Wolf-Hirschhorn Syndrome
Chromosomes Humains De La Paire 14
Hypertélorisme
Récepteur Fgfr5
Malformations Multiples
Microcéphalie
Acétate Potassium
Translocation, Genetic
Intellectual Disability
Maladies Chromosomiques
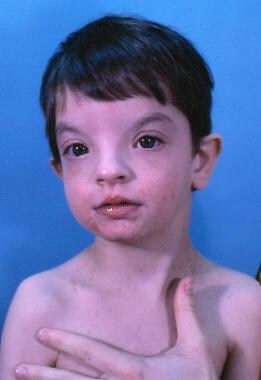
Le syndrome de Wolf-Hirschhorn est un trouble génétique rare caractérisé par une gamme de symptômes, y compris un retard de développement sévère, un visage distinct avec une fente labiale et/ou palatine, une croissance insuffisante, des problèmes cardiaques congénitaux, des anomalies squelettiques et des problèmes rénaux. Ce syndrome est causé par une délétion partielle du chromosome 4, ce qui entraîne la perte de matériel génétique dans la région 4p16.3. Les symptômes et leur gravité peuvent varier considérablement d'un individu à l'autre, en fonction de la quantité de matériel génétique perdu. Le syndrome de Wolf-Hirschhorn est généralement découvert à la naissance ou dans les premiers mois de vie et nécessite une prise en charge multidisciplinaire tout au long de la vie.
Les chromosomes humains de la paire 14, également connus sous le nom de chromosomes 14, sont des structures composées de ADN et protéines qui contiennent des gènes et se trouvent dans le noyau de chaque cellule du corps. Les chromosomes 14 sont une paire de chromosomes homologues, ce qui signifie qu'ils ont la même taille, la même forme et contiennent des gènes similaires aux mêmes emplacements le long de la chromosome.
Chaque personne a 23 paires de chromosomes dans chaque cellule de leur corps, pour un total de 46 chromosomes. Les chromosomes 14 sont la 14ème paire de ces chromosomes, et ils sont numérotés de 14 à 14, ce qui signifie qu'ils sont présents deux fois dans chaque cellule du corps.
Les chromosomes 14 contiennent des centaines de gènes qui fournissent des instructions pour la production de protéines et d'autres produits génétiques importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions, telles que les troubles neurodégénératifs, les cancers et les maladies héréditaires.
En résumé, les chromosomes humains de la paire 14 sont des structures composées d'ADN et de protéines qui contiennent des gènes importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions.
L'hypertélorisme est un terme utilisé en ophtalmologie et en médecine générale pour décrire une condition anatomique où les distances entre les deux yeux (interpupillaires) sont plus éloignées que la normale. Cette distance est mesurée en millimètres à partir du milieu de chaque pupille. Chez les personnes sans hypertélorisme, cette distance est généralement d'environ 28 à 35 mm. Cependant, chez les personnes atteintes d'hypertélorisme, cette distance peut dépasser 35 mm.
Cette condition peut être isolée (appelée hypertélorisme simple) ou associée à d'autres anomalies congénitales du visage et du crâne, telles que la fente labiale ou palatine, les yeux écartés (exophtalmie), le nez large et aplati, et une fontanelle antérieure plus grande que d'habitude. L'hypertélorisme est souvent associé à certaines maladies génétiques, telles que le syndrome de Noonan, le syndrome de Waardenburg, le syndrome de Crouzon et le syndrome de Pfeiffer.
Il est important de noter que l'hypertélorisme peut être un signe révélateur d'une maladie sous-jacente plus grave, il est donc essentiel de consulter un médecin si vous ou votre enfant présentez des signes d'hypertélorisme.
Le récepteur FGFR5 (Fibroblast Growth Factor Receptor 5) est une protéine qui se trouve sur la surface des cellules et joue un rôle crucial dans la régulation de divers processus biologiques, tels que la croissance, la différenciation et la survie cellulaire. Il s'agit d'un membre de la famille des récepteurs aux facteurs de croissance fibroblastique (FGFR).
Le récepteur FGFR5 est codé par le gène FGFR5 et se lie spécifiquement au ligand FGF23 (Facteur de croissance des fibroblastes 23). Lorsque le ligand FGF23 se lie au récepteur FGFR5, il active une cascade de signalisation intracellulaire qui régule l'homéostasie minérale et la phosphaturie rénale.
Des mutations dans le gène FGFR5 ont été associées à certaines maladies humaines, telles que l'hypophosphatémie liée à l'X (XLH) et la tumorigenèse autosomique dominante de l'ostéogénèse imparfaite (ADTD). Ces mutations peuvent entraîner une activation ou une inhibition anormale du récepteur FGFR5, ce qui peut perturber les processus biologiques régulés par ce récepteur et entraîner des maladies.
En résumé, le récepteur FGFR5 est un membre important de la famille des récepteurs aux facteurs de croissance fibroblastique qui joue un rôle clé dans la régulation de divers processus biologiques et dont les mutations peuvent être associées à certaines maladies humaines.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
Les malformations multiples, également connues sous le nom de malformations congénitales multiples, se réfèrent à la présence de deux ou plusieurs anomalies congénitales affectant différents organes ou systèmes du corps. Ces anomalies sont présentes dès la naissance et peuvent être causées par des facteurs génétiques, environnementaux ou une combinaison des deux.
Les malformations multiples peuvent affecter n'importe quelle partie du corps et peuvent varier en gravité, allant de légères à graves. Elles peuvent également affecter la fonctionnalité des organes touchés et dans les cas les plus sévères, peuvent être fatales.
Les exemples courants de malformations multiples comprennent le syndrome de Down (trisomie 21), qui est caractérisé par un retard mental, une apparence faciale distinctive et souvent d'autres anomalies telles que des problèmes cardiaques congénitaux ; le syndrome de Di George, qui affecte la croissance et le développement et peut causer des problèmes cardiaques, immunitaires et de développement du cerveau ; et le spina bifida, une anomalie de la colonne vertébrale qui peut causer des problèmes de mouvement et de sensation dans les jambes.
Le diagnostic et le traitement des malformations multiples dépendent du type et de la gravité des anomalies présentes. Les soins peuvent inclure une combinaison de chirurgie, de médicaments, de thérapies et de soutien de développement pour aider à gérer les symptômes et améliorer la qualité de vie de l'enfant affecté.
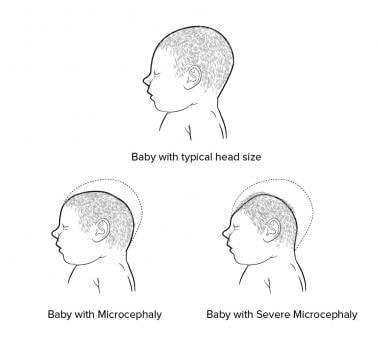
La microcéphalie est une condition médicale dans laquelle le périmètre crânien d'un individu est inférieur à deux écarts-types de la moyenne pour l'âge, le sexe et la race. Cela se traduit généralement par une tête anormalement petite et souvent accompagnée d'un retard mental variable, allant de léger à sévère. La microcéphalie peut être causée par des facteurs génétiques ou environnementaux, tels que des infections maternelles pendant la grossesse, une exposition à des toxines ou un manque d'oxygène pendant le développement du fœtus. Dans certains cas, la cause de la microcéphalie est inconnue. Il s'agit d'une affection rare, et son incidence varie selon les populations et les critères diagnostiques utilisés.
L'acétate de potassium est un composé chimique qui est souvent utilisé dans les applications médicales. Il se compose d'ions potassium (K+) et d'ions acétate (C2H3O2-).
Dans le contexte médical, l'acétate de potassium est souvent utilisé comme source de potassium pour remplacer les électrolytes perdus dans le corps en raison de diverses affections médicales, telles que la diarrhée sévère, les vomissements ou les désordres rénaux. Il peut également être utilisé pour aider à équilibrer les niveaux d'électrolytes dans le sang chez les personnes souffrant d'insuffisance cardiaque congestive ou de cirrhose du foie.
L'acétate de potassium est disponible sous forme de comprimés, de solutions orales et de solutions injectables. Les effets secondaires courants peuvent inclure des nausées, des vomissements, une diarrhée ou des douleurs abdominales. Dans de rares cas, une hyperkaliémie (niveaux élevés de potassium dans le sang) peut survenir, en particulier chez les personnes souffrant d'insuffisance rénale ou ayant d'autres problèmes de santé sous-jacents.
Il est important de suivre les instructions posologiques de votre fournisseur de soins de santé lorsque vous prenez de l'acétate de potassium, car une surdose peut entraîner des effets indésirables graves. Si vous avez des questions ou des préoccupations concernant l'utilisation de ce médicament, il est important de consulter votre fournisseur de soins de santé.
Une délétion chromosomique est un type d'anomalie chromosomique qui se produit lorsqu'une partie d'un chromosome est manquante ou absente. Cela se produit lorsque des segments du chromosome se cassent et que les morceaux perdus ne sont pas correctement réintégrés. Les délétions chromosomiques peuvent être héréditaires ou spontanées, et leur taille et leur emplacement varient considérablement.
Les conséquences d'une délétion chromosomique dépendent de la taille et de l'emplacement de la région déléguée. Les petites délétions peuvent ne provoquer aucun symptôme, tandis que les grandes délétions peuvent entraîner des anomalies congénitales graves, un retard mental et d'autres problèmes de santé.
Les délétions chromosomiques peuvent être détectées avant la naissance par le biais de tests prénataux tels que l'amniocentèse ou le prélèvement de villosités choriales. Les nouveau-nés atteints d'une délétion chromosomique peuvent présenter des caractéristiques physiques uniques, telles qu'un visage allongé, une petite tête, des yeux largement séparés et des oreilles bas situées.
Le traitement d'une délétion chromosomique dépend de la gravité des symptômes et peut inclure une thérapie physique, une thérapie occupationnelle, une éducation spécialisée et d'autres interventions de soutien. Dans certains cas, les personnes atteintes d'une délétion chromosomique peuvent mener une vie relativement normale avec un traitement et un soutien appropriés.
La translocation génétique est un type d'anomalie chromosomique où des segments entiers de deux chromosomes différents changent de place. Il existe deux types principaux de translocations génétiques : les translocations réciproques et les translocations Robertsoniennes.
Les translocations réciproques se produisent lorsque des segments de deux chromosomes différents sont échangés l'un avec l'autre. Ces translocations peuvent être équilibrées, ce qui signifie qu'aucun matériel génétique n'est ni gagné ni perdu dans le processus, ou déséquilibrée, ce qui entraîne une perte ou un gain de matériel génétique.
Les translocations Robertsoniennes, quant à elles, se produisent lorsque la partie distale (la partie la plus éloignée du centromère) de deux chromosomes acrocentriques (qui comprennent les chromosomes 13, 14, 15, 21 et 22) est interchangée, entraînant la fusion des deux chromosomes à leur centromère commun. Cela entraîne la formation d'un seul chromosome avec deux bras courts (p) et aucun bras long (q). Les translocations Robertsoniennes sont le plus souvent équilibrées, mais lorsqu'elles ne le sont pas, elles peuvent entraîner des anomalies génétiques et des troubles du développement.
Les translocations génétiques peuvent être héritées ou spontanées (de novo). Lorsqu'elles sont héritées, elles peuvent être asymptomatiques ou causer des problèmes de santé dépendamment de la façon dont les gènes affectés sont exprimés. Cependant, lorsqu'elles sont spontanées, elles peuvent entraîner des anomalies chromosomiques telles que le syndrome de Down (translocation entre les chromosomes 21 et un autre chromosome) ou le syndrome de Patau (translocation entre les chromosomes 13 et un autre chromosome).
En résumé, les translocations génétiques sont des réarrangements chromosomiques qui peuvent entraîner des problèmes de santé et des anomalies du développement. Elles peuvent être héritées ou spontanées et peuvent affecter n'importe quel chromosome. Les translocations Robertsoniennes sont un type spécifique de translocation qui implique la fusion de deux chromosomes à leur centromère commun, entraînant la formation d'un seul chromosome avec deux bras courts et aucun bras long.
L'intelligence désigne les capacités d'une personne à apprendre, à raisonner, à résoudre des problèmes, à faire preuve de jugement et de pensée abstraite. Un handicap intellectuel, également connu sous le nom de déficience intellectuelle ou retard mental, est un trouble du développement qui affecte ces capacités intellectuelles et la capacité d'une personne à fonctionner de manière indépendante dans la vie quotidienne.
Il est généralement diagnostiqué avant l'âge de 18 ans et peut varier de léger à sévère. Les personnes atteintes de handicap intellectuel peuvent avoir des difficultés à acquérir et à appliquer de nouvelles connaissances, à communiquer efficacement, à prendre soin d'elles-mêmes, à établir des relations sociales et à faire face aux situations stressantes.
Les causes du handicap intellectuel peuvent être génétiques, environnementales ou résulter de complications pendant la grossesse ou la naissance. Il est important de noter que les personnes atteintes de handicap intellectuel ont des capacités et des besoins uniques, et qu'un diagnostic précoce et une intervention appropriée peuvent améliorer considérablement leur qualité de vie et leurs perspectives d'avenir.
Les maladies chromosomiques sont des troubles médicaux causés par des anomalies dans le nombre ou la structure des chromosomes. Les chromosomes sont des structures situées dans le noyau des cellules qui contiennent nos gènes, les unités de base de l'hérédité. Normalement, chaque cellule humaine a 46 chromosomes répartis en 23 paires, sauf les spermatozoïdes et les ovules qui n'en ont qu'une seule de chaque.
Les maladies chromosomiques peuvent résulter d'une absence (délétion), d'un surplus (duplication) ou d'une mauvaise position (translocation) d'un segment chromosomique, ou encore d'un nombre anormal de chromosomes. Par exemple, la trisomie 21, également connue sous le nom de syndrome de Down, est une maladie chromosomique courante causée par la présence d'un chromosome supplémentaire à la paire 21, ce qui donne un total de 47 chromosomes.
Ces anomalies chromosomiques peuvent se produire pendant la formation des ovules ou des spermatozoïdes (méiose) ou pendant le développement embryonnaire (segmentation). Elles peuvent entraîner une grande variété de symptômes, selon la région du chromosome affectée et l'ampleur de l'anomalie.
Les maladies chromosomiques comprennent des affections bien connues telles que le syndrome de Down, le syndrome d'Edwards (trisomie 18), le syndrome de Patau (trisomie 13), la syndactylie (doigts ou orteils collés ensemble) et le Turner et le syndrome de Klinefelter. Ces maladies peuvent entraîner une variété de problèmes de santé, notamment des anomalies physiques, des retards de développement, des déficiences intellectuelles et des problèmes de croissance.
Les malformations de la bouche, également connues sous le nom de fentes labiales et palatines ou simplement fentes orofaciales, sont des anomalies congénitales qui se produisent lors du développement embryonnaire. Elles affectent la formation des lèvres et/ou du palais (toit de la bouche).
Une fente labiale est une ouverture ou une fente dans la lèvre supérieure, qui peut varier en largeur et en position, allant d'une petite encoche à une fente s'étendant jusqu'à la narine. Une fente palatine est une division ou une ouverture dans le palais, ce qui peut affecter la capacité de l'enfant à manger, à parler et à respirer normalement.
Ces malformations peuvent se produire séparément ou ensemble, on les appelle alors des fentes labiales et palatines complètes. Elles sont causées par une combinaison de facteurs génétiques et environnementaux et peuvent être associées à d'autres anomalies congénitales. Le traitement implique généralement une intervention chirurgicale précoce, suivie d'une thérapie de réadaptation pour aider à la correction des problèmes fonctionnels et esthétiques.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
 Syndrome de Wolf-Hirschhorn
Syndrome de Wolf-Hirschhorn
Hirschhorn
Chromosome
Pitt
Chromosome 4 humain
Cassure chromosomique
Hypertélorisme
Microcéphalie
Maladie congénitale
Liste de maladies rares
CIM-10 Chapitre 17 : Malformations congénitales et anomalies chromosomiques
Micrognathie
Syndrome de Wolf-Hirschhorn - Wikipedia
 Syndrome de Wolf-Hirschhorn - Problèmes de santé infantiles - Manuels MSD pour le grand public
Syndrome de Wolf-Hirschhorn - Problèmes de santé infantiles - Manuels MSD pour le grand public
 Utilisation d'équipement de transfert et de manipulation pour faciliter les interventions thérapeutiques | Guldmann France
Utilisation d'équipement de transfert et de manipulation pour faciliter les interventions thérapeutiques | Guldmann France
 6 conseils pour améliorer la qualité du sommeil - yes, therapy helps!
6 conseils pour améliorer la qualité du sommeil - yes, therapy helps!
 Une grande famille unie autour du petit Kelyan - La Région
Une grande famille unie autour du petit Kelyan - La Région
 Le Syndrome de Broadway, Centre d'art contemporain, Pougues-les-Eaux
Le Syndrome de Broadway, Centre d'art contemporain, Pougues-les-Eaux
 Base de données - Soins | LUCAS BALOUP - Avocats à la Cour de Paris
Base de données - Soins | LUCAS BALOUP - Avocats à la Cour de Paris
Coaching et leadership 2023
Présentation des anomalies des chromosomes sexuels - Problèmes de santé infantiles - Manuels MSD pour le grand public
 RESPONSABILITE MEDICALE : caractérisation de la faute - Nicolas Bonnet - Avocat Lyon et Villeurbanne, divorce, droit médical,...
RESPONSABILITE MEDICALE : caractérisation de la faute - Nicolas Bonnet - Avocat Lyon et Villeurbanne, divorce, droit médical,...
Syndrome de Down et anomalies chromosomiques Revues en libre acc&
Base de données - SNIR | LUCAS BALOUP - Avocats à la Cour de Paris
 DeCS
DeCSChromosomique3
- Délétion de la région proximale du bras court du 4 Les autres syndromes à éliminer : Syndrome de Seckel Syndrome CHARGE Syndrome de Smith-Lemli-Opitz Syndrome de Williams Syndrome de Rett Syndrome d'Angelman Syndrome de Smith-Magenis Syndrome de Malpuech Syndrome de Lowry-Maclean L'analyse chromosomique des parents recherchera une translocation intéressant la région critique. (wikipedia.org)
- Présentation des syndromes de délétion chromosomique Les syndromes de délétion chromosomique surviennent lorsqu'une partie d'un chromosome est absente. (msdmanuals.com)
- Le syndrome de Down est une maladie génétique au niveau chromosomique. (longdom.org)
Diagnostic du syndrome1
- Le diagnostic du syndrome de Wolf-Hirschhorn peut être suspecté lors d'analyses chromosomiques avant la naissance ou évoqué par les caractéristiques physiques de l'enfant après la naissance. (msdmanuals.com)
Chromosome4
- Syndrome 4p Délétion 4p Monosomie 4p Syndrome de Pitt-Rogers-Danks 4p- Délétion de la portion distale du bras court du chromosome 4 impliquant la bande 4p16 (région critique WHCR) 75 % des monosomies 4p sont des délétions de novo (chromosome paternel le plus souvent) Translocation familiale est retrouvée chez 5 à 13 % des patients. (wikipedia.org)
- Dans le syndrome de Wolf-Hirschhorn, une partie du chromosome 4 est absente. (msdmanuals.com)
- Journal of Down syndrome and chromosome anomalies est une revue à comité de lecture, au service de la communauté scientifique internationale, offrant une plateforme en libre accès aux auteurs pour publier les résultats de leurs recherches. (longdom.org)
- Journal of Down syndrome and chromosome anomalies est l'une des meilleures revues en libre accès qui vise à publier la source d'information la plus complète et la plus fiable sur les découvertes et les développements actuels sous la forme d'articles originaux, d'articles de synthèse, de rapports de cas, de communications courtes, etc. dans ce domaine et fournir un accès en ligne sans aucune restriction ni abonnement aux chercheurs du monde entier. (longdom.org)
Anomalies1
- Les anomalies des chromosomes sexuels sont fréquentes et provoquent des syndromes associés à divers problèmes physiques et développementaux. (merckmanuals.com)