN et ribose dans le cas de l'ARN), d'un groupe phosphate et d'une base azotée. Les bases azotées peuvent être adénine (A), guanine (G), cytosine (C) et thymine (T) dans l'ADN, tandis que dans l'ARN, la thymine est remplacée par l'uracile (U).
La séquence nucléotidique d'une molécule d'ADN ou d'ARN contient des informations génétiques cruciales qui déterminent les caractéristiques et les fonctions de tous les organismes vivants. La décodage de ces séquences, appelée génomique, est essentiel pour comprendre la biologie moléculaire, la médecine et la recherche biologique en général.
Les tumeurs de l'abdomen se réfèrent à toute croissance anormale et non planifiée des cellules dans la cavité abdominale. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs abdominales peuvent se développer dans n'importe quel organe de l'abdomen, y compris le foie, le pancréas, les reins, les intestins, la rate et l'estomac.
Les symptômes des tumeurs de l'abdomen dépendent de leur emplacement, de leur taille et s'ils sont bénins ou malins. Les signes et les symptômes peuvent inclure une masse palpable dans l'abdomen, une douleur abdominale, des nausées, des vomissements, une perte d'appétit, une perte de poids involontaire, des ballonnements, des changements dans les habitudes intestinales et du sang dans les selles.
Le diagnostic des tumeurs abdominales implique généralement plusieurs étapes, y compris un examen physique, des analyses de sang, des tests d'imagerie tels que les tomodensitogrammes (TDM), les imageries par résonance magnétique (IRM) et les échographies. Dans certains cas, une biopsie peut être nécessaire pour confirmer le diagnostic et déterminer le type de tumeur.
Le traitement des tumeurs abdominales dépend du type, de la taille, de l'emplacement de la tumeur et de son stade. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces traitements. Dans certains cas, une surveillance attentive peut être recommandée si la tumeur est bénigne et ne cause pas de symptômes.
En médecine, le terme "pronostic" se réfère à la prévision du résultat ou de l'issue attendue d'une maladie ou d'une blessure dans le corps humain. Il s'agit essentiellement d'une estimation de la probabilité du rétablissement complet, de l'amélioration continue, de l'évolution vers une invalidité permanente ou du décès d'un patient atteint d'une certaine maladie ou blessure.
Le pronostic est généralement fondé sur les antécédents médicaux du patient, les résultats des tests diagnostiques, l'étendue de la maladie ou de la lésion, la réponse au traitement et d'autres facteurs pertinents. Il peut être exprimé en termes généraux ou spécifiques, tels qu'un pronostic favorable, défavorable ou incertain.
Il est important de noter que le pronostic n'est pas une garantie et ne doit pas être considéré comme tel. Il s'agit simplement d'une estimation basée sur des données probantes et l'expérience clinique, qui peut varier d'un patient à l'autre. Les médecins doivent communiquer clairement le pronostic aux patients et à leur famille, en s'assurant qu'ils comprennent les risques, les avantages et les incertitudes associés au traitement et à la maladie sous-jacente.
La kératine 17 est une protéine structurelle qui appartient à la famille des kératines de type I. Elle est principalement exprimée dans les cellules épithéliales stratifiées, en particulier dans les couches basales de l'épiderme et dans les glandes sudoripares eccrines. La kératine 17 joue un rôle important dans la différenciation des kératinocytes, la régulation de la prolifération cellulaire et la réponse inflammatoire.
Dans l'épiderme, la kératine 17 s'associe à la kératine 5 pour former un complexe hétérodimérique qui confère aux kératinocytes une résistance mécanique et protège les cellules contre le stress environnemental. Des mutations dans le gène de la kératine 17 ont été associées à des maladies génétiques telles que le pemphigus foliaceus bulleux, une forme de pemphigus caractérisée par la formation de bulles cutanées douloureuses.
En plus de son rôle structurel, la kératine 17 est également impliquée dans la régulation de la transcription génique et de la signalisation cellulaire. Elle peut interagir avec des facteurs de transcription et des coactivateurs pour moduler l'expression des gènes impliqués dans la différenciation épidermique, la réponse immunitaire et la carcinogenèse.
Dans les glandes sudoripares eccrines, la kératine 17 est exprimée dans les cellules myoépitheliales et joue un rôle important dans la régulation de la sécrétion de sueur et de l'homéostasie ionique. Des études ont montré que des niveaux élevés de kératine 17 dans les glandes sudoripares eccrines peuvent être associés à une augmentation de la transpiration et à une susceptibilité accrue aux infections bactériennes.
En résumé, la kératine 17 est une protéine structurelle et fonctionnelle importante qui joue un rôle crucial dans la différenciation épidermique, la réponse immunitaire et la carcinogenèse. Elle est exprimée dans les cellules épithéliales de la peau et des glandes sudoripares eccrines et peut interagir avec d'autres protéines pour réguler l'expression des gènes et la signalisation cellulaire. Des anomalies dans l'expression de la kératine 17 peuvent entraîner des maladies telles que le pemphigus foliaceus bulleux et une susceptibilité accrue aux infections bactériennes.
Le sarcome des cellules de Langerhans est un type rare et agressif de cancer qui affecte les cellules dendritiques, qui sont un type de globule blanc présent dans la peau et les muqueuses. Les cellules de Langerhans sont responsables de la présentation des antigènes aux lymphocytes T, ce qui est une partie importante du système immunitaire.
Le sarcome des cellules de Langerhans se développe à partir de ces cellules et peut se propager à d'autres parties du corps. Les symptômes peuvent inclure des lésions cutanées ou muqueuses, des gonflements, des douleurs et une fatigue générale. Le diagnostic est généralement posé par biopsie et analyse histopathologique des tissus affectés.
Le traitement du sarcome des cellules de Langerhans peut inclure la chirurgie, la radiothérapie et la chimiothérapie, en fonction de l'étendue de la maladie et de son stade au moment du diagnostic. Malheureusement, le pronostic pour cette forme de cancer est généralement mauvais, avec un taux de survie à cinq ans inférieur à 20%. Des recherches sont en cours pour développer de nouveaux traitements plus efficaces pour cette maladie rare et agressive.
Les tumeurs vasculaires sont des growths anormaux qui se développent dans les vaisseaux sanguins ou lymphatiques. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs vasculaires bénignes comprennent les hémangiomes, qui sont des growths faits de vaisseaux sanguins anormaux, et les lymphangiomes, qui sont des growths faits de vaisseaux lymphatiques anormaux. Les tumeurs vasculaires malignes comprennent le sarcome angiosarcome, qui est un cancer rare des vaisseaux sanguins.
Les tumeurs vasculaires peuvent se produire n'importe où dans le corps, mais elles sont les plus fréquentes dans la peau, le cerveau et le foie. Les symptômes dépendent de l'emplacement et de la taille de la tumeur. Certaines tumeurs vasculaires ne causent aucun symptôme et sont découvertes par hasard lors d'examens médicaux de routine. D'autres peuvent causer des douleurs, des saignements, des ecchymoses faciles, une pression sur les organes voisins ou des problèmes de fonctionnement des organes.
Le traitement dépend du type, de la taille, de l'emplacement et de l'étendue de la tumeur. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces traitements. Dans certains cas, une observation attentive peut être recommandée si la tumeur est petite et ne cause aucun symptôme. Il est important de consulter un médecin si vous remarquez des changements dans votre peau ou si vous présentez des symptômes qui pourraient indiquer une tumeur vasculaire.
Les tumeurs rétropéritonéales se réfèrent à un groupe hétérogène de lésions tumorales qui se développent dans la région située derrière (rétro) la cavité abdominale (péritonéal). Le rétropéritoine est un espace anatomique situé entre le péritoine, qui tapisse l'intérieur de l'abdomen, et les muscles du dos. Il contient des organes et des tissus importants tels que les reins, les glandes surrénales, les vaisseaux sanguins rétropéritonéaux, le plexus nerveux sympathique et la graisse.
Les tumeurs rétropéritonéales peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes sont généralement plus rares que les tumeurs malignes et ont tendance à se développer plus lentement. Elles peuvent inclure des lipomes, des schwannomes, des fibromes et des néoplasmes à cellules granuleuses.
Les tumeurs malignes rétropéritonéales comprennent un large éventail de cancers qui se développent dans les organes rétropéritonéaux ou qui s'y propagent à partir d'autres sites du corps. Les types courants de tumeurs malignes rétropéritonéales incluent:
1. Sarcomes: Ce sont des cancers des tissus mous, y compris les graisses, les muscles, les vaisseaux sanguins, les nerfs et les fibres conjonctives. Les sarcomes rétropéritonéaux peuvent inclure des leiomyosarcomes, des liposarcomes et des synoviosarcomes.
2. Carcinomes: Ce sont des cancers qui se développent à partir des cellules épithéliales tapissant les organes internes. Les carcinomes rétropéritonéaux peuvent inclure des adénocarcinomes du côlon, du pancréas et de l'estomac, ainsi que des cancers du rein et du foie.
3. Lymphomes: Ce sont des cancers qui se développent dans le système immunitaire, principalement à partir des cellules appelées lymphocytes. Les lymphomes rétropéritonéaux peuvent inclure des lymphomes non hodgkiniens et des lymphomes de Hodgkin.
4. Tumeurs neuroendocrines: Ce sont des cancers qui se développent à partir des cellules du système nerveux et des glandes endocrines. Les tumeurs neuroendocrines rétropéritonéales peuvent inclure des carcinoïdes, des phéochromocytomes et des paragangliomes.
Les symptômes des tumeurs malignes rétropéritonéales dépendent de leur taille, de leur emplacement et de la propagation de la maladie. Les symptômes courants comprennent une douleur abdominale ou pelvienne persistante, un gonflement abdominal, des nausées, des vomissements, une perte d'appétit, une perte de poids involontaire et une fatigue extrême. Dans certains cas, les tumeurs peuvent se propager à d'autres parties du corps, provoquant des symptômes supplémentaires tels qu'un essoufflement, une toux persistante, des douleurs osseuses ou des ecchymoses inhabituelles.
Le diagnostic des tumeurs malignes rétropéritonéales implique généralement plusieurs tests d'imagerie, tels que des tomodensitogrammes (TDM), des imageries par résonance magnétique (IRM) et des échographies. Ces tests peuvent aider à déterminer la taille, l'emplacement et la propagation de la tumeur. Une biopsie peut également être effectuée pour confirmer le diagnostic et déterminer le type de cancer.
Le traitement des tumeurs malignes rétropéritonéales dépend du type et de l'étendue du cancer, ainsi que de la santé générale du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses à l'aide de rayonnements, une chimiothérapie pour tuer les cellules cancéreuses à l'aide de médicaments ou une thérapie ciblée pour attaquer spécifiquement les cellules cancéreuses. Dans certains cas, une combinaison de ces traitements peut être utilisée pour obtenir les meilleurs résultats possibles.
Il est important de noter que le pronostic des tumeurs malignes rétropéritonéales dépend de plusieurs facteurs, notamment du type et de l'étendue du cancer, de la santé générale du patient et de la réponse au traitement. Les patients atteints de ce type de cancer doivent travailler en étroite collaboration avec leur équipe médicale pour élaborer un plan de traitement personnalisé et obtenir les meilleurs résultats possibles.
Je suis désolé, mais la dénomination médicale "embryon de poulet" n'est pas exacte ou largement reconnue dans le domaine de l'anatomie ou de l'embryologie. Cependant, il est possible que vous cherchiez à comprendre le développement embryonnaire d'un œuf de poule, qui est un sujet d'étude courant en biologie du développement.
Un œuf de poule contient un blastodisque, qui est une masse cellulaire discoïdale située sur la surface interne de l'oeuf. Le blastodisque est composé de deux parties : le disque germinal (ou area opaca) et le disque épiblastique (ou area pellucida). L'embryon se développe à partir du disque germinal, qui est la partie centrale et plus opaque du blastodisque.
Environ 48 heures après la fertilisation de l'oeuf, le début du développement embryonnaire devient visible sous forme d'un petit renflement au centre du disque germinal, appelé blastoderme primitif. Ce blastoderme primitif se développe progressivement pour former tous les tissus et organes de l'embryon de poulet.
Par conséquent, si vous cherchiez une définition médicale ou scientifique du développement embryonnaire dans un œuf de poule, j'espère que cette explication vous aura été utile.
Une seconde tumeur primitive (STP) est définie comme une nouvelle tumeur maligne qui survient chez un patient pendant ou après la prise en charge d'une tumeur maligne précédente, lorsque cette nouvelle tumeur n'est pas considérée comme une récidive locale, régionale ou à distance de la tumeur initiale et qu'elle n'est pas non plus considérée comme un métastase de la tumeur initiale.
En d'autres termes, il s'agit d'une tumeur distincte et indépendante qui se développe chez un patient atteint d'un cancer préexistant. Les STP peuvent survenir simultanément ou séquentiellement par rapport à la tumeur initiale. Elles peuvent être diagnostiquées avant, pendant ou après le traitement de la tumeur primitive initiale.
Le diagnostic et la prise en charge des STP sont complexes, car elles peuvent être influencées par les antécédents de traitement du cancer initial, y compris la chirurgie, la radiothérapie et la chimiothérapie. De plus, certaines thérapies contre le cancer peuvent augmenter le risque de développer des STP en raison de leur effet mutagène sur les cellules saines. Par conséquent, une évaluation approfondie est nécessaire pour déterminer le plan de traitement optimal pour chaque patient atteint d'une STP.
En médecine et en santé mentale, l'issue du traitement, également appelée résultat du traitement ou issue de la prise en charge, se réfère au changement dans l'état de santé d'un patient après avoir reçu des soins médicaux, des interventions thérapeutiques ou des services de santé mentale. Il s'agit de l'effet global ou du bénéfice obtenu grâce à ces procédures, qui peuvent être mesurées en termes d'amélioration des symptômes, de réduction de la douleur, de prévention de complications, de restauration des fonctions corporelles ou mentales, d'augmentation de la qualité de vie et de réadaptation sociale. L'issue du traitement peut être évaluée en utilisant différents critères et outils d'évaluation, selon la nature de la maladie, des lésions ou des troubles en question. Elle est généralement déterminée par une combinaison de facteurs objectifs (tels que les tests de laboratoire ou les mesures physiologiques) et subjectifs (tels que les auto-évaluations du patient ou les observations du clinicien). Une issue favorable du traitement est considérée comme un résultat positif, tandis qu'une issue défavorable ou négative indique l'absence d'amélioration ou la détérioration de l'état de santé du patient.
La dactinomycine est un antibiotique antinéoplasique, qui est utilisé dans le traitement de divers types de cancer. Elle est isolée à partir du champignon filamenteux Streptomyces parvulus et agit en se liant à l'ADN, inhibant ainsi la synthèse des acides nucléiques et entraînant la mort cellulaire.
La dactinomycine est principalement utilisée dans le traitement du cancer du testicule, du choriocarcinome gestationnel, du sarcome de Kaposi et du cancer du col de l'utérus. Elle peut être administrée par injection intraveineuse ou sous forme de gel pour une application topique dans certaines conditions.
Les effets secondaires courants de la dactinomycine comprennent des nausées, des vomissements, une perte d'appétit, une fatigue, une alopécie (perte de cheveux) et une irritation au site d'injection. Des effets secondaires plus graves peuvent inclure une suppression de la moelle osseuse, entraînant une anémie, une leucopénie et une thrombocytopénie, ainsi qu'une toxicité pulmonaire et cardiaque.
La dactinomycine est généralement administrée sous surveillance médicale étroite en raison de ses effets secondaires potentiellement graves.
Le récepteur de l'insuline-like growth factor de type 1 (IGF-1R) est un type de récepteur transmembranaire qui se lie spécifiquement au facteur de croissance insulinique-1 (IGF-1) et au facteur de croissance similaire à l'insuline-2 (IGF-2). Il s'agit d'un membre de la famille des récepteurs à tyrosine kinase qui joue un rôle crucial dans la transduction des signaux intracellulaires régulant la croissance, la différenciation et la survie cellulaire.
Lorsque l'IGF-1 ou l'IGF-2 se lie au récepteur IGF-1R, il subit un changement conformationnel qui active sa tyrosine kinase intrinsèque. Cela entraîne une phosphorylation de certains résidus de tyrosine sur le récepteur et la recrutation d'autres protéines adaptatrices, ce qui déclenche une cascade de signalisation complexe via les voies PI3K/AKT et RAS/MAPK. Ces voies régulent divers processus cellulaires tels que la croissance, la division cellulaire, l'apoptose, la migration et la métabolisme cellulaire.
Des mutations ou des variations dans le gène du récepteur IGF-1R ont été associées à certaines affections médicales, telles que le syndrome de Laron, un trouble rare caractérisé par une résistance à l'hormone de croissance et une petite taille. De plus, des études suggèrent que le récepteur IGF-1R pourrait être impliqué dans la pathogenèse de divers cancers, car il favorise la survie, la prolifération et la migration des cellules tumorales. Par conséquent, le récepteur IGF-1R est considéré comme une cible thérapeutique potentielle pour le traitement du cancer.
Le sarcome des cellules dendritiques folliculaires (FDCS) est un type rare de cancer qui affecte les cellules dendritiques, qui sont un type de globule blanc présent dans le système immunitaire. Les cellules dendritiques jouent un rôle crucial dans la reconnaissance et la présentation des antigènes aux lymphocytes T, ce qui déclenche une réponse immunitaire.
Le FDCS se développe à partir de cellules dendritiques anormales qui se trouvent dans les follicules lymphoïdes, qui sont des structures spécialisées trouvées dans les ganglions lymphatiques et la rate. Ces cellules dendritiques anormales peuvent se multiplier de manière incontrôlable et former une tumeur maligne.
Le FDCS peut se manifester n'importe où dans le corps, mais il a tendance à affecter les ganglions lymphatiques du cou, de la poitrine ou de l'abdomen. Les symptômes peuvent inclure des gonflements douloureux des ganglions lymphatiques, de la fièvre, des sueurs nocturnes et une perte de poids inexpliquée.
Le diagnostic du FDCS repose sur une biopsie des tissus affectés, suivie d'une analyse immunohistochimique pour confirmer la présence de marqueurs spécifiques des cellules dendritiques. Le traitement dépend de l'étendue de la maladie et peut inclure une chirurgie, une radiothérapie, une chimiothérapie ou une thérapie ciblée.
Il est important de noter que le FDCS est un cancer rare et que les informations disponibles sur cette maladie peuvent être limitées. Les patients atteints de FDCS devraient consulter des spécialistes expérimentés dans le traitement des tumeurs rares pour obtenir des soins optimaux.
Un virus auxiliaire, également connu sous le nom de virus satellite ou déféctif, est un type de virus qui ne peut pas compléter son cycle de réplication sans l'aide d'un autre virus appelé virus helper ou virus aidant. Ce virus helper fournit les fonctions essentielles à la réplication du virus auxiliaire, telles que l'encapsidation (emballage de l'acide nucléique viral dans une capside protéique) et la libération du virus auxiliaire des cellules infectées.
Les virus auxiliaires peuvent modifier le phénotype du virus helper, par exemple en augmentant sa virulence ou en modifiant ses propriétés antigéniques. Ils peuvent également interférer avec la réplication du virus helper et entraîner une diminution de sa production.
Les virus auxiliaires sont souvent étudiés dans le contexte de la virologie expérimentale, car ils peuvent être utilisés pour étudier les interactions entre les virus et les cellules hôtes, ainsi que pour comprendre les mécanismes de réplication des virus.
La récidive tumorale locale est un terme médical utilisé pour décrire la réapparition d'une tumeur maligne (cancer) dans la même région où elle s'est développée initialement, après un traitement initial qui avait apparemment réussi à éliminer la tumeur. Cela signifie que des cellules cancéreuses sont restées dans le corps, inaperçues par les médecins, et ont recommencé à se multiplier pour former une nouvelle tumeur au même endroit.
Une récidive tumorale locale peut survenir des mois ou même des années après le traitement initial. Elle est souvent associée à un pronostic moins favorable que lors du diagnostic initial, car les cellules cancéreuses peuvent avoir développé une résistance aux thérapies précédemment utilisées. Toutefois, la prise en charge d'une récidive dépend du type de cancer, de son stade au moment du diagnostic de la récidive et des antécédents thérapeutiques du patient.
La polychimiothérapie antinéoplasique est un type de traitement du cancer qui implique l'utilisation de plusieurs médicaments chimotherapiques différents. Le terme "polychimiothérapie" signifie simplement l'utilisation de plusieurs agents chimiques, tandis que "antinéoplasique" se réfère aux médicaments utilisés pour traiter les néoplasies, ou tumeurs anormales.
Ce protocole est couramment utilisé dans le traitement des cancers avancés ou métastatiques, où il peut être difficile de éradiquer complètement la tumeur avec un seul médicament. En combinant plusieurs médicaments, les médecins peuvent augmenter les chances d'éliminer toutes les cellules cancéreuses et réduire le risque de résistance aux médicaments.
Les protocoles de polychimiothérapie antinéoplasique sont soigneusement planifiés et surveillés par une équipe de spécialistes du cancer, y compris des oncologues médicaux, des infirmières en oncologie et d'autres professionnels de la santé. Les médicaments sont généralement administrés par voie intraveineuse dans un cadre hospitalier ou clinique, bien que certains régimes puissent être administrés sur une base ambulatoire.
Les effets secondaires de la polychimiothérapie antinéoplasique dépendent du type et de la dose des médicaments utilisés, mais peuvent inclure la nausée, les vomissements, la perte de cheveux, la fatigue, la douleur et une augmentation du risque d'infection. Les patients peuvent également éprouver des effets secondaires à long terme, tels que la neuropathie périphérique, la cardiotoxicité et la toxicité pulmonaire.
Dans l'ensemble, les protocoles de polychimiothérapie antinéoplasique sont un traitement important pour de nombreux types de cancer et peuvent contribuer à améliorer les taux de survie et la qualité de vie des patients atteints de cancer. Cependant, ils comportent également des risques et nécessitent une surveillance et une gestion attentives des effets secondaires.
Les points de rupture des chromosomes (CBs) se réfèrent à des sites spécifiques sur les chromosomes où ils peuvent être fragiles et susceptibles de se casser pendant le processus de recombinaison génétique ou en raison d'une exposition à des agents mutagènes. Ces CBs sont souvent associés aux réarrangements chromosomiques structuraux, tels que les délétions, les duplications, les inversions et les translocations.
Les CBs peuvent être hérités ou acquérir de novo en raison d'erreurs pendant la méiose ou la mitose. Les mutations génétiques qui affectent la stabilité des chromosomes, telles que les mutations dans les gènes de réparation de l'ADN, peuvent augmenter la fréquence et la gravité des CBs.
Les CBs sont importants en médecine clinique car ils peuvent être associés à des maladies génétiques héréditaires ou sporadiques, telles que les troubles neurodéveloppementaux, les cancers et les maladies cardiovasculaires. Les techniques de cytogénétique moléculaire, telles que l'hybridation in situ fluorescente (FISH) et la séquençage de nouvelle génération (NGS), peuvent être utilisées pour identifier et caractériser les CBs à des fins diagnostiques et pronostiques.
Une lignée cellulaire est un groupe homogène de cellules dérivées d'un seul type de cellule d'origine, qui se divisent et se reproduisent de manière continue dans des conditions de culture en laboratoire. Ces cellules sont capables de maintenir certaines caractéristiques spécifiques à leur type cellulaire d'origine, telles que la forme, les fonctions et les marqueurs moléculaires, même après plusieurs générations.
Les lignées cellulaires sont largement utilisées dans la recherche biomédicale pour étudier divers processus cellulaires et moléculaires, tester de nouveaux médicaments, développer des thérapies et comprendre les mécanismes sous-jacents aux maladies humaines. Il est important de noter que certaines lignées cellulaires peuvent présenter des anomalies chromosomiques ou génétiques dues à leur manipulation en laboratoire, ce qui peut limiter leur utilisation dans certains contextes expérimentaux ou cliniques.
Les tumeurs musculaires sont des growths anormaux qui se développent dans les tissus musculaires du corps. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs musculaires bénignes comprennent par exemple les lipomes, les leiomyomes et les rhabdomyomes. Ces tumeurs ne se propagent généralement pas à d'autres parties du corps et peuvent être traitées par une intervention chirurgicale mineure.
Les tumeurs musculaires malignes, également appelées sarcomes des tissus mous, sont plus rares mais aussi plus graves. Elles se développent rapidement et ont tendance à se propager à d'autres parties du corps. Les sous-types de sarcomes des tissus mous comprennent le leiomyosarcome, le liposarcome et le rhabdomyosarcome. Le traitement dépend du type, de la taille, de l'emplacement et de l'étendue de la tumeur, mais peut inclure une combinaison de chirurgie, de radiothérapie et de chimiothérapie.
Les cellules cancéreuses en culture sont des cellules cancéreuses prélevées sur un être humain ou un animal, qui sont ensuite cultivées et multipliées dans un laboratoire. Ce processus est souvent utilisé pour la recherche médicale et biologique, y compris l'étude de la croissance et du comportement des cellules cancéreuses, la découverte de nouveaux traitements contre le cancer, et les tests de sécurité et d'efficacité des médicaments et des thérapies expérimentales.
Les cellules cancéreuses en culture sont généralement prélevées lors d'une biopsie ou d'une intervention chirurgicale, puis transportées dans un milieu de culture spécial qui contient les nutriments et les facteurs de croissance nécessaires à la survie et à la reproduction des cellules. Les cellules sont maintenues dans des conditions stériles et sous observation constante pour assurer leur santé et leur pureté.
Les cultures de cellules cancéreuses peuvent être utilisées seules ou en combinaison avec d'autres méthodes de recherche, telles que l'imagerie cellulaire, la génomique, la protéomique et la biologie des systèmes. Ces approches permettent aux chercheurs d'étudier les mécanismes moléculaires du cancer à un niveau granulaire, ce qui peut conduire à une meilleure compréhension de la maladie et au développement de nouveaux traitements plus efficaces.
La transplantation tumorale, également connue sous le nom de greffe de tumeur, est un processus expérimental dans le domaine de la recherche biomédicale. Il s'agit d'une procédure dans laquelle des cellules cancéreuses ou une tumeur entière sont prélevées sur un organisme donneur et transplantées dans un organisme receveur. Cette technique est généralement utilisée dans les études de recherche pour comprendre comment les tumeurs se développent, progressent et répondent au traitement.
Cependant, il est important de noter que la transplantation tumorale n'est pas une forme de traitement clinique standard pour le cancer. En effet, cela peut être éthiquement controversé car il existe un risque de propager le cancer chez le receveur. Par conséquent, cette procédure est strictement réglementée et ne doit être effectuée que dans des cadres de recherche très contrôlés et avec un consentement éclairé complet des deux parties concernées.
L'appareil locomoteur est un terme médical qui fait référence au système complexe d'organes et de structures du corps humain qui travaillent ensemble pour permettre la mobilité et le soutien du corps. Il est composé de deux sous-systèmes principaux :
1. Le système musculo-squelettique : il comprend les os, les articulations, les ligaments, les tendons, les bourses séreuses et les disques intervertébraux. Ces structures travaillent en synergie pour fournir un cadre stable et une surface sur laquelle les muscles peuvent s'attacher et se contracter pour produire le mouvement.
2. Le système nerveux périphérique : il comprend les nerfs qui transmettent les signaux entre le cerveau, la moelle épinière et les muscles. Ces signaux permettent de contrôler et de coordonner les mouvements du corps.
L'appareil locomoteur est responsable de la capacité du corps à se déplacer, à maintenir une posture et à effectuer des tâches quotidiennes telles que soulever des objets, marcher, courir et sauter. Des problèmes au niveau de l'appareil locomoteur peuvent entraîner des douleurs, des raideurs, une perte de fonction et une diminution de la qualité de vie. Les affections courantes qui affectent l'appareil locomoteur comprennent l'arthrite, les blessures sportives, les maladies dégénératives des disques intervertébraux et les troubles neuromusculaires.
Les facteurs de transcription sont des protéines qui régulent l'expression des gènes en se liant aux séquences d'ADN spécifiques, appelées éléments de réponse, dans les régions promotrices ou enhancers des gènes. Ces facteurs peuvent activer ou réprimer la transcription des gènes en recrutant ou en éloignant d'autres protéines impliquées dans le processus de transcription, y compris l'ARN polymérase II, qui synthétise l'ARN messager (ARNm). Les facteurs de transcription peuvent être régulés au niveau de leur activation, de leur localisation cellulaire et de leur dégradation, ce qui permet une régulation complexe et dynamique de l'expression des gènes en réponse à différents signaux et stimuli cellulaires. Les dysfonctionnements des facteurs de transcription ont été associés à diverses maladies, y compris le cancer et les maladies neurodégénératives.
L'Avian Leukosis Virus (ALV) est un virus appartenant à la famille des Retroviridae et au genre Alpharetrovirus. Il est connu pour causer une variété de maladies néoplasiques et non néoplasiques chez les oiseaux, en particulier les volailles domestiquées telles que les poulets et les dindes. Les souches de ce virus peuvent être exogènes ou endogènes.
Les souches exogènes sont transmises horizontalement par contact avec des sécrétions infectées, comme le sang ou les fientes, tandis que les souches endogènes sont hébergées dans le génome de l'oiseau et peuvent être transmises verticalement de parent à descendant.
Les maladies associées à l'ALV comprennent la leucose, la myéloblastose, la sarcome, la myélocytomatose et d'autres tumeurs malignes. Ces maladies peuvent entraîner une baisse de production d'œufs, une décoloration des coquilles d'œufs, une croissance anormale, une faiblesse, une diminution de l'appétit et, finalement, la mort.
Il n'existe actuellement aucun traitement spécifique pour les infections à ALV. Les mesures préventives comprennent des programmes de biosécurité stricts, y compris l'isolement des oiseaux infectés, le contrôle des mouvements d'oiseaux et de matériel contaminé, la désinfection régulière et l'utilisation de vaccins pour certaines souches du virus.
Une souris « nude » est un type spécifique de souche de souris utilisée dans la recherche biomédicale. Ces souris sont appelées « nude » en raison de leur apparence physique distinctive, qui comprend une fourrure clairsemée ou absente et l'absence de vibrisses (moustaches).
La caractéristique génétique la plus importante des souris nude est leur déficience immunitaire congénitale sévère. Elles manquent de thymus et ont donc un système immunitaire considérablement affaibli, en particulier en ce qui concerne le système immunitaire adaptatif. Cela signifie qu'elles ne peuvent pas rejeter les greffes de tissus étrangers aussi efficacement que les souris normales.
Cette caractéristique fait des souris nude un outil précieux dans la recherche biomédicale, en particulier dans le domaine de l'immunologie et de la recherche sur le cancer. Les chercheurs peuvent greffer des tissus humains ou des cellules cancéreuses sur ces souris pour étudier la façon dont ils se comportent et réagissent dans un organisme vivant. Cela permet aux scientifiques d'en apprendre davantage sur le développement du cancer, les traitements potentiels et la réaction du système immunitaire humain à divers stimuli sans mettre en danger des sujets humains.
L'étoposide est un médicament anticancéreux utilisé dans le traitement de divers types de cancer, y compris le cancer du poumon à petites cellules, le lymphome de Hodgkin, le cancer testiculaire et certains types de leucémie. Il appartient à une classe de médicaments appelés inhibiteurs de la topoisomérase II, qui fonctionnent en interférant avec l'action d'une enzyme appelée topoisomérase II, ce qui entraîne des dommages à l'ADN des cellules cancéreuses et empêche leur croissance et leur division.
L'étoposide est disponible sous forme de solution injectable ou de capsule orale et est généralement administré en combinaison avec d'autres médicaments de chimiothérapie. Les effets secondaires courants de l'étoposide comprennent la suppression des cellules sanguines, entraînant une augmentation du risque d'infections, de saignements et de fatigue, ainsi que des nausées, des vomissements, des diarrhées et des dommages aux nerfs périphériques.
Il est important de noter que l'étoposide ne doit être administré que sous la supervision d'un professionnel de la santé qualifié et que les patients doivent suivre attentivement les instructions posologiques pour minimiser les risques d'effets secondaires graves.
Un marqueur biologique tumoral, également connu sous le nom de biomarqueur tumoral, est une substance ou un signe que l'on peut détecter dans le sang, d'autres fluides corporels, ou des tissus qui peuvent indiquer la présence d'une tumeur cancéreuse ou d'un processus pathologique spécifique. Ces marqueurs peuvent être des protéines, des gènes, des hormones ou d'autres molécules produites par les cellules cancéreuses ou par l'organisme en réponse à la présence de la tumeur.
Les marqueurs biologiques tumoraux sont souvent utilisés pour aider au diagnostic, au staging (détermination du degré d'avancement) et au suivi du traitement du cancer. Cependant, il est important de noter que ces marqueurs ne sont pas spécifiques à un seul type de cancer et peuvent être présents dans d'autres conditions médicales. Par conséquent, ils doivent être utilisés en combinaison avec d'autres tests diagnostiques pour confirmer le diagnostic de cancer.
Exemples courants de marqueurs biologiques tumoraux comprennent l'antigène prostatique spécifique (PSA) pour le cancer de la prostate, l'alpha-fœtoprotéine (AFP) pour le cancer du foie, et l'antigène carcinoembryonnaire (CEA) pour le cancer colorectal.
L'hémangiosarcome est un type rare et agressif de cancer qui se développe à partir des cellules endothéliales, qui tapissent les parois des vaisseaux sanguins. Ce cancer peut survenir dans divers organes du corps, tels que le foie, la rate, le cœur, les poumons, les reins et la peau.
Les hémangiosarcomes se développent généralement sans symptômes précoces et peuvent atteindre une taille considérable avant d'être détectés. Les signes et symptômes de cette maladie dépendent de l'emplacement et de la taille de la tumeur, mais ils peuvent inclure :
* Une masse ou gonflement dans l'abdomen ou la poitrine
* Douleurs abdominales ou thoraciques
* Faiblesse, fatigue, essoufflement et pâleur due à une anémie (liée à des saignements internes)
* Perte d'appétit et perte de poids
* Vomissements et diarrhée
Le diagnostic de l'hémangiosarcome repose sur des examens d'imagerie tels que la tomodensitométrie (TDM), l'imagerie par résonance magnétique (IRM) ou l'échographie, ainsi que sur une biopsie de la tumeur pour analyse histopathologique.
Le traitement de cette maladie dépend du stade et de l'emplacement de la tumeur. Les options thérapeutiques peuvent inclure la chirurgie, la radiothérapie et/ou la chimiothérapie. Malheureusement, le pronostic pour les patients atteints d'hémangiosarcome est généralement mauvais, en raison de sa nature agressive et de sa tendance à se propager rapidement dans tout l'organisme.
Les antinéoplasiques sont une classe de médicaments utilisés dans le traitement du cancer. Ils fonctionnent en ciblant et en détruisant les cellules cancéreuses ou en arrêtant leur croissance et leur division. Ces médicaments peuvent être administrés par voie orale, intraveineuse ou intramusculaire, selon le type de cancer traité et la voie d'administration recommandée.
Les antinéoplasiques comprennent plusieurs sous-catégories, telles que les chimiothérapies, les thérapies ciblées, l'immunothérapie et la hormonothérapie. Chacune de ces sous-catégories fonctionne de manière différente pour cibler et détruire les cellules cancéreuses.
Les chimiothérapies sont des médicaments qui interfèrent avec la division cellulaire, ce qui entraîne la mort des cellules cancéreuses. Cependant, ils peuvent également affecter les cellules saines à division rapide, comme les cellules du sang et du système digestif, entraînant des effets secondaires tels que la fatigue, la nausée et la perte de cheveux.
Les thérapies ciblées sont conçues pour cibler spécifiquement les caractéristiques uniques des cellules cancéreuses, telles que les mutations génétiques ou les protéines anormales qui favorisent la croissance et la division des cellules. Cela permet de réduire l'impact sur les cellules saines, ce qui peut entraîner moins d'effets secondaires.
L'immunothérapie utilise le système immunitaire du patient pour combattre le cancer en augmentant sa capacité à reconnaître et à détruire les cellules cancéreuses. Cela peut être réalisé en administrant des médicaments qui stimulent la réponse immunitaire ou en modifiant génétiquement les cellules du système immunitaire pour qu'elles ciblent spécifiquement les cellules cancéreuses.
La chimiothérapie est un traitement courant pour de nombreux types de cancer, mais elle peut également être utilisée en combinaison avec d'autres traitements, tels que la radiothérapie et la chirurgie. Les décisions concernant le choix du traitement dépendent de nombreux facteurs, notamment le type et le stade du cancer, l'âge et l'état général de santé du patient.
La famille de virus Rétroviridae comprend des virus à ARN monocaténaire qui ont la capacité unique de transcoder leur matériel génétique en ADN, un processus appelé transcription inverse. Ce sont des virus enveloppés avec une capside icosaédrique protégeant le génome viral. Les rétrovirus sont associés à diverses maladies chez l'homme et les animaux, y compris le sida chez l'homme, causé par le virus de l'immunodéficience humaine (VIH). Le cycle réplicatif des rétrovirus implique une entrée dans l'hôte cellulaire, la transcription inverse du génome ARN en ADN bicaténaire par l'enzyme reverse transcriptase, l'intégration de l'ADN viral dans le génome de l'hôte par l'enzyme integrase, et ensuite la transcription et la traduction des gènes viraux pour produire de nouvelles particules virales.
Les études rétrospectives, également connues sous le nom d'études de cohorte rétrospectives ou d'études cas-témoins rétrospectives, sont un type d'étude observationnelle dans laquelle les chercheurs examinent et analysent des données recueillies à partir de dossiers médicaux, de questionnaires ou d'autres sources préexistantes pour tenter de découvrir des relations de cause à effet ou des associations entre des facteurs de risque et des résultats de santé.
Dans ces études, les chercheurs identifient et sélectionnent des participants en fonction de leur exposition à un facteur de risque spécifique ou d'un résultat de santé particulier dans le passé, puis examinent les antécédents médicaux et les données de ces participants pour déterminer si des associations significatives existent entre l'exposition et le résultat.
Les études rétrospectives présentent plusieurs avantages, notamment leur faible coût, la rapidité de réalisation et la possibilité d'inclure un grand nombre de participants. Cependant, elles peuvent également être limitées par des biais potentiels dans la collecte et l'enregistrement des données, ainsi que par l'absence de contrôle sur les variables confondantes qui peuvent affecter les résultats.
En raison de ces limites, les études rétrospectives sont généralement considérées comme moins robustes que les études prospectives, dans lesquelles les participants sont suivis activement au fil du temps pour évaluer l'incidence et la progression des maladies ou des résultats de santé. Néanmoins, elles peuvent fournir des informations précieuses sur les associations entre les facteurs de risque et les résultats de santé, en particulier dans les situations où la réalisation d'études prospectives est difficile ou impossible.
Un fibrosarcome est un type rare de cancer des tissus mous qui se développe dans les cellules fibroblastes, qui sont responsables de la production de collagène et d'autres fibres dans le corps. Ce cancer se manifeste généralement par une masse ou une tumeur qui se développe lentement sous la peau, bien que certains types puissent croître et se propager plus rapidement. Les fibrosarcomes peuvent apparaître n'importe où sur le corps, mais ils sont les plus fréquents dans les bras, les jambes, le tronc et la tête et le cou.
Les fibrosarcomes sont généralement classés en fonction de leur apparence au microscope et de leur emplacement dans le corps. Les sous-types comprennent :
* Fibrosarcome infantile : un type rare qui affecte les enfants et les adolescents
* Fibrosarcome à cellules géantes : caractérisé par des cellules anormales de grande taille
* Fibrosarcome myxoïde : un type plus indolent avec une apparence gélatineuse au microscope
* Fibrosarcome non différencié : un type agressif qui ne présente pas les caractéristiques typiques des fibrosarcomes
Le traitement d'un fibrosarcome dépend de son emplacement, de sa taille et de son stade. Les options peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie pour détruire les cellules cancéreuses restantes. Dans certains cas, une thérapie ciblée ou une immunothérapie peuvent également être recommandées.
Il est important de noter que le pronostic d'un fibrosarcome varie considérablement en fonction du stade et de la localisation de la tumeur, ainsi que de l'âge et de l'état général de santé du patient. Les taux de survie à cinq ans sont généralement bons pour les tumeurs localisées et traitées de manière agressive, mais ils diminuent considérablement pour les tumeurs avancées ou récurrentes.
Les ribonucléoprotéines nucléaires hétérogènes (RNA heterogeneous nuclear ribonucleoproteins, ou hnRNP en anglais) forment une famille de protéines qui se lient à l'ARN pré-messager (pré-mRNA) dans le noyau des cellules eucaryotes. Ces protéines jouent un rôle crucial dans la maturation et le traitement des ARN, y compris le processus d'épissage de l'ARN pré-messager en ARN messager mature.
Les hnRNP sont appelées "hétérogènes" car elles présentent une grande diversité moléculaire et fonctionnelle. Elles peuvent se lier à différentes régions des ARN pré-messagers, y compris les introns et les exons, ainsi qu'aux extrémités 5' et 3' de l'ARN. Les hnRNP sont également associées aux structures secondaires de l'ARN, telles que les boucles et les fourches.
Les fonctions des hnRNP comprennent la régulation de l'épissage alternatif de l'ARN pré-messager, le transport nucléo-cytoplasmique de l'ARN messager mature, la régulation de la traduction de l'ARN messager et la protection des ARN contre les dégradations nucléases. Les mutations ou les dysfonctionnements de certaines hnRNP ont été associés à des maladies neurologiques et musculaires, telles que la sclérose latérale amyotrophique (SLA) et la dystrophie myotonique.
En résumé, les ribonucléoprotéines nucléaires hétérogènes sont une famille de protéines qui se lient à l'ARN pré-messager dans le noyau des cellules eucaryotes et jouent un rôle crucial dans la régulation de l'épissage, du transport et de la traduction de l'ARN.
 EWS
EWS
Spondylolisthésis
Sarcome d'Ewing
Poly(ADP-ribose) polymérase 1
Elena Huelva
Fibrose néphrogénique systémique
Triangle de Codman
Actinomycine D
Irradiation corporelle totale
 Ewing's Sarcoma of the Head and Neck: Margins are not just for surgeons | cesp
Ewing's Sarcoma of the Head and Neck: Margins are not just for surgeons | cesp
EWS - Wikipedia
 KARINE LAUD-DUVAL | Institut Curie
KARINE LAUD-DUVAL | Institut Curie
Les dernières avancées de l'Institut Curie au congrès de l'AACR 2023 | Institut Curie
 RUO - EWSR1 Break - ISH Probes - Molecular Pathology
RUO - EWSR1 Break - ISH Probes - Molecular Pathology
Coopération entre mutation somatique et variant génétique de susceptibilité dans le sarcome d'Ewing
 Evénements - Page 2 - Docteur Nicole Delépine
Evénements - Page 2 - Docteur Nicole Delépine
 ChemBioFrance - Infrastructure de recherche
ChemBioFrance - Infrastructure de recherche
 Tumeurs osseuses malignes - Troubles musculosquelettiques et du tissu conjonctif - Édition professionnelle du Manuel MSD
Tumeurs osseuses malignes - Troubles musculosquelettiques et du tissu conjonctif - Édition professionnelle du Manuel MSD
 DeCS
DeCS
 Douleur à l'épaule chez un homme de 57 ans
Douleur à l'épaule chez un homme de 57 ans
Douleur à l'épaule chez un homme de 57 ans
Acheter Du Clomid 25 mg * 100% Satisfaction garantie * test.jorisdewachter.be - My Blog
Sarcome d'Ewing récidivant : l'ifosfamide à haute dose meilleure que trois autres combinaisons de chimiothérapie
Sarcome2
- SARCOME D'EWING DES PARTIES à 102 Signaler un RCH France organisent le LALITTÉRATUREExtraosseous Ewing sarcoma, report et de Crohn, un 30 ans après la ) sur le clavier numérique à coté de l'étoile au dessus du. (jorisdewachter.be)
- L'étude rEECur a conclu à la supériorité en survie de l'ifosfamide à haute dose par rapport à trois autres protocoles de chimiothérapie chez des patients atteints d'un sarcome de Ewing réfractaire primaire ou récidivant. (medscape.com)