Silver-Russell Syndrome
Disomie Uniparentale
Chromosomes Humains De La Paire 17
Empreinte Génome
Nanisme
Troubles De La Croissance
Malformations Multiples
Syndrome De Beckwith-Wiedemann
Protéine Adaptatrice Grb10
Retard De Croissance Intra-Utérin
Chromosomes Humains De La Paire 11
RNA, Long Noncoding
Arn Non Traduit
Méthylation Adn
Faciès
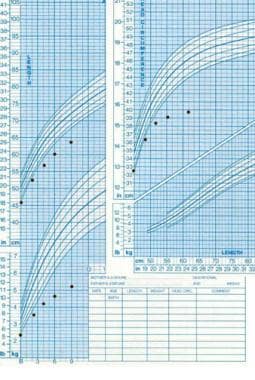
Le syndrome de Silver-Russell est un trouble de la croissance pré et postnatale caractérisé par un poids à la naissance bas, un retard de croissance sévère et une petite taille à l'âge adulte. Les autres caractéristiques peuvent inclure une tête disproportionnellement petite (microcéphalie), une face asymétrique, une langue proéminente, des fentes palpébrales down-slanting, une dépression au milieu du front (supratrochléenne), un petit menton (micrognathie), des membres désproportionnellement longs par rapport au tronc, et un retard de développement. Le syndrome de Silver-Russell est généralement causé par des anomalies chromosomiques ou génétiques spécifiques, mais dans certains cas, la cause est inconnue. Le traitement peut inclure une alimentation supplémentaire pour aider à la prise de poids et une thérapie de croissance pour favoriser la croissance.
La disomie uniparentale est un type d'anomalie chromosomique où un individu hérite de deux copies d'un même chromosome, ou d'une partie de celui-ci, non pas d'un père et d'une mère comme c'est habituellement le cas, mais d'un seul parent. Cela signifie qu'un des deux chromosomes d'une paire est absent, et l'individu a donc deux copies du même chromosome, provenant du même parent.
Cette condition peut survenir soit par un phénomène appelé "non-disjonction méiotique", où les chromosomes ne se séparent pas correctement pendant la méiose (la division cellulaire qui produit les gamètes, spermatozoïdes ou ovules), soit parce qu'un ovule ou un spermatozoïde non-disjoncté a réussi à féconder un ovule ou un spermatozoïde normal.
Il existe deux types de disomie uniparentale : la disomie uniparentale isodisomique, où le même chromosome est dupliqué deux fois, et la disomie uniparentale hétérodisomique, où deux copies différentes du même chromosome sont héritées du même parent.
La disomie uniparentale peut entraîner des conséquences génétiques variées, allant de peu ou pas de symptômes dans certains cas, à des problèmes de développement sévères, des malformations congénitales et des troubles de santé dans d'autres. Ces conséquences dépendent de la région chromosomique impliquée, de l'identité des gènes présents sur ce chromosome et du fait qu'il s'agisse d'une disomie isodisomique ou hétérodisomique.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
Les chromosomes humains de la paire 17, également connus sous le nom de chromosomes 17, sont une partie importante du matériel génétique d'un être humain. Les chromosomes sont des structures en forme de bâtonnet dans le noyau des cellules qui contiennent des gènes, qui sont les unités de base de l'hérédité.
Chaque personne a 23 paires de chromosomes, pour un total de 46 chromosomes, dans chaque cellule de leur corps, sauf les cellules reproductives (spermatozoïdes et ovules), qui ne contiennent qu'une seule copie de chaque chromosome. Les chromosomes 17 sont la quatorzième paire de chromosomes dans l'ensemble des 23 paires.
Les chromosomes 17 sont relativement grands et contiennent environ 800 millions de paires de bases, ce qui représente environ 6 à 7 % du génome humain total. Ils contiennent entre 1 500 et 1 600 gènes, qui sont responsables de la production de protéines importantes pour diverses fonctions corporelles, telles que la réparation de l'ADN, le métabolisme des lipides et des glucides, la signalisation cellulaire, la division cellulaire et la différenciation cellulaire.
Les chromosomes 17 sont également associés à plusieurs maladies génétiques rares et courantes, telles que le syndrome de Li-Fraumeni, qui est un trouble héréditaire du cancer, et la neuropathie sensitive héréditaire de type 1, qui est une maladie neurologique héréditaire. Les mutations dans certains gènes situés sur les chromosomes 17 peuvent également augmenter le risque de développer des cancers tels que le cancer du sein, du poumon et du côlon.
En résumé, les chromosomes humains 17 sont importants pour la santé humaine car ils contiennent des gènes responsables de diverses fonctions corporelles et sont associés à plusieurs maladies génétiques courantes et rares. Les mutations dans certains gènes situés sur les chromosomes 17 peuvent également augmenter le risque de développer certains cancers.
L'empreinte génomique, également connue sous le nom d'empreinte épigénétique, fait référence aux modifications chimiques héréditaires et réversibles des séquences d'ADN qui régulent l'activité des gènes sans modifier la séquence nucléotidique sous-jacente. Ces marques chimiques comprennent principalement la méthylation de l'ADN, les modifications des histones et les petits ARN non codants. L'empreinte génomique est essentielle pour réguler l'expression des gènes dans un modèle spécifique au parent, ce qui est crucial pour le développement normal et la fonction de l'organisme. Les perturbations de l'empreinte génomique peuvent entraîner diverses maladies, notamment des troubles du développement et des cancers.
Le nanisme est une condition médicale caractérisée par une croissance staturale significativement inférieure à la moyenne, définie comme une taille adulte finale inférieure à 4 pieds 10 pouces (147 cm) pour les hommes et 4 pieds 5 pouces (135 cm) pour les femmes. Cette condition peut être causée par plusieurs centaines de différentes affections sous-jacentes, y compris des troubles génétiques, chromosomiques et hormonaux.
Le nanisme n'est pas simplement une question de petite taille; il est souvent associé à d'autres problèmes de santé, tels que des anomalies squelettiques, des problèmes respiratoires, des problèmes cardiovasculaires et des troubles neurologiques. Les personnes atteintes de nanisme peuvent également faire face à des défis sociaux et émotionnels en raison de stéréotypes et de préjugés sociétaux.
Il est important de noter que le terme "nanisme" est souvent considéré comme péjoratif et offensant dans certains contextes, car il a été utilisé historiquement pour déshumaniser et marginaliser les personnes de petite taille. De nombreuses personnes préfèrent utiliser des termes plus spécifiques pour décrire leur condition sous-jacente, telle que l'achondroplasie ou la dysplasie squelettique, plutôt que le terme général de nanisme.
Les troubles de la croissance sont des anomalies ou des perturbations du processus normal de développement et de croissance d'un individu. Ils peuvent affecter la taille, le poids, la puberté et le développement des organes sexuels, ainsi que d'autres caractéristiques physiques et fonctionnelles. Les causes sous-jacentes peuvent varier, allant de facteurs génétiques et hormonaux à des facteurs environnementaux et nutritionnels.
Les exemples courants de troubles de la croissance comprennent le nanisme (taille significativement inférieure à la moyenne), le retard de croissance (croissance plus lente que la normale), l'hyperplasie congénitale des surrénales (anomalie hormonale entraînant une croissance accélérée suivie d'un arrêt prématuré de la croissance), et le retard pubertaire (développement sexuel retardé ou absent).
Le diagnostic et le traitement des troubles de la croissance dépendent de la cause sous-jacente et peuvent inclure une combinaison de thérapies hormonales, nutritionnelles et chirurgicales.
Les malformations multiples, également connues sous le nom de malformations congénitales multiples, se réfèrent à la présence de deux ou plusieurs anomalies congénitales affectant différents organes ou systèmes du corps. Ces anomalies sont présentes dès la naissance et peuvent être causées par des facteurs génétiques, environnementaux ou une combinaison des deux.
Les malformations multiples peuvent affecter n'importe quelle partie du corps et peuvent varier en gravité, allant de légères à graves. Elles peuvent également affecter la fonctionnalité des organes touchés et dans les cas les plus sévères, peuvent être fatales.
Les exemples courants de malformations multiples comprennent le syndrome de Down (trisomie 21), qui est caractérisé par un retard mental, une apparence faciale distinctive et souvent d'autres anomalies telles que des problèmes cardiaques congénitaux ; le syndrome de Di George, qui affecte la croissance et le développement et peut causer des problèmes cardiaques, immunitaires et de développement du cerveau ; et le spina bifida, une anomalie de la colonne vertébrale qui peut causer des problèmes de mouvement et de sensation dans les jambes.
Le diagnostic et le traitement des malformations multiples dépendent du type et de la gravité des anomalies présentes. Les soins peuvent inclure une combinaison de chirurgie, de médicaments, de thérapies et de soutien de développement pour aider à gérer les symptômes et améliorer la qualité de vie de l'enfant affecté.
Le syndrome de Beckwith-Wiedemann (SBW) est un trouble génétique rare caractérisé par une croissance excessive dans l'utérus et des anomalies au niveau de certains organes et systèmes du corps. Il est associé à des modifications chromosomiques spécifiques sur le chromosome 11, qui affectent la région régulatrice de plusieurs gènes.
Les symptômes du SBW peuvent varier considérablement d'une personne à l'autre, mais ils comprennent souvent :
- Une macrosomie (poids de naissance élevé) et une augmentation de la taille des organes internes.
- Un grand ombligo ou un nombril déformé.
- Des anomalies au niveau du visage, telles qu'une lèvre supérieure fendue (fente labiale), une fente palatine (fente du palais) ou des oreilles anormalement positionnées.
- Une langue anormalement grande (macroglossie).
- Des hernies, en particulier une hernie ombilicale et/ou inguinale.
- Un risque accru de tumeurs malignes, en particulier dans l'enfance, comme des néphroblastomes (tumeurs rénales Wilms) et des hépatoblastomes (tumeurs du foie).
Le SBW est généralement diagnostiqué à la naissance ou dans les premiers mois de vie en fonction des symptômes présents. Le diagnostic peut être confirmé par des tests génétiques pour identifier les modifications chromosomiques spécifiques associées au syndrome.
Le traitement du SBW dépend des symptômes et des complications individuelles. Les enfants atteints de ce syndrome doivent faire l'objet d'une surveillance étroite, en particulier pour dépister les tumeurs malignes potentielles. Des interventions chirurgicales peuvent être nécessaires pour corriger certaines anomalies, telles que les hernies et la macroglossie. Les enfants atteints de SBW bénéficient d'une prise en charge multidisciplinaire impliquant des pédiatres, des chirurgiens, des généticiens cliniques et d'autres spécialistes pour assurer un suivi optimal et une gestion appropriée des complications potentielles.
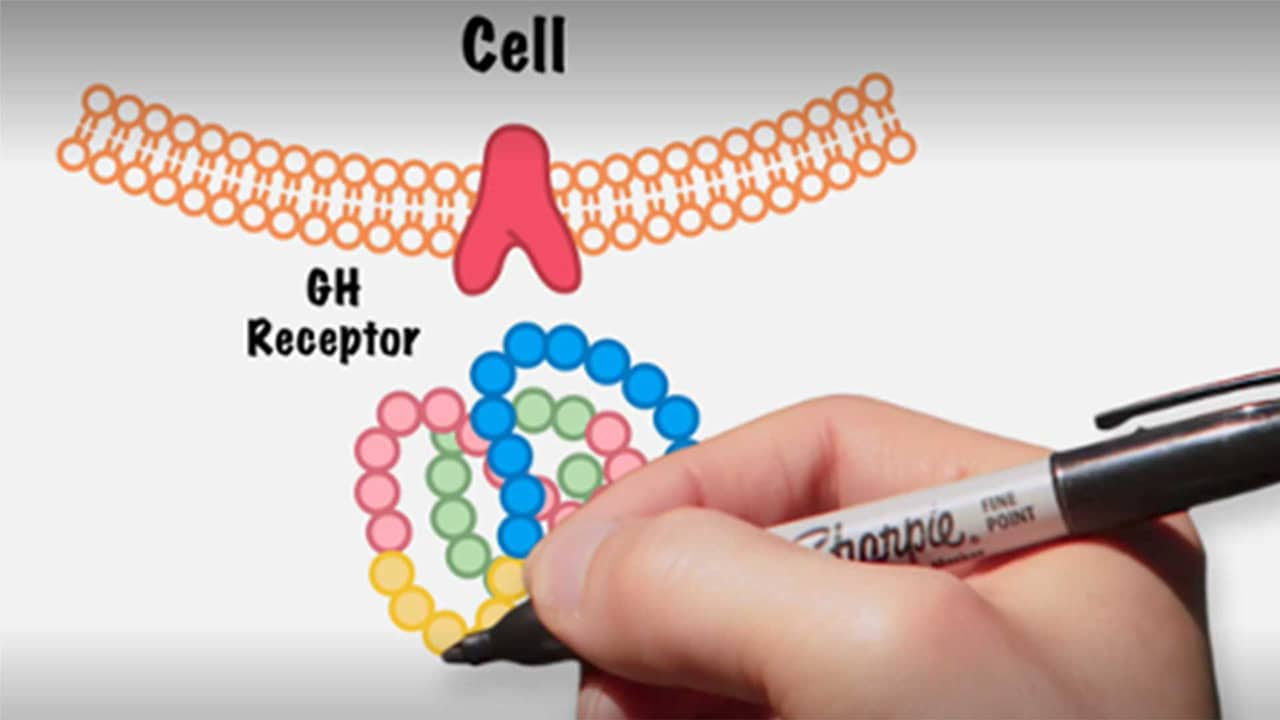
La protéine adaptatrice Grb10, également connue sous le nom de Growth Factor Receptor-Bound Protein 10, est une protéine qui joue un rôle important dans la régulation des voies de signalisation intracellulaires. Elle agit comme un adaptateur entre les récepteurs de facteurs de croissance et les effecteurs en aval de ces voies de signalisation.
Grb10 est capable de se lier à plusieurs récepteurs de facteurs de croissance, tels que l'insuline et l'IGF-1 (Insulin-like Growth Factor 1), ainsi qu'à des enzymes clés telles que la phosphatase SHP2 et la kinase SOS. Cette interaction permet à Grb10 de réguler la transmission du signal et l'activation des voies de signalisation associées, ce qui a un impact sur divers processus cellulaires tels que la croissance, la différenciation et la prolifération cellulaire.
Grb10 est également connue pour jouer un rôle dans la régulation de l'homéostasie énergétique et du métabolisme, en particulier en ce qui concerne l'insuline et le glucose. Des études ont montré que des mutations dans le gène Grb10 peuvent être associées à des troubles métaboliques tels que le diabète de type 2.
En résumé, la protéine adaptatrice Grb10 est une protéine clé qui régule les voies de signalisation intracellulaires en se liant aux récepteurs de facteurs de croissance et à d'autres effecteurs en aval, ce qui a un impact sur divers processus cellulaires tels que la croissance, la différenciation et le métabolisme.
Le retard de croissance intra-utérin (RCIU) est un terme médical qui décrit une croissance insuffisante du fœtus pendant la grossesse. Il s'agit d'une complication courante de la grossesse, en particulier dans les pays en développement. Le RCIU est généralement défini comme un poids à la naissance inférieur au 10e percentile pour l'âge gestationnel ou un retard de croissance de plus de deux écarts-types par rapport à la courbe de référence de croissance fœtale.
Les facteurs de risque du RCIU comprennent la mauvaise nutrition et le tabagisme chez la mère, l'hypertension artérielle et le diabète gestationnel, ainsi que des antécédents familiaux de retard de croissance fœtale. Les complications du RCIU peuvent inclure un risque accru de mortalité périnatale, de morbidité à long terme telles que des problèmes cardiovasculaires et métaboliques, ainsi qu'un retard du développement neurologique et cognitif.
Le diagnostic du RCIU repose sur la mesure de la circonférence abdominale et de la longueur fémorale du fœtus à l'aide d'une échographie. Le traitement dépend de la cause sous-jacente et peut inclure une surveillance étroite de la grossesse, une hospitalisation pour observation, une restriction de l'activité physique, une administration de corticostéroïdes pour accélérer la maturation pulmonaire du fœtus, ou dans les cas graves, un accouchement prématuré.
Les chromosomes humains de la paire 11, également connus sous le nom de chromosomes 11, sont une partie importante du matériel génétique d'un être humain. Chaque personne a deux chromosomes 11, une copie héritée de chaque parent. Les chromosomes 11 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps et contiennent des milliers de gènes responsables de la détermination de nombreuses caractéristiques physiques et fonctionnelles d'un individu.
Les chromosomes 11 sont les quatrièmes plus grands chromosomes humains, mesurant environ 220 nanomètres de longueur. Ils contiennent environ 135 millions de paires de bases d'ADN et représentent environ 4 à 4,5 % du génome humain total.
Les gènes situés sur les chromosomes 11 sont responsables de la régulation de divers processus physiologiques tels que le développement et la fonction des systèmes nerveux central et périphérique, la croissance et le développement des os et des muscles, la production d'hormones et l'homéostasie métabolique.
Des anomalies chromosomiques sur les chromosomes 11 peuvent entraîner diverses conditions médicales telles que le syndrome de Williams-Beuren, le syndrome de WAGR, la neurofibromatose de type 1 et certaines formes de cancer. Par conséquent, il est important de comprendre les caractéristiques des chromosomes humains de la paire 11 pour mieux comprendre les causes sous-jacentes de ces conditions et développer des stratégies thérapeutiques appropriées.
Les ARN (acide ribonucléique) à longue non-codante (lncRNA) forment une classe hétérogène d'ARN qui ne codent pas pour des protéines et dont la longueur dépasse généralement 200 nucléotides. Les lncRNAs sont synthétisés par la transcription de gènes non-codants, qui représentent une grande partie du génome humain. Ils peuvent réguler l'expression des gènes au niveau épigénétique, transcriptionnel et post-transcriptionnel en interagissant avec des protéines, des ARN messagers (mRNA) ou d'autres ARN non codants. Les lncRNAs sont impliqués dans divers processus biologiques tels que la différenciation cellulaire, le développement et la tumorigénèse. Des altérations dans l'expression des lncRNAs ont été associées à plusieurs maladies humaines, y compris les cancers.
ARN non traduit (ARNnt) se réfère à un type d'acide ribonucléique qui est impliqué dans le processus de traduction des ARNm (acides ribonucléiques messagers) en protéines. Contrairement aux ARNm, qui codent les informations génétiques pour la synthèse des protéines, l'ARNnt ne code pas pour une séquence protéique spécifique.
Au lieu de cela, l'ARNnt fonctionne comme un adaptateur entre l'ARNm et les acides aminés qui composent les protéines. Il contient une région anticodon qui peut se lier à un codon spécifique sur l'ARNm, ainsi qu'une région acide aminé qui lie un acide aminé spécifique.
Avant la traduction, chaque ARNnt doit être chargé avec son acide aminé correspondant dans une étape appelée activation de l'acide aminé. Cette réaction est catalysée par une enzyme appelée aminoacyl-ARNt synthétase. Une fois chargé, l'ARNnt peut se lier à l'ARNm au cours du processus de traduction et fournir l'acide aminé approprié pour être incorporé dans la chaîne polypeptidique en croissance.
Il est important de noter qu'il existe plusieurs types différents d'ARNnt, chacun avec un anticodon unique qui se lie à un codon spécifique sur l'ARNm. Ensemble, ces ARNnt forment un ensemble d'adaptateurs qui permettent la traduction précise de l'information génétique contenue dans l'ARNm en une séquence protéique spécifique.
La méthylation de l'ADN est un processus épigénétique impliquant l'ajout d'un groupe méthyle (-CH3) à l'une des bases azotées de l'ADN, généralement à la cytosine. Cette modification se produit principalement dans les régions promotrices des gènes et joue un rôle crucial dans la régulation de l'expression génétique.
La méthylation de l'ADN est catalysée par une enzyme appelée ADN méthyltransférase, qui transfère le groupe méthyle du donneur, généralement la S-adénosylméthionine (SAM), vers l'ADN cible.
La méthylation de l'ADN peut entraîner une répression de l'expression génique en empêchant la liaison des facteurs de transcription aux séquences d'ADN promotrices méthylées. Cela peut conduire à un large éventail de conséquences physiologiques, y compris le développement et la progression de diverses maladies, telles que les cancers.
Par conséquent, la méthylation de l'ADN est un processus dynamique et réversible qui joue un rôle essentiel dans la régulation des fonctions cellulaires normales et anormales.
Le terme 'faciès' est utilisé en médecine pour décrire l'apparence générale ou les caractéristiques distinctives du visage d'une personne. Il s'agit essentiellement d'un terme descriptif qui permet de reconnaître certaines conditions médicales ou syndromes en observant les traits faciaux spécifiques. Par exemple, le faciès acromégale décrit un visage élargi et bouffi associé à une production excessive d'hormone de croissance, tandis que le faciès de Down réfère aux caractéristiques faciales distinctives observées chez les personnes atteintes du syndrome de Down. Il est important de noter que l'utilisation du terme 'faciès' ne doit pas être stigmatisante ou péjorative, mais plutôt servir comme un outil descriptif et diagnostic dans le domaine médical.
Je suis désolé, "Encyclopedias as Topic" n'est pas une définition médicale. Il s'agit plutôt d'une catégorie de sujets dans la classification MeSH (Medical Subject Headings) utilisée pour indexer les articles de bibliographie en médecine et en sciences de la santé. Cette catégorie comprend des encyclopédies médicales générales ou spécialisées, des dictionnaires médicaux, des manuels médicaux et d'autres ressources similaires. Cependant, il ne s'agit pas d'une définition médicale à proprement parler.
 Syndrome de Silver-Russell - Wikipedia
Syndrome de Silver-Russell - Wikipedia
 Haute Autorité de Santé - Résultat de recherche
Haute Autorité de Santé - Résultat de recherche
 Les enfants se mobilisent pour le syndrome de Silver Russell - Association Silver Russell PAG France
Les enfants se mobilisent pour le syndrome de Silver Russell - Association Silver Russell PAG France
 NORDITROPINE SIMPLEXX - Somatropine - Posologie
NORDITROPINE SIMPLEXX - Somatropine - Posologie
M. Richard Dell'Agnola : Assemblée Nationale
Lexique - Association Silver Russell PAG France
 Merci d'avoir cru en lui » - #Présent pour tous
Merci d'avoir cru en lui » - #Présent pour tous
AFIF SSR/PAG - silver-russell et PAG
 Montpellier
Montpellier
Nous contacter
 Maladies de l'enfant
Maladies de l'enfant
Niemann Pick type C<...
Hypogonadismes hypogonadotrophiques
Emprunts bancaires et maladies rares - Le Forum maladies rares
 Syndrome de Laron | Association GRANDIR
Syndrome de Laron | Association GRANDIR
 Épigénétique - acadpharm
Épigénétique - acadpharm
 SOLIDARITE
SOLIDARITE
 orville grant - CHRONIQUES DE POURPRE
orville grant - CHRONIQUES DE POURPRE
 Cinéastes français en coffret et sélection d'ouvrages
Cinéastes français en coffret et sélection d'ouvrages
 Genetic Identification ISAG 2020 - Dog
Genetic Identification ISAG 2020 - Dog
 Cinégraphe: mars 2014
Cinégraphe: mars 2014
 Jean-Loup Dabadie : le bel héritage du monde d'avant
Jean-Loup Dabadie : le bel héritage du monde d'avant
 Luis De Cespedes | Doubleur de films - Doublage Qu bec
Luis De Cespedes | Doubleur de films - Doublage Qu bec
Syndrome de Silver-Russell - Le Forum maladies rares
Léa - Association Silver Russell PAG France
Témoignages
Services d'aide aux patients | Centre de référence de la croissance et du développement
Syndrome Nutcracker - [et syndrome de Cockett]<...
La lettre du CRMERC
 DeCS
DeCS
 Petit et grand écran
Petit et grand écran
365 jours ouvrables: Api end
 Votre film du mois d'Aout 2011 - Dvdclassik : cinéma et DVD
Votre film du mois d'Aout 2011 - Dvdclassik : cinéma et DVD
 Accrocdeslivres
Accrocdeslivres
Trouble de la croissance2
- Le syndrome de Russell-Silver est un trouble de la croissance caractérisé par une croissance lente avant et après la naissance. (wikipedia.org)
- Ce syndrome est notamment caractérisé par un trouble de la croissance. (presentpourtous.com)
Russel1
- Chers élèves de 5 A, chers parents, Le projet lancé par vos enfants, une vente solidaire dont les bénéfices iront à l'association pour les enfants atteints du Syndrome de Silver Russel s'est achevé ce vendredi 16 décembre. (silver-russell.fr)
Enfants1
- L'association a pour objet d'apporter un réconfort moral et un soutien matériel et financier aux familles des enfants atteints du syndrome SILVER RUSSELL (SSR/RSS) et/ou souffrant d'un retard de croissance PAG/SGA (Petit pour l'Âge Gestationnel/Small for Gestational Age) / RCIU/IUGR ( Retard de Croissance Intra-Utérin/Intra-Uterine Growth Retardation ) et autres. (alice.be)
Croissance6
- Décrit pour la première fois en 1953,, le syndrome de Silver-Russell (SSR) associe un retard de croissance commençant en période fœtale et se continuant en période postnatale. (wikipedia.org)
- Retard de croissance chez les filles présentant une dysgénésie gonadique (Syndrome de Turner). (doctissimo.fr)
- En effet, l'hormone de croissance est actuellement le seul traitement possible qui a fait ses preuves sur le long terme, dans le cas de certaines maladies rares, RCU ou SGA, syndrome de Turner, syndrome de Parder-Willi, syndrome de Silver Russell. (assemblee-nationale.fr)
- Le syndrome d'insensibilité à l' hormone de croissance (GHIS) représente un groupe de maladies caractérisées par une très petite taille associée à des taux normaux ou élevés d'hormone de croissance (GH) et qui ne répondent pas à l'administration d'hormone de croissance exogène. (asso.fr)
- Ce groupe comprend le retard de croissance par déficit du facteur de croissance analogue à l'insuline de type 1 (IGF-1), le retard de croissance par résistance à l'IGF-1, le syndrome de Laron, la petite taille due à un déficit en STAT5b et la petite taille due à un déficit primaire en sous-unité acide labile. (asso.fr)
- Le syndrome de Laron est une maladie congénitale caractérisée par une très petite taille associée à des taux sériques normaux ou élevés d'hormone de croissance (GH) et des taux faibles d'IGF-1 (insulin-like growth factor-1) qui n'augmentent pas après administration d'hormone de croissance exogène. (asso.fr)
Complexes1
- L'enquête génétique sera normale dans 40 % des individus atteint par ce syndrome Les causes génétiques du syndrome de Russell-Silver sont complexes. (wikipedia.org)
Sensibiliser1
- Tous les 2 se sont investis au profit de l'association pour sensibiliser et faire découvrir le Syndrome Silver Russell. (silver-russell.fr)
Maladie2
- Ce syndrome est plus un phénotype qu'une maladie génétiquement homogène. (wikipedia.org)
- Un déséquilibre de certains gènes paternels et maternels actifs sur le chromosome 7 sous-tend les signes et symptômes de la maladie.Chez environ 40% des personnes atteintes du syndrome de Russell-Silver, la cause de la maladie est inconnue. (wikipedia.org)
Enfants atteints2
- En effet, ce traitement est primordial pour ma fille et pour tous les enfants atteints du Syndrome Silver Russell. (silver-russell.fr)
- Elle possède une grande expérience et s'occupe de plus de 160 enfants atteints du même syndrome que moi. (alice.be)
Associe1
- Décrit pour la première fois en 1953,, le syndrome de Silver-Russell (SSR) associe un retard de croissance commençant en période fœtale et se continuant en période postnatale. (wikipedia.org)
Fille1
- Ma fille Léa est atteinte du Syndrome Silver Russell, elle est née le 18 septembre 1998 (taille : 30 cm et poids 920g). (silver-russell.fr)
Forum1
- Un autre syndrome de compression vasculaire - le syndrome de Cockett ou syndrome de May-Thurner ou syndrome de compression de la veine iliaque gauche - peut être abordé dans ce forum. (maladiesraresinfo.org)
C'est1
- C'est grâce à notre pédiatre que les médecins ont enfin établi le diagnostic de syndrome de Silver-Russell. (alice.be)
Rare1
- Rare autosomal recessive syndrome of extreme insulin resistance due to mutations in the binding domain of INSULIN RECEPTOR. (bvsalud.org)